A Streamlined and Standardized Procedure for Generating High-Titer, High-Quality Adeno-Associated Virus Vectors Utilizing a Cell Factory Platform
In This Article
Summary
As the field of gene therapy continues to evolve, there is a growing need for innovative methods that can address these challenges. Here, a unique method is presented, which streamlines the process of generating high-yield and high-purity AAV vectors using a cell factory platform, meeting the quality standards for in vivo studies.
Abstract
Preclinical gene therapy research, particularly in rodent and large animal models, necessitates the production of AAV vectors with high yield and purity. Traditional approaches in research laboratories often involve extensive use of cell culture dishes to cultivate HEK293T cells, a process that can be both laborious and problematic. Here, a unique in-house method is presented, which simplifies this process with a specific cell factory (or cell stacks, CF10) platform. An integration of polyethylene glycol/aqueous two-phase partitioning with iodixanol gradient ultracentrifugation improves both the yield and purity of the generated AAV vectors. The purity of the AAV vectors is verified through SDS-PAGE and silver staining, while the ratio of full to empty particles is determined using transmission electron microscopy (TEM). This approach offers an efficient cell factory platform for the production of AAV vectors at high yields, coupled with an improved purification method to meet the quality demands for in vivo studies.
Introduction
Adeno-associated virus (AAV) vectors have become an indispensable tool in gene therapy research, offering a unique combination of efficacy and safety for gene delivery1. Traditional methods for generating AAVs in laboratory settings have been pivotal in advancing our understanding and application of gene therapy2. However, these methods, while foundational, exhibit certain limitations and challenges, especially in terms of yield, time efficiency, and the quality of the vectors produced, notably the ratio of full to empty particles3.
The conventional procedure for AAV production primarily involves the transfection of HEK293 cells4. This process, typically conducted in cell culture dishes, requires the cells to be transfected with a plasmid containing the gene of interest along with helper plasmid and AAV capsid plasmid5,6. Following transfection, the cells produce AAV particles, which are then harvested and purified5,6. The purification process often involves ultracentrifugation, a critical step in obtaining high-purity AAV vectors7. Ultracentrifugation, particularly using cesium chloride (CsCl) or iodixanol gradient, is a standard method for separating AAV particles from cellular debris and other impurities8. This step is crucial for achieving the desired purity and concentration of AAV vectors, which directly impacts their efficacy in gene delivery8. Despite its widespread use, traditional ultracentrifugation has its drawbacks. For example, the yield of AAV vectors from this method can be variable and often low, which poses significant challenges when large quantities of high-titer vectors are needed, particularly for in vivo studies or large animal models9.
Another critical aspect of AAV vector quality is the ratio of full to empty particles10. AAV preparations often contain a mixture of these particles; however, only the full particles contain the therapeutic genetic material. The presence of a high proportion of empty particles can significantly reduce the efficiency of gene delivery10. Assessing and optimizing the ratio of full to empty particles is thus a key parameter in evaluating the efficacy of AAV vectors. Traditional methods, while capable of producing AAV vectors, often struggle to control this ratio consistently, leading to variations in vector potency10.
Here, a unique method is presented, which streamlines the process of generating high-yield and high-purity AAV vectors using a cell factory platform free from using labor-intensive HEK293T cell cultures in cell dishes, integrating polyethylene glycol/aqueous two-phase partitioning with iodixanol gradient ultracentrifugation. The AAV vector purity is confirmed via SDS-PAGE and silver staining, and the full-to-empty particle ratio is determined using transmission electron microscopy (TEM), meeting the quality standards for in vivo studies11.
Protocol
The details of the reagents, plasmids, and equipment used in the study are listed in the Table of Materials. The composition of the buffers used is provided in Supplementary File 1.
1. Plasmid preparation
- Transform plasmids (pAAV-GOI, pHelper, pAAV-Cap) in E. coli.
NOTE: Any E.coli strain can be used for plasmid amplification. For some plasmids, special competent cells such as NEB stable can improve the plasmid yield. The three plasmids used in this protocol are pAAV-GOI: pAAV-CMV-GFP, pHelper: pAdDeltaF6, and pAAV-Cap: pAAV-RC6. - Grow bacteria in LB media containing the appropriate selective antibiotics in optimal volume at 37 °C for 16-18 h.
NOTE: When using NEB stable cells, culture at 30 °C for 18-20 h. - Harvest plasmid DNA by centrifuging the E. coli. culture medium at 4000 x g, 4 °C, for 15 min.
- Carefully pour the supernatant liquid from the centrifuge bottle into a waste container and purify the plasmid DNA from the pellet with a large-scale endotoxin-free plasmid purification kit.
NOTE: Ensure to pour it slowly and avoid splashing. - Measure the DNA concentration and purity using a spectrophotometer.
NOTE: Check the DNA purity by checking OD260/OD280 ratio. The ratio for pure DNA is about 1.8. DNA concentration should be above 1 µg/µL for the later co-transfection step. Make bacterial glycerol stocks by adding 500 µL of the overnight culture to 500 µL of 50% glycerol in a cryovial and store at -80 °C.
2. Preparing HEK293T cells
- Seed HEK293T cells with DMEM high glucose media containing 10% FBS into a 10-layer cell factory (CF10) and let the cells grow in the incubator overnight.
NOTE: Passages of the 293T cells should be less than 10 to have the optimal condition for robust AAV production. If possible, do not add antibiotics to the culture media at this time. CF10 has approximately 42 times the growth surface area of a 150 mm cell culture dish. Seed the same cell density in a 150 mm cell culture dish to allow checking the cell growth under a microscope. Prepare 1075 mL cell suspension, add 25 mL cell suspension to a 150 mm dish, and add the remaining into the CF10. - Check cell confluency the next day before the transfection.
NOTE: Cells should reach 80%-90% confluency at the time of transfection by observing under a microscope.
3. Triple-transfection of AAV plasmids
- Calculate the amount of each plasmid needed (pAAV-GOI, pHelper, pAAV-serotype) to have a 1.2: 1: 1 molar ratio with 2.5-5 mg of total DNA per CF10.
NOTE: The three plasmids' molar ratio might be variable for the optimal transfection efficacy. It is better to test it in a small-scale setting before moving to this CF10 setting. pAAV-CMV-GFP, pAdDeltaF6, and pAAV-RC6 are used to make the AAV6-CMV-GFP virus as an example. The PEI calculator formula is listed in Table 1. - Aliquot 350 mL of OptiMEM into a sterile 500 mL bottle.
- Add all three plasmid DNA into the bottle containing the Opti-MEM. Mix well.
- Add 15 mL of 1 mg/mL polyethylenimine (PEI) solution (1:3 µg of DNA to µg PEI ratio). Shake the OptiMEM/DNA/PEI mixture up and down vigorously for 30 s.
NOTE: It is okay to make bubbles. - Incubate the mixture at room temperature for 10-15 min.
NOTE: Longer incubation can reduce transfection efficiency. - Add the OptiMEM/DNA/PEI solution to the 1 L bottle containing 700 mL of DMEM low glucose with 10 mM HEPES and 2% FBS. Mix well.
NOTE: In this study, the low glucose in the DMEM showed better AAV vector production after co-transfection. The addition of HEPES helps to maintain the cells in a better condition. Mix by rotating the bottle slowly. Avoid creating too many bubbles. - Take the CF10 out of the incubator and vacuum the media out into a waste container.
NOTE: Put down the CF10 on the surface inside the hood and gradually lift one side up, slowly vacuum the media out without detaching the cells. - Carefully add the transfectant mixture to the CF10. Ensure that all ten layers are covered with media.
NOTE: Add 25 mL of transfectant mixture into the 150 mm dish. Monitor the cells daily until the harvest. Add the remaining transfectant mixture into the CF10 by pouring slowly. Rotate the CF10 very carefully and ensure equal volume for each layer. - Return the CF10 to the incubator for 72-96 h.
NOTE: Check the 150 mm monitor dish to decide when to harvest the AAV vectors. When the cells become very easily detached with a full load of AAV, it is time to harvest.
4. Harvesting AAV vectors
- Harvest the virus 3-4 days after transfection by shaking the CF10 vigorously. Pour the media into the 250 mL conical tubes and spin down the virus at 4000 x g for 20 min at 4 °C.
NOTE: For the AAV6 serotype used in this protocol, 80% AAV will remain mostly bound to cellular material in the pellet. And the 20% AAV were secreted into the media. These proportions may differ for other AAV serotypes. - Filter clarified supernatant with 0.45 µm filter unit and save it for precipitation.
- Rinse the CF10 with 500 mL of DPBS 1x buffer and centrifuge at 4000 x g for 20 min at 4 °C.
- Discard the supernatant using a vacuum system and an aspirating pipette. Add 20 mL of AAV lysis buffer per CF10 at -80 °C until purification.
NOTE: Transfer AAV lysate to a 50 mL conical tube for later AAV extraction. - Take out the filtered supernatant, add 40% PEG-2.5 M NaCl solution for a final concentration of 8% PEG, and incubate in a cold room on an orbital rotator for at least 3 h or overnight.
NOTE: For 100 mL supernatant, add 25 mL 40% PEG-2.5 M NaCl solution. - Precipitate the virus by centrifuging at 4000 x g for 30 min at 4 °C.
NOTE: Transfer the mixture to 250 mL conical tubes for centrifugation. - Aspirate to remove supernatant and add 5-10 mL of AAV HEPES resuspension buffer to suspend the pellets. Transfer into a 50 mL conical tube to continue the downstream purification, or store it at -80 °C.
5. AAV extraction
- Thaw the virus pellet in a 37 °C water bath for 10-15 min with a shaker.
NOTE: Vortex the AAV lysate to fully melt. - Freeze in -80 °C 100% ethanol bath for 20-30 min.
NOTE: Alternatively, the freeze cycle can be done with a cold 100% ethanol beaker surrounded by dry ice. - Perform freeze/thaw 3-4 times, and vortex in between if necessary.
NOTE: This cycle can be paused at the freeze step. Store the AAV lysate at -80 °C until the downstream work. - Thaw AAV lysate (from step 5.3) and resuspended AAV (from step 4.7)” in a 37 °C water bath for 10-15 min with a shaker, and vortex.
NOTE: Ensure to melt completely and mix it. - Add Benzonase nuclease (250 units/µL) to AAV lysate and resuspended AAV for a final concentration of 50 units/mL. Vortex the solution.
- Incubate in a 37 °C water bath with shaking for 30 min.
NOTE: Take out 10% sodium deoxycholate aliquots in a water bath to allow time to dissolve, warm up, and mix thoroughly. - Add 10% sodium deoxycholate for a final concentration of 0.5% and incubate at 37 °C with a shaker on for 30 min.
NOTE: Sodium deoxycholate was used to enhance AAV release from the cells. - Distribute the raw AAV solution in 2 mL centrifuge tubes and centrifuge at 14,000 x g for 10 min at 4 °C.
- Transfer the supernatant into a 50 mL conical tube.
NOTE: Use a 1 mL pipette to transfer the supernatant. Discard the pellets. - Add an equal amount of chloroform and extract AAV by vortexing for 1-2 min.
NOTE: The solution should become opaque, white, milk-like. - Transfer the milky solution into 2 mL centrifuge tubes and distribute evenly. Centrifuge at 14,000 x g for 10 min at 4 °C.
- Carefully pipette out the top pink layer and transfer it in a new 50 mL conical tube.
NOTE: Do not disturb the chloroform/protein layers. - Measure the final volume using a pipette. According to the raw AAV-PEG-Sulfate volume chart (Table 2), mix appropriate volumes of 50% (NH4)2SO4 and 40% PEG solution. Vortex for 2 min.
- Transfer the mixture to 2 mL centrifuge tubes to distribute evenly. Centrifuge at 14,000 x g for 10 min at 4 °C
- Collect the bottom layer by using a 22 G needle and a 3 mL syringe. Collect the AAV in a 50 mL conical tube and store it at 4 °C for later iodixanol gradients ultracentrifugation.
6. AAV purification by iodixanol gradient ultracentrifugation
- Prepare the iodixanol gradients freshly before loading the AAV according to the number of ultracentrifugation tubes used (Table 3).
NOTE: Phenol red was used to observe the layers. - Overlay each solution into a round-top polypropylene ultracentrifuge quick-seal tube slowly using a 10 mL syringe attached with a 16 G punted long needle. Avoid bubbles.
- Carefully add up to 17 mL of AAV solution on top of the gradient with a 22 G syringe. Use AAV dialysis buffer to top off the tube.
NOTE: Add the AAV solution by loading drops against the wall in the ultracentrifuge tube. Don't disturb the gradient layers. - Seal the ultracentrifuge tubes with an electric sealer.
NOTE: Balance the tubes before sending them to ultracentrifuge with ±0.2 g difference. - Centrifuge at 350,000 x g for 2 h in a Ti70 rotor at 4 °C.
- Carefully remove the tubes from the rotor and place them in the ultracentrifuge tubes rack.
NOTE: Ensure that the gradients are not disturbed. - Collect AAV fractions following the steps below:
- Stabilize the tube on a stand holder.
- Puncture the ultracentrifuge tubes slightly below the 40%-60% interface with a 19 G needle attached to a 10 mL syringe.
NOTE: The opening of the needle should be up, facing the 40% gradient. - Punch a hole in the top of the tube with a 16 G needle.
- Collect up to 5 mL per tube. Avoid collecting the protein contamination at the 25%-40% interface.
- Repeat for each ultracentrifuge tube.
NOTE: Transfer the AAV fractions to a 50 mL conical tube.
7. Second round iodixanol gradients ultracentrifugation
NOTE: This step is optional. This step is to reduce the empty AAV ratio for a higher quality full AAV capsid.
- Dilute AAV >1:1 with AAV dialysis buffer.
NOTE: AAV collected from the first round of ultracentrifugation was at 40% iodixanol layer. It should be diluted by at least 50% to be able to be loaded on top of the 30% iodixanol layer. - Overlay each solution (Table 4) into an ultracentrifugation tube using a 16 G punted needle and slowly attach a 10 mL syringe. Avoid bubbles.
- Carefully add 20 mL of diluted AAV solution on top of the gradient with a 22 G syringe. Use AAV dialysis buffer to top off the tube.
- Repeat the steps 6.4 to 6.7.
8. AAV dialysis and concentration
- Transfer the AAV virus into the dialysis cassette using a new 18 G needle and 10 mL syringe. Slowly inject the virus into the cassette and remove air from it.
- Add a stir bar to the beaker containing AAV dialysis buffer and place it on a stir plate.
- Put the dialysis cassette into the dialysis buffer with a float buoy. Stir at 4 °C. Change 2-3 times the AAV dialysis buffer.
NOTE: Use enough buffer to allow the cassette to float and perform the buffer exchange. Cover the beaker with aluminum foil. - Using a 10 mL syringe attached with an 18 G needle, collect the virus from the cassette into a centrifugal filter unit.
- Centrifuge at 4,000 x g for 15-30 min at 4 °C.
NOTE: Rince the AAV with AAV dialysis buffer 2-3 times. Concentrate the AAV until ~200 µL of remaining is on top of the filter. - Collect the concentrated AAV from the filter unit. Rinse the filter with another 300 µL of AAV dialysis buffer and transfer it to the concentrated AAV.
- Filter the virus through a 0.22 µm syringe filter using a 1 mL tuberculin luer slip syringe. To ensure the least amount of volume loss, use the 10 mL syringe to push air through the filter 2-3 times.
- Store the purified AAV virus at 4 °C temporally for titration.
NOTE: Titrate the virus, aliquot, and store at -80 °C within 1 week.
9. AAV virus titration
NOTE: TaqMan quantitative polymerase chain reaction (qPCR) was used to titrate purified AAV.
- Treat AAV vectors with DNase I and incubate at 37 °C for 30 min, then incubate at 95 °C for 10 min.
NOTE: As an example of 10 µL reaction, 2 µL AAV virus sample, 1 µL DNase, 1 µL DNase buffer, and 6 µL H2O is used. It can be performed in a thermocycler with PCR tubes. - Prepare the primer sets targeting the AAV insert region.
NOTE: Targets could be the promoter, transgene, and reporter in the AAV construct. The primers are listed in Table 5. - Prepare DNA plasmid standards (10, 1, 0.1, 0.01, 0.001, and 0.0001 pg/µL) and a negative control (H2O).
- Prepare the qPCR master mix, including primers, according to the manufacturer's instructions.
- Place the standards and samples in triplicate on a PCR plate. Add the qPCR mix into the standards and samples. Seal the plate and perform the qPCR reaction according to the manufacturer's instructions.
NOTE: The reaction conditions depend on the materials/reagents and instrument. Fast and conventional PCR cycles are applicable. - Calculate AAV virus titer.
NOTE: The sample concentration in this protocol is 1/10 dilution from the original samples. - Aliquot the AAV virus in 1.5 mL tubes and store at 4 °C for up to one month and store at -80 °C for the long term.
NOTE: Avoid freeze/thaw cycles for the storage. When used for in vivo experiments, do not use thaw over twice aliquots.
10. Quality control of AAV
NOTE: AAV viruses were characterized for purity by SDS-PAGE silver stain using an SDS-PAGE gel and stained using a commercially available staining kit.
- Denature 3 x 109 vg of AAV virus sample with Laemmli buffer.
NOTE: Use a reference AAV virus as a control. The AAV6-CMV-GFP reference full capsid is used. - Load the denatured AAV on a 4%-20% gradient SDS-PAGE gel and run at 180 V for 50 min.
- Remove the gel from the electrophoresis plate. Stain the gel with a silver staining kit according to the manufacturer's instructions.
- Check the AAV capsid proteins VP1, VP2, and VP3.
NOTE: Pure AAV virus contains only VP1, VP2, and VP3 capsid protein. If not pure, other protein bands would be visible on the gel.
NOTE: Transmission electron microscopy (TEM) of negative-stained recombinant AAV virions was performed to assess morphological integrity and full/empty ratio. - Pick up an EM grid with a tweezer and set down on the bench with the shiny side of the grid facing up.
- Pipet 5 µL of AAV sample onto the grid and allow it to dry by evaporation.
NOTE: Dilute the AAV if needed. 1012 vg/mL is an average titer. It might take 30-60 min to dry the grid. - Wash the grid by pipetting, drop by drop, with about 200 µL of H2O onto the grid.
- Remove the excess water by slowly placing a chromatography paper vertically next to the grid.
- Pipet 5 µL of 2% Uranyl Acetate solution onto the grid. Incubate for 5 min and wick off as mentioned above. Allow the grid to dry.
- Visualize the AAV particles under a transmission electron microscope (at 50,000-fold magnification). Viral capsids with a viral genome will appear as homogeneous white hexagons, while empty capsids will appear as hexagons with a white rim but a dark center.
- Count randomly at least 100 particles to determine the approximate percentage of full vs. empty AAV particles.
Representative Results
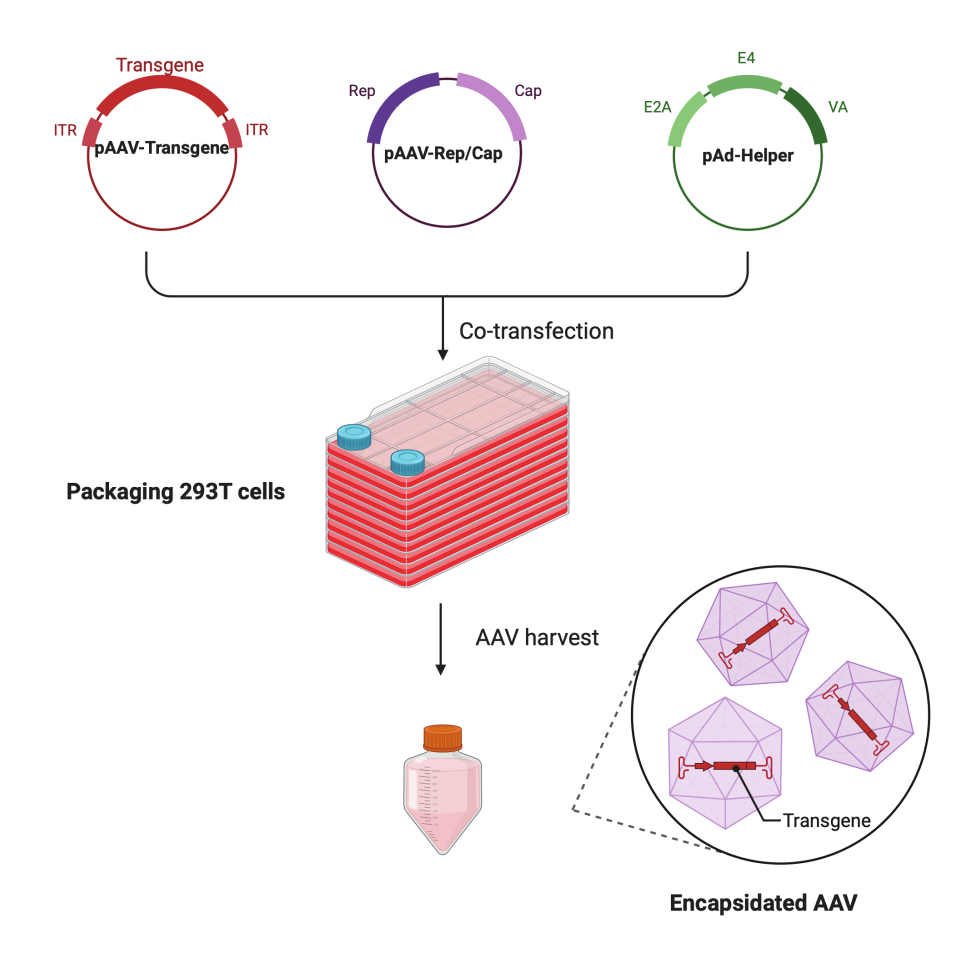
In this detailed step-by-step protocol, a standardized platform is demonstrated to make high-titer and high-quality AAV virus with the CF10 in a large-scale research lab setting. Compared with conventional cell culture dishes, the CF10 provides a convenient way to culture large amounts of cells and produce AAV virus (Figure 1). Several culture conditions were tested to determine whether cells in an optimal environment can promote viral production. A low glucose DMEM supplemented with 10 mM HEPES and 2% FBS showed the best AAV production.
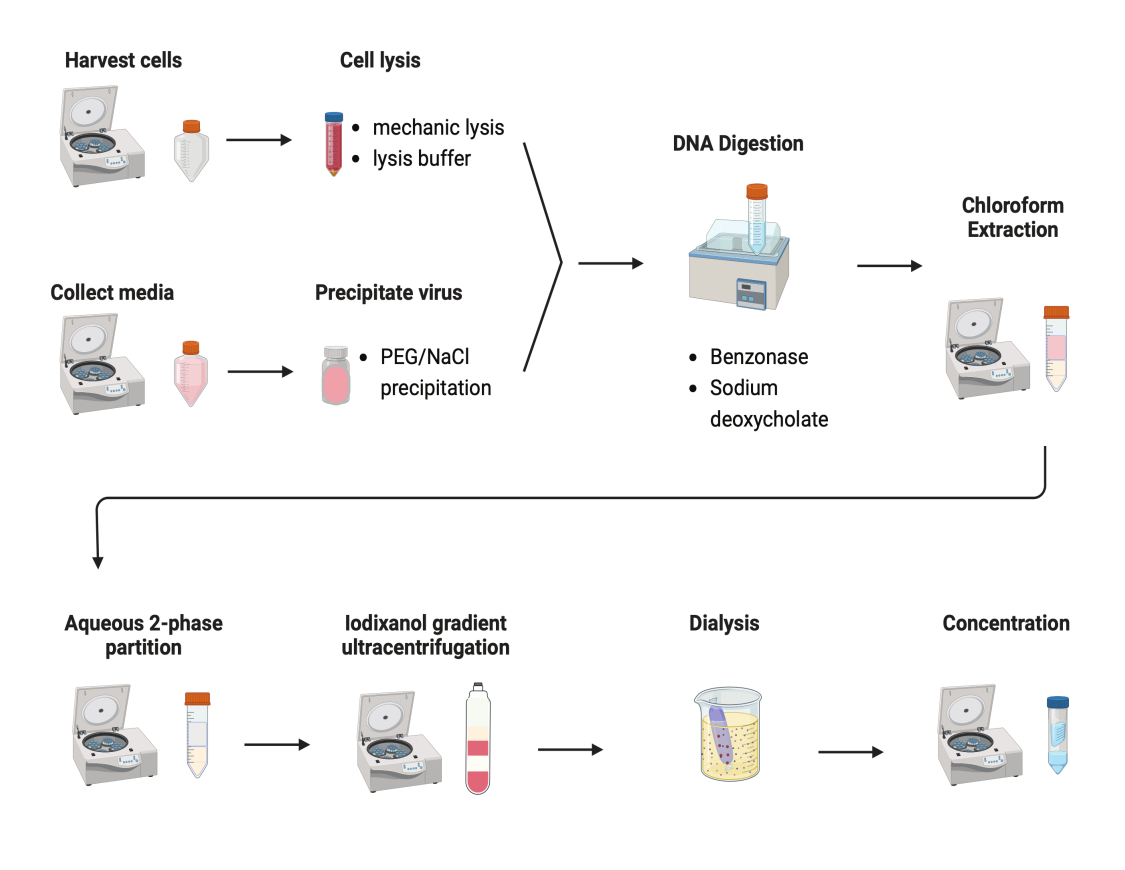
Several protocols were tested to purify AAVs. Most procedures have low virus yields and impurity of AAV capsids. Here, a revised purification protocol was developed, combining the AAV from both cell pellets and culture media (Figure 2). We have found that 80% of AAV was in the cells, and another 20% of AAV was in the culture media, which were secreted from the cells. Both parts of AAV were treated with DNase to remove the free DNA. Sodium deoxycholate was used to further release AAV from the cells. AAVs were then extracted with chloroform extraction followed by an aqueous two-phase partitioning. These steps allowed most protein contaminants to be removed. AAV remains soluble in the ammonium sulfate phase.
The remaining contaminants were removed with a discontinuous iodixanol gradient ultracentrifugation (Figure 3A). The gradient was also helpful in removing the empty AAV capsids, especially with the second round of iodixanol gradient ultracentrifugation.
The purity of the AAV virus was determined by silver staining. When three major bands corresponding to AAV capsid protein, VP1, VP2, and VP3, with a purity greater than 90%, were obtained, the AAV virus was suitable for in vivo use (Figure 3B). The AAV full capsid to empty capsid ratio was accessed by TEM (Figure 3C). Only a full capsid with a transgene insert would allow the expression of transgene in the targeted tissue. A high portion of the empty capsid could also induce an immune response to the AAV capsid. These quality checks are necessary for each AAV virus that is produced and purified before use.

Figure 1: Schematic illustration of the AAV production by HEK293T cells triple-transfection method. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Schematic illustration of the AAV purification. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: AAV purification by iodixanol gradient ultracentrifugation and AAV virus purity verification. (A) Iodixanol gradient layers and the position of the needle for harvesting. (B) Representative gel assessing capsid content and purity. M: molecular marker; R: reference AAV6 full capsid; S1: in-house made AAVDJ capsid with one round of ultracentrifugation; S2: in-house made AAVDJ capsid with two rounds of ultracentrifugation; S3: in-house made AAV6 capsid. (C) Electron microscopy image of AAV. Virus collected after purification. Viral capsids containing a viral genome appear as homogeneous white hexagons, while empty capsids appear as hexagons with a white rim but a dark center. Scale bar: 100 nm. Please click here to view a larger version of this figure.
{kind=link}
Table 1: PEI calculator for AAV packaging. Please click here to download this Table.
Table 2: Raw AAV-PEG-Sulfate volume chart. Please click here to download this Table.
Table 3: Preparation of iodixanol gradient. Please click here to download this Table.
Table 4: Preparation of second round iodixanol gradient. Please click here to download this Table.
Table 5: AAV titration primers. Please click here to download this Table.
Supplementary File 1: Compositions of the buffers used for the study. Please click here to download this File.
Discussion
Here, a technically advanced protocol for large-scale production of high-titer and high-quality AAV vectors using a cell factory platform (CF10) is introduced, representing a significant improvement over conventional cell culture dish methods. The use of cell factories simplifies the process of cultivating large volumes of cells, facilitating the production of AAV viruses more efficiently1. Also, by optimizing the culture conditions, particularly with low glucose DMEM supplemented with 10 mM HEPES and 2% FBS, a significantly enhanced viral production was confirmed, indicating the crucial role of the cellular environment in virus yield.
The revised purification protocol, which combines AAV from both cell pellets and culture media, addresses the common issue of low virus yields and impurity seen in many existing protocols. The steps of chloroform extraction and aqueous two-phase partitioning effectively remove most protein contaminants, with AAV remaining soluble in the ammonium sulfate phase. The improvement in both the yield and purity of AAV vectors using the PEG/aqueous two-phase partitioning combined with iodixanol gradient ultracentrifugation, as opposed to traditional gradient ultracentrifugation methods, may be attributed to enhanced initial separation with PEG/aqueous two-phase partitioning, refined purity with iodixanol gradient ultracentrifugation and reduction in contaminant co-purification12. First, the introduction of PEG/aqueous two-phase partitioning before ultracentrifugation significantly improves the initial separation of AAV particles from cellular debris and other contaminants. PEG, a high molecular weight polymer, when mixed with an aqueous solution, creates two distinct phases13. AAV vectors have a propensity to partition preferentially into one of these phases (commonly the PEG-rich phase), while many contaminants and impurities are partitioned into the other13. This selective partitioning effectively concentrates the AAV particles and removes a substantial portion of impurities even before ultracentrifugation, thereby increasing the yield and reducing the contaminant load entering the ultracentrifugation step13. Second, iodixanol gradient ultracentrifugation further refines the purity of AAV vectors. Iodixanol, a non-ionic, iso-osmolar gradient medium, allows for a more gentle and controlled separation compared to traditional CsCl gradients14. In this gradient, AAV particles migrate to a position in the gradient that corresponds to their buoyant density14. Importantly, this process is effective in separating full AAV capsids (containing the genetic payload) from empty capsids (lacking genetic material), which is a crucial determinant of vector quality. Iodixanol's iso-osmolar nature also preserves the integrity of the AAV capsids better than hyperosmolar agents like CsCl, potentially leading to higher yields of intact, functional vectors14. Finally, traditional ultracentrifugation methods, especially those using CsCl gradients, can sometimes co-purify contaminants that have similar buoyant densities to AAV vectors15. By using PEG partitioning as a preliminary step, the load of such contaminants is greatly reduced before ultracentrifugation13. This reduction in contaminant load means that the iodixanol gradient can work more effectively and selectively in purifying AAV vectors, leading to higher purity15.
The purity and quality of the AAV vectors are rigorously assessed through silver staining and TEM11. The observation of three major bands corresponding to AAV capsid proteins VP1, VP2, and VP3, with a purity exceeding 90%, indicates the suitability of these AAV vectors for in vivo use. The TEM analysis for determining the full-to-empty capsid ratio is particularly crucial, as a high proportion of empty capsids can lead to reduced gene delivery efficiency and potential immune responses11. This quality check, although essential, adds to the procedural complexity and may require additional technical expertise.
In conclusion, the protocol offers significant technical advancements in the production of AAV vectors, particularly in terms of scalability and purity. However, the complexities associated with the purification process and the need for specialized equipment and expertise may still be a minor limitation for its application in certain research settings. Further refinement and simplification of these techniques could make this approach more accessible and widely applicable in the field of gene therapy research.
Acknowledgements
TZ designed the experiments. TZ, VD, SB, and JP performed the experiments. TZ and VD generated data and analyzed the data. TZ and YX wrote the manuscript. TZ and GG revised the manuscript. This work was supported by UPMC Children's Hospital of Pittsburgh.
Materials
| Name | Company | Catalog Number | Comments |
| 293T/17 cells | ATTC | CRL-11268 | |
| (NH4)2SO4 | Millipore Sigma | 1.01217.1000 | |
| 0.5 M EDTA | MilliporeSigma | 324506-100ml | |
| 1 mL Henke-Ject syringe | Fisher Scientific | 14-817-211 | |
| 10% pluronic F68 solution | Fisher Scientific | 24-040-032 | |
| 10x Tris/Glycine/SDS Buffer | Biorad | 1610732 | |
| 1M HEPES | Fisher Scientific | 15-630-080 | |
| 2% Uranyl Acetate Solution | Electron Microscopy Sciences | 22400-2 | |
| 4%–20% Precast Protein Gels | biorad | 4561094 | |
| 40% PEG solution | Sigma | P1458-50ML | |
| AAV6 reference full capsids | Charles River Laboratories | RS-AAV6-FL | |
| Accutase Cell Detachment Solution | Fisher Scientific | A6964-100ML | |
| Benzonase | Sigma | E1014-25KU | |
| BioLite Cell Culture Treated Dishes 150 mm | Fisher Scientific | 12-556-003 | |
| Centrifugal Filter Unit | MilliporeSigma | UFC905024 | |
| Corning PES Syringe Filters | Fisher Scientific | 09-754-29 | |
| Dialysis Cassettes, 10 K MWCO | Fisher Scientific | PI66810 | |
| Disposable PES Filter Units 1 L 0.2 µm | Fisher Scientific | FB12566506 | |
| Disposable PES Filter Units 1 L 0.45 µm | Fisher Scientific | FB12566507 | |
| Disposable PES Filter Units 500 mL 0.2 µm | Fisher Scientific | FB12566504 | |
| DMEM high glucose | Fisher Scientific | 10-569-044 | |
| DMEM low glucose | Fisher Scientific | 10567022 | |
| DNase | NEB | M0303S | |
| DPBS 1x | Fisher Scientific | 14-190-250 | |
| Fetal Bovin Serum (FBS) | Biowest | S1620 | |
| Formvar/Carbon 300 Mesh, Cu | Electron Microscopy Sciences | FCF300-Cu-50 | |
| glycerol | Sigma | G5516-1L | |
| KCl | Sigma | P9541-500G | |
| LB agar | Sigma | L2897-250G | |
| LB broth | Fisher Scientific | BP9732-500 | |
| MgCL2·6H2O | Sigma | M9272-100G | |
| NEB stable competent cells | NEB | C3040H | |
| Nest Biofactory 10 chamber | MidSci | 771302 | |
| NucleoBond Xtra Maxi EF | Macherey-Nagel | 740424 | |
| Opti-MEM | Fisher Scientific | 31-985-088 | |
| OptiPrep Density Gradient Medium | Millipore Sigma | D1556-250ml | |
| pAAV-CMV-GFP | Addgene | 105530 | |
| pAAV-DJ | Cell BioLab | VPK-420-DJ | |
| pAAV-RC6 | Cell BioLab | VPK-426 | |
| pAdDeltaF6 | Addgene | 112867 | |
| PEG 8000 | Promega | V3011 | |
| PEI Max | Polysciences, Inc | 49553-93-7 | |
| Pen-Strep | Fisher Scientific | 15-140-163 | |
| Phenol red | Millipore Sigma | 1.07242.0100 | |
| Pierce Silver Stain Kit | Thermo Fisher Scientific | 24612 | |
| QuickSeal tube | Fisher Scientific | NC9144589 | |
| Sodium Chloride | Sigma | 1162245000 | |
| sodium deoxycholate | Millipore Sigma | D6750-100G | |
| Taqman Fast Advanced Master Mix | Thermo Fisher Scientific | 4444557 | |
| Type 70 Ti Fixed-Angle Titanium Rotor | Beckman Coulter | 337922 | |
| Western Blotting Substrate | ThermoFisher | 32209 |
References
- Arjomandnejad, M., Dasgupta, I., Flotte, T. R., Keeler, A. M. Immunogenicity of recombinant adeno-associated virus (AAV) vectors for gene transfer. BioDrugs. 37 (3), 311-329 (2023).
- Liu, Y., Siriwon, N., A Rohrs, J., Wang, P. Generation of targeted adeno-associated virus (AAV) vectors for human gene therapy. Curr Pharm Des. 21 (22), 3248-3256 (2015).
- Bilal, A. S., et al. Optimization of large-scale Adeno-Associated Virus (AAV) production. Curr Protoc. 3 (5), e757 (2023).
- Rashnonejad, A., Chermahini, G. A., Li, S., Ozkinay, F., Gao, G. Large-scale production of adeno-associated viral vector serotype-9 carrying the human survival motor neuron gene. Mol Biotechnol. 58, 30-36 (2016).
- Challis, R. C., et al. Systemic AAV vectors for widespread and targeted gene delivery in rodents. Nat Protoc. 14 (2), 379-414 (2019).
- Challis, R. C., et al. Publisher Correction: Systemic AAV vectors for widespread and targeted gene delivery in rodents. Nat Protoc. 14 (8), 2597-2597 (2019).
- Mueller, C., Ratner, D., Zhong, L., Esteves-Sena, M., Gao, G. Production and discovery of novel recombinant adeno-associated viral vectors. Curr Protoc Microbiol. 26 (1), 14 (2012).
- Guo, P., Wiersch, J., Xiao, X., Gittes, G. Simplified purification of AAV and delivery to the pancreas by intraductal administration. Methods Mol Biol. 1950, 373-387 (2019).
- Berns, K. I., Srivastava, A. Next generation of adeno-associated virus vectors for gene therapy for human liver diseases. Gastroenterol Clin North Am. 48 (2), 319-330 (2019).
- Khasa, H., Kilby, G., Chen, X., Wang, C. Analytical band centrifugation for the separation and quantification of empty and full AAV particles. Mol Ther Methods Clin Dev. 21, 585-591 (2021).
- Chen, H. Comparative observation of the recombinant adeno-associated virus 2 using transmission electron microscopy and atomic force microscopy. Microsc Microanal. 13 (5), 384-389 (2007).
- Burnham, B., et al. Analytical ultracentrifugation as an approach to characterize recombinant adeno-associated viral vectors. Hum Gene Ther Methods. 26 (6), 228-242 (2015).
- Kato, M., et al. In situ-formable, dynamic crosslinked poly (ethylene glycol) carrier for localized adeno-associated virus infection and reduced off-target effects. Commun Biol. 6 (1), 508 (2023).
- Sena-Esteves, M., Gao, G. Enrichment of fully packaged virions in column-purified recombinant adeno-associated virus (rAAV) preparations by iodixanol gradient centrifugation followed by anion-exchange column chromatography. Cold Spring Harb Protoc. 2020 (2), 095638 (2020).
- Matsumoto, M., Wangelin, J. R., Murphy, M. L. Purification of avian encephalomyelitis virus by ultracentrifugation in a nonlinear cesium chloride gradient. Avian Dis. 22 (3), 496-502 (1978).
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved