
Method Article
Isolation and Identification of Porcine Bone Marrow Mesenchymal Stem Cells and their Derived Extracellular Vesicles
In This Article
Summary
This article elaborates a method to isolate and identify porcine bone marrow mesenchymal stem cells (pBM-MSCs) and extracellular vesicles (EVs) derived from them, providing a methodological basis for the pre-clinical evaluation of transplantation efficacy of BM-MSCs and their derived EVs.
Abstract
With the development of stem cell therapy in translational research and regenerative medicine, bone marrow mesenchymal stem cells (BM-MSCs), as a kind of pluripotent stem cells, are favored for their instant availability and proven safety. It has been reported that transplantation of BM-MSCs is of great benefit to repairing injured tissues in various diseases, which might be related to modulating the immune and inflammatory responses via paracrine mechanisms. Extracellular vesicles (EVs), featuring a double-layer lipid membrane structure, are considered to be the main mediators of the paracrine effects of stem cells. Recognized for their crucial roles in cell communication and epigenetic regulation, EVs have already been applied in vivo for immunotherapy. However, similar to its maternal cells, most of the studies on the efficacy of transplantation of EVs still remain at the level of small animals, which is not enough to provide essential evidence for clinical translation. Here, we use density-gradient centrifugation to isolate bone marrow cells (BMC) from porcine bone marrow at first, and get porcine BM-MSCs (pBM-MSCs) by cell culture subsequently, identified by the results of observation under the microscope, induced differentiation assay, and flow cytometry. Furthermore, we isolate EVs derived from pBM-MSCs in cell supernatant by ultracentrifugation, proved by the techniques of transmission electron microscopy (TEM), nanoparticle tracking analysis (NTA), and western blotting successfully. Overall, pBM-MSCs and their derived EVs can be isolated and identified effectively by the following protocols, which might be widely used in pre-clinical studies on the transplantation efficacy of BM-MSCs and their derived EVs.
Introduction
Over the past 10 years, stem cell therapy has promised great benefits for patients suffering from a variety of diseases and injuries, such as trauma, respiratory, and cardiovascular diseases. With progress in the field, bone marrow mesenchymal stem cells (BM-MSCs) are gradually being favored by people for their accessibility and few ethical disputes1, which have been considered the gold standard for clinical research despite other cell types2. Therapies based on BM-MSCs are also attractive to more and more researchers due to their unique ability to modulate immune and inflammatory responses and repair injured tissues via differentiation or paracrine mechanisms3.
Extracellular vesicles (EVs), as the International Society for Extracellular Vesicles (ISEV) endorses4, refer to the total particles with a lipid bilayer structure that are naturally released from cells. With the recent discoveries of various contents such as proteins, lipids, and genetic materials (e.g., miRNA, mRNA, DNA molecules, as well as long noncoding RNAs) in EVs from different cell types5, their crucial roles in cell communication and epigenetic regulation have been recognized6. As a novel substitute for maternal cells, EVs have been applied in immunotherapy and regenerative medicine with studies in vivo, which serve as the basis for the ongoing pre-clinical research and follow-up clinical trials7.
However, at present, most of the studies on the efficacy of transplantation of BM-MSCs and their derived EVs still remain at the level of small animals, which are not enough to provide the necessary evidence for clinical translation. Consequently, it is extremely urgent to carry out pre-clinical research on the transplantation of BM-MSCs and their derived EVs at the level of large animals such as swine.
It has been reported that MSCs are present in extremely low numbers in the bone marrow, accounting for only 0.01% to 0.001% of the total cells8. However, pre-clinical administration of BM-MSCs requires a large number of cells (≥107 per animal)9; the amount of EVs required is even greater, the median dose of which is 0.25 mg of protein per kilogram bodyweight in swine10. To achieve these large numbers, there is an urgent need for a safe and effective method to isolate and culture MSCs from porcine bone marrow to achieve their massive expansion in vitro and acquire their EVs with high protein concentration subsequently.
So far, there are various methods for isolating BM-MSCs and their derived EVs. The current methods for isolating BM-MSCs include direct planting of bone marrow cells (BMCs)11, density-gradient centrifugation, cell surface molecular label sorting, and flow cytometry screening. It has been reported that the cell surface molecular label sorting and flow cytometry screening result in a decrease in cell adhesion rate, an increase in 24 h mortality, and proliferation inhibition12, while direct culture of BMCs can result in a high number of mixed hematopoietic cells. Therefore, density-gradient centrifugation is now commonly used to obtain BM-MSCs. Current methods for isolating EVs from cell supernatants include ultracentrifugation, ultrafiltration, polymer precipitation, and size exclusion13. Compared with other methods, ultracentrifugation has the advantage of low cost, ease of use, and compatibility with large volume preparation without complicated pretreatment, which has been the "gold standard" for EV separation14. However, a large heterogeneity exists in reagents and techniques across different labs during the process3,15, which might be misleading to readers. This article explains a series of sequential steps to isolate pBM-MSCs and EVs derived from them in detail, and subsequent identification results prove that the method is feasible to obtain pBM-MSCs and their EVs for further analysis in pre-clinical research. We hope this systematic work can provide a methodological basis for researchers engaged in the pre-clinical evaluation of transplantation of pBM-MSCs and their derived EVs, so that clinical trials can be carried out as soon as possible.
Protocol
According to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health, USA, all the experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC), Fuwai Hospital, Chinese Academy of Medical Sciences.
1. Preoperative preparation for the animals
- Obtain adult male Chinese minipigs (30 ± 5 kg) at about 12 months old from the Institute of Zoology, Chinese Academy of Sciences, and house them in the facilities of Animal Experimental Center, Fuwai Hospital, at least 2 weeks in advance. Carry out preoperative inspections such as blood routine examinations to ensure that the animals are healthy.
- Clean and shave the skin of the thigh area of the minipig the day before the operation. To avoid aspiration, fast the minipig for 12 h before bone marrow extraction.
2. Preparation for cell isolation and cultivation
- Use Percoll (1.130 g/mL) to isolate mesenchymal stem cells from bone marrow. Mix the stock solution with 10x concentrated PBS at a ratio of 9:1 to obtain an isotonic medium. Then mix the isotonic medium with PBS at a ratio of 3:2 to acquire 60% Percoll solution (1.077 g/mL), which can be used as the final separating solution to isolate pBM-MSCs.
- Prepare Iscove's Modified Dulbecco's Medium (IMDM) with 10% fetal bovine serum and 1.0% penicillin-streptomycin to obtain a complete medium. Preheat the complete medium and PBS in a 37 °C water bath for subsequent cell cultivation.
3. Anesthetization for the animals
- Administer general anesthesia with ketamine (10 mg/kg) and xylazine (2 mg/kg) intramuscularly. Carry out endotracheal intubation quickly when the minipig has slow respiration and fewer limb activity, and preserve spontaneous breathing to prevent failed intubation.
- Perform maintenance of anesthesia by inhalation of 2% isoflurane, with oxygen (1.5 L/min) as the carrier gas. During anesthesia, monitor heart rate, respiration, and blood oxygen saturation of the minipig in real-time.
4. Extracting bone marrow from the minipig
- Place the minipig in a lateral position. To ensure a sufficient amount of bone marrow for subsequent cell culture, locate the bone marrow puncture point at the proximal femur of the minipig (Figure 1). Disinfect and drape the skin of the puncture area before the operation is applied.
- Tighten the skin around the puncture point with the non-dominant hand, and pierce the bone marrow biopsy needle vertically at the point with the dominant hand. When feeling the needle in contact with the cortical bone, gently rotate the handle left and right to drill the needle. When the needle enters the bone marrow cavity, there is often a feeling of losing resistance.
- After the puncture is in place, withdraw the core needle. Then attach a disposable sterile 50 mL syringe to the end of the outer needle, and rinse the inner wall of the syringe with heparin in advance.
- Extract 20 mL of bone marrow slowly and transfer them to a sterile 50 mL centrifuge tube carefully.
- When the procedure is complete, pull out the needle and remove the drape. Disinfect the puncture site and press it for 20 min for hemostasis.
- Extubate the minipig after restoring spontaneous breathing. When it is fully awake with free movement of limbs, return the minipig to the cage to continue feeding.
5. Isolating mesenchymal stem cells from the bone marrow
- Add an equal volume of preheated PBS to the bone marrow and mix them thoroughly. Then use a sterile pipette to transfer 20 mL of the diluted bone marrow above the 60% density gradient solution level at a 1:1 volume ratio in a sterile 50 mL centrifuge tube carefully.
- Centrifuge the tube at 600 x g ( acceleration (ACC) = 5, deceleration (DEC) = 5) for 20 min at room temperature (RT).
NOTE: Four phases are formed in the tube after centrifugation, including the serum phase, mononuclear cell phase, density gradient medium phase, and precipitation phase from top to bottom. The mononuclear cell phase is a thin flocculent layer between the serum and the density gradient medium. Mesenchymal stem cells derived from the bone marrow are located in this phase. - Draw the mononuclear cell phase into a sterile 15 mL centrifuge tube, and wash twice with PBS at 800 x g for 5 min.
- Resuspend the washed cells in 2 mL of complete medium and plant the resuspended cells in a 175 cm2 cell culture flask at a density of 3-5 x 105/mL.
NOTE: All the above procedures for isolating mesenchymal stem cells can be seen in Figure 2.
6. Cultivating mesenchymal stem cells in vitro
- Incubate the culture flask at 37 °C in a saturated humidified atmosphere of 5% CO2. Shake the culture flask gently every 24 h to prevent adherent growth of the precipitated hematopoietic stem cells, and observe cell growth, morphology, and contamination under a microscope.
- Replace the culture medium for the first time after 3 days, and then change the medium every 2-3 days. When the cell colonies reach 80%-90% confluence, subculture the cells at a ratio of 1:2.
7. Adipogenic, osteogenic, and chondrogenic differentiation of pBM-MSCs
- Adipogenic differentiation assay
- Prepare adipogenic differentiation medium A (Medium A) and B (Medium B) for BM-MSCs according to the kit instructions. Specific information about the kit can be found in the Table of Materials.
- Add 1 mL of 0.1% gelatin to the six-well plate and shake gently so that it can cover the bottom of each well evenly. Then place the six-well plate in a clean bench or CO2 incubator for at least 30 min.
- After 30 min, aspirate the gelatin and add 2 mL of general complete medium to each well. Then plant the pBM-MSCs in the six-well plate at a cell density of 2 x 104 cells/cm2. After that, incubate the plate at 37 °C in saturated humidity of 5% CO2.
- When the cells reach 100% confluence, remove the complete medium carefully and add 2 mL of Medium A to each well of the plate. After 3 days, remove Medium A from the plate and add 2 mL of Medium B to each well.
- After maintaining for 1 day, remove Medium B and replace it with Medium A for induction. According to the manner of "Medium A for 3 days, Medium B for 1 day", use Medium A and B sequentially for induction.
- Observe the cell state every day during the period. If the cells shrink or die during the induction process of Medium A, replace it with Medium B in time until the cell state recovers.
- Repeat the induction and maintenance process, and prepare for staining when sufficient lipid droplets of suitable size are observed under the microscope.
- Remove the medium for adipogenic differentiation in the six-well plate, and wash with 1x PBS gently. Add 2 mL of 4% paraformaldehyde solution to each well and fix for 30 min at RT.
- Remove the paraformaldehyde fixative and wash with 1x PBS two or three times to ensure that the fixative is removed thoroughly. Add 2 mL of Oil Red O dye to each well and stain for 30 min at RT.
- Remove the Oil Red O dye and wash with 1x PBS two or three times. Add 2 mL of 1x PBS to each well, and then observe the effect of adipogenic differentiation under a microscope.
- Osteogenic differentiation assay
- Prepare the complete medium for osteogenic differentiation according to the kit instructions.
- Follow steps 7.1.2-7.1.3 to plant and culture pBM-MSCs.
- When the cells reach 70% confluence, remove the general complete medium carefully and add 2 mL of medium for osteogenic differentiation to each well of the plate.
- Change to fresh osteogenic differentiation medium every 3 days. Continue the induction for 2-4 weeks, and prepare to stain with Alizarin Red dye when obvious calcium nodules appear during osteogenesis.
- Remove the medium for osteogenic differentiation in the six-well plate, and wash with 1x PBS gently. Add 2 mL of 4% paraformaldehyde solution to each well and fix for 30 min at RT.
- Remove the paraformaldehyde fixative and wash with 1x PBS two or three times to ensure that the fixative is cleaned thoroughly. Add 2 mL of Alizarin Red dye to each well and stain for 10 min at RT.
- Remove the Alizarin Red dye and wash with 1x PBS two or three times. Add 2 mL of 1x PBS to each well, and then observe the effect of osteogenic differentiation under a microscope.
- Chondrogenic differentiation assay
- Prepare the premix for chondrogenic differentiation according to the kit instructions.
- Transfer 3-4 x 105 pBM-MSCs to a sterile 15 mL centrifuge tube. Centrifuge at 250 x g for 4 min at 20 °C.
- Remove the supernatant and add 0.5 mL of premix to resuspend the pellet obtained by centrifugation in the previous step, and then centrifuge at 150 x g for 5 min at 20 °C. Repeat this step to wash the cells again.
- Prepare the complete medium for chondrogenic differentiation.
- Resuspend the cell obtained in the previous step with 0.5 mL of complete medium and centrifuge at 150 x g for 5 min at 20 °C.
- Unscrew the cap of the centrifuge tube to facilitate gas exchange. Place it upright in an incubator at 37 °C, 5% CO2, and saturated humidity.
- When the cells appear to aggregate (usually after 24-48 h, depending on the actual situation), flick the bottom of the centrifuge tube to make the cartilage balls detach from the bottom and suspend in the medium.
- Change to fresh complete medium for chondrogenic differentiation every 2-3 days. Continue the induction until cartilage balls with a diameter of 1.5-2 mm are formed in the tube, and then prepare sections for staining.
- Prepare paraffin sections of cartilage balls according to the routine steps of pathological experiments. Add Alicia blue dye to the de-waxed sections and stain at 37 °C for 1 h.
- Rinse the slide with running water for 5 min, and then observe the staining effect of Alicia blue under a microscope after drying.
8. Identification of cell phenotype by flow cytometry
- When the cell colonies reach 80%-90% confluence at passages 3-5, remove the culture medium and wash the cells twice with preheated PBS. Then digest the cells with 3-4 mL of 0.25% trypsin/EDTA and incubate them at 37 °C in a saturated humidified atmosphere of 5% CO2 for 2-3 min until they are detached from the bottom of the flask under a microscope.
- Harvest the cells with a 10 mL complete medium and transfer the cell suspension into a sterile 15 mL centrifuge tube. Centrifuge the cell suspension at 800 x g for 5 min at RT. Discard the supernatant and wash the cells with 4 °C PBS.
- Resuspend the cells to 10 mL with 4 °C PBS. The cell suspension is divided into nine groups with a volume of 1 mL in each 1.5 mL microtube, named negative control, FITC isotype control, PE isotype control, APC isotype control, CD105, CD29, CD90, CD14, and CD45 group, respectively. Ensure that the number of cells in each microtube is between 1 x 105 and 1 x 106.
- Centrifuge the suspension at 800 x g for 5 min at 4 °C, and resuspend the cells in each microtube with 100 µL of 4 °C PBS again. Except for the negative control group, add 5 µL of the corresponding isotype control (FITC, PE, and APC Mouse IgG1 kappa Isotype Control) and antibodies (CD105, CD29, CD90, CD14, and CD45 Monoclonal Antibody) for flow cytometry to each microtube according to the order in step 8.3. Mix gently and incubate for 1.5 h at 4 °C in the dark.
- Add 1 mL of 4 °C PBS to each microtube and centrifuge at 300 x g for 10 min at 4 °C. Discard the supernatant and add 200 µL of 4 °C PBS to resuspend the cells.
- Test at least 10,000 cells on the flow cytometry after filtering the cell suspension16 and analyze the data using the flow cytometry software.
9. Isolating extracellular vesicles (EVs) derived from porcine bone marrow mesenchymal stem cells
- When the pBM-MSCs confluence reaches 80%-90%, discard the supernatant and wash cells with PBS 2x. Then add 25 mL of serum-free IMDM to each culture flask and continue to incubate at 37 °C in a humidified atmosphere of 5% CO2 for 48 h.
- Collect the cell supernatant (conditioned medium, CM) into a 50 mL centrifuge tube and centrifuge at 300 x g for 10 min at 4 °C to remove the cell debris.
- Collect the supernatant again into another 50 mL centrifuge tube. Carry out the isolation of EVs as soon as possible after collecting the supernatant. For long-term storage, store the supernatant in the refrigerator at -80 °C to prevent the loss of EVs.
- Centrifuge the supernatant in step 9.3 at 2,000 x g for 20 min at 4 °C.
- Transfer the supernatant to a sterile tube used for high-speed centrifuge and centrifuge at 16,500 x g for 30 min at 4 °C.
- Transfer the supernatant again to an ultracentrifuge tube, and centrifuge at 120,000 x g for at least 70 min at 4 °C, with a fixed angle rotor.
- Discard the supernatant completely. Add 1 mL of 4 °C PBS to each ultracentrifuge tube and resuspend the precipitation with a micropipette. Mix the solution from the same group into an ultracentrifuge tube, and then add 4 °C PBS to make the volume more than 3/4 of the tube.
- Centrifuge at 120,000 x g for 60 min at 4 °C and remove the supernatant as much as possible.
- Resuspend the precipitant with sterile PBS again, and store the EVs in a -80 °C refrigerator.
NOTE: The amount of PBS for resuspending can be determined as 100 µL for the precipitation from every two 175 cm2 culture flasks. All above steps of isolating EVs are shown in Figure 3 systematically.
10. Identification of EVs by transmission electron microscopy (TEM), nanoparticle tracking analysis (NTA), and western blotting
- Place the EVs sample on ice after thawing it in a 25 °C water bath, and then dilute to half of the previous concentration with PBS for NTA detection17.
- Pipette 10 µL of the sample on the copper mesh, allow it to settle for 1 min, and absorb the floating liquid with filter paper. Then, add 10 µL of phosphotungstic acid to the copper mesh, allow it to stand for 1 min, and absorb the excess liquid.
- After drying for a few minutes at RT, perform electron microscopy imaging under the condition of 100 KV accelerating voltage18.
- Resuspend the EVs sample from step 9.9 in RIPA lysis buffer (25 mM Tris·HCl (pH 7.6), 150 mM sodium chloride [NaCl], 1% nonyl phenoxypolyethoxylethanol (NP-40), 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 1 mM Phenylmethanesulfonyl fluoride (PMSF), 1x protease inhibitor), and detect the expression of specific markers for EVs such as Alix, TSG101, CD81, and CD63 by western blotting19.
Results
Establishment of porcine bone marrow mesenchymal stem cells
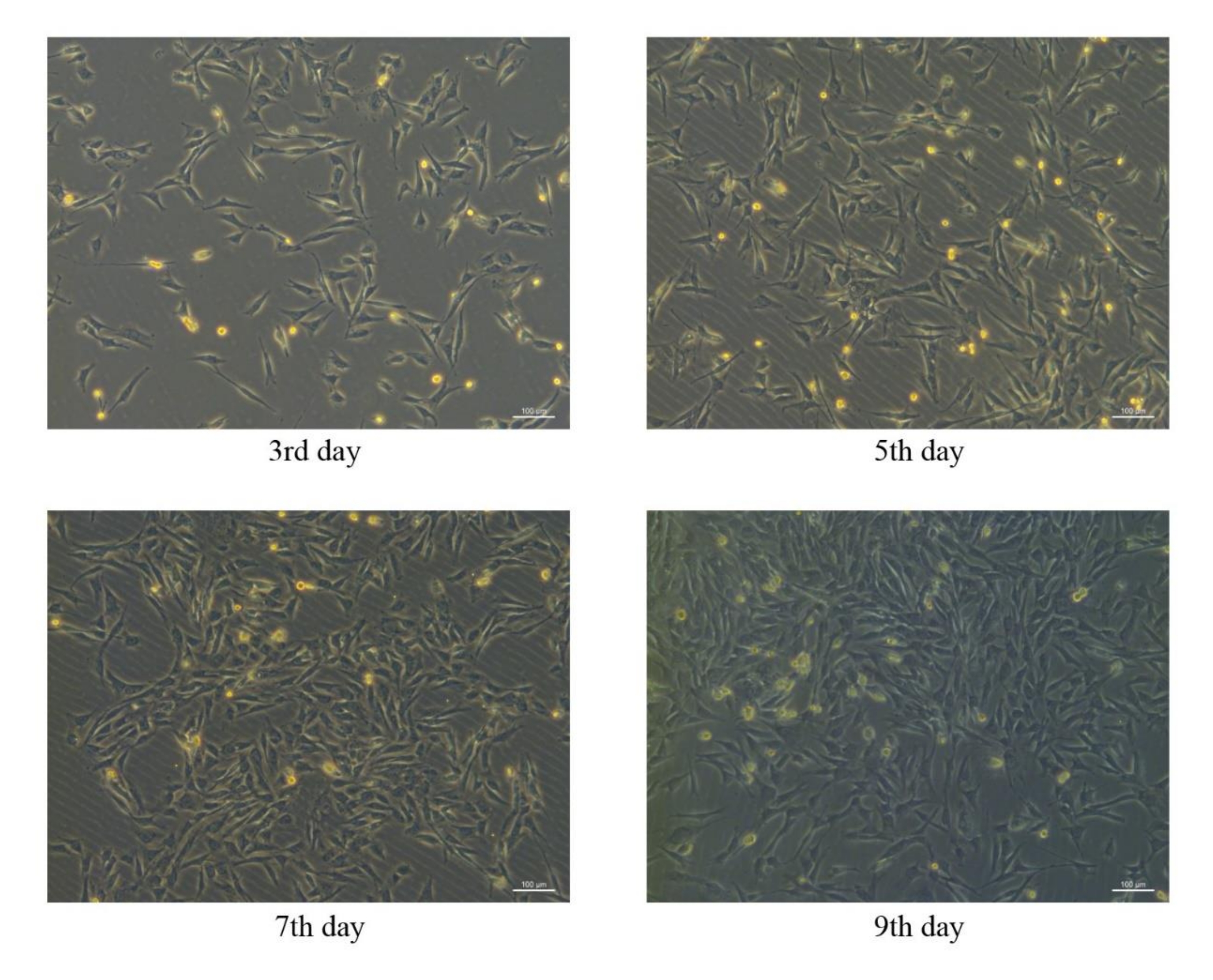
Mesenchymal stem cells derived from porcine bone marrow were successfully isolated and cultured in vitro, and the morphology of pBM-MSCs on different days can be seen in Figure 4. In the primary culture of pBM-MSCs, microscopic observation showed that cell adherence occurred one day after planting, and the adherent cells were usually round in shape. The primary pBM-MSCs generally remained at the quiescent phase for 3 days after planting, and cell proliferation began on the 4th day. The cell morphology changed from round to spindle, multilateral, or star type after proliferation, and the nuclei are central, with double nucleoli in some cells. Cell colonies were formed 7-9 days after the initiation of cell proliferation, and 80%-90% cell confluence could be achieved at 12-14 days. The microscopic observation showed that adherent cells grew as scattered colonies and were arranged in a swirling pattern.
Cell proliferation was significantly accelerated after passaging, and 80%-90% confluence could be reached in a week. The cell morphology was homogeneous spindle-shaped from the second passage, resembling fibroblasts, with a length to width ratio of about 2-3:1. If the cells were differentiated, they might appear polygonal or star-shaped. After passaging, the cells no longer grew as scattered colonies, but evenly and radially in a parallel arrangement.
Identification of cell differentiation potential by staining
In the adipogenic differentiation assay, Oil Red O staining showed that round orange-red lipid droplets of different sizes appeared around the nucleus (Figure 5A); In the osteogenic differentiation assay, Alizarin Red staining showed red nodules on the cell surface (Figure 5B), which was caused by the color reaction with calcium salts deposited by osteoblasts differentiated from pBM-MSCs. In the chondrogenic differentiation assay, Alicia blue staining showed that the whole tissue section was blue (Figure 5C), which was caused by the staining of endo-acidic mucopolysaccharide in cartilage balls.
Identification of cell phenotype by flow cytometry
Assays of the cell surface markers were performed to create a phenotype of pBM-MSCs. From the flow cytometry results (Figure 6), three positive markers such as CD105, CD29, and CD90 were expressed significantly on the surface of pBM-MSCs, accounting for 96.5%, 99.8%, and 92%, respectively (Figure 6A-C). However, the expression of CD14 and CD45 was negative (Figure 6D,E). Meanwhile, the results of corresponding isotype controls were all negative, which has already been overlaid in the figure, ruling out the possibility of non-specific binding of antibodies.
Identification of EVs derived from pBM-MSCs by NTA, TEM, and western blotting
The result of NTA showed that the median particle size was 126.9 nm, which was within the range of EVs; besides, the original concentration of the EVs sample was 1.5 x 1010 particles/mL, and the accurate value assigned to the size can be found in Figure 7A. The particle trajectory diagram is shown in Figure 7B, illustrating that the particles were in irregular Brownian motion. Furthermore, the discoid vesicle, as the classic structure of EVs, could be seen clearly under the electron microscope at magnifications of 50,000x (Figure 7C). Also, the expression of specific markers for EVs such as Alix, TSG101, CD81, and CD63 was detected in the sample by western blotting (Figure 7D).

Figure 1: Bone marrow puncture point of the minipig. The red area shows the puncture point of extracting bone marrow, located at the proximal femur of the minipig. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Isolating mesenchymal stem cells from porcine bone marrow. The process of isolating mesenchymal stem cells from porcine bone marrow is shown in the flow chart, and four liquid phases are illustrated clearly after density-gradient centrifugation. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Isolating EVs derived from pBM-MSCs. The schematic diagram demonstrates specific steps to isolate EVs from the conditioned medium by ultracentrifugation. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Morphological characteristics of pBM-MSCs on different days. Similar morphological characteristics of pBM-MSCs can be seen on the 3rd, 5th, 7th, and 9th day after planting under the 100x microscopic field, and cell colonies have been formed on the 9th day. Please click here to view a larger version of this figure.
{kind=link}

Figure 5. Identification of differentiation potential of pBM-MSCs by staining. (A) Adipogenic, (B) osteogenic, and (C) chondrogenic differentiation assay of pBM-MSCs, respectively. The differentiation potential of pBM-MSCs can be identified by these staining results. Please click here to view a larger version of this figure.
{kind=link}

Figure 6: Identification results of pBM-MSCs by flow cytometry. CD105, CD29, and CD90 are expressed significantly on the surface of pBM-MSCs, accounting for 96.5%, 99.8%, and 92.0%, respectively, whereas the expression of CD14 and CD45 is negative. Please click here to view a larger version of this figure.
{kind=link}

Figure 7: Identification results of EVs derived from pBM-MSCs by morphology and molecular biology. (A) NTA result of EVs derived from pBM-MSCs, with particle size distribution graph and (B) particle trajectory diagram, respectively; (C) TEM image taken at magnifications of 50,000x, and the white arrow shows the classic structure of discoid vesicles. (D) Expression of specific markers for EVs by western blotting. Please click here to view a larger version of this figure.
{kind=link}
Discussion
Traditional bone marrow puncture point of minipigs was positioned at the iliac crest20. Although it is easy to locate, the amount of bone marrow extraction is limited21 (only about 5 mL in general), so it is difficult to meet the requirement of a large number of expansions in vitro for the transplantation in vivo. In this method, we repositioned the bone marrow puncture point to the proximal femur, and at least 20 mL of bone marrow can be extracted from this site, guaranteeing a sufficient amount of pBM-MSCs for subsequent cell culture.
The two main separating solutions used for isolating BM-MSCs by density-gradient centrifugation are Percoll and Ficoll. Percoll is composed of siliconized polyvinylpyrrolidone (PVP), which is a novel non-toxic and non-irritating density gradient centrifugal separating agent. The low diffusion constant of Percoll results in a relatively stable density gradient; therefore, satisfactory cell separation can usually be achieved within tens of minutes at low centrifugal forces (200-1000 x g). The method for isolation of pBM-MSCs using Ficoll has been reported previously21. Compared with Ficoll, Percoll has been gradually used due to its advantages of easy isopermeability, low viscosity, non-toxicity, and not causing cell aggregation, which can complement the existing methods for isolating pBM-MSCs.
In isolating and culturing pBM-MSCs, some critical steps cannot be ignored. Firstly, successful stratification of different liquid phases after density-gradient centrifugation is the key to isolating purified pBM-MSCs. BM-MSCs, as a type of bone marrow mononuclear cells (BM-MNCs), have a specific gravity similar to that of lymphocytes and monocytes, around 1.075 g/mL. The original density of Percoll is 1.130 g/mL, and to successfully obtain the cell layer containing BM-MSCs after density-gradient centrifugation, 60% isotonic density gradient separating solution (1.077 g/mL) needs to be configured in advance according to the Percoll density-concentration relationship22. Furthermore, appropriate centrifugation conditions also contribute to successful stratification. Considering the low diffusion constant of Percoll, we centrifuged the extracted bone marrow at 600 x g for 20 min at relatively low acceleration/deceleration levels (ACC = 5, DEC = 5), which achieved a good stratification effect. Secondly, appropriate planting density is also essential for cell culture. In order to acquire a sufficient number of MSCs (usually more than 107 per animal9) for subsequent transplantation, we use 175 cm2 culture flasks for cell culture. In a previous study20, the obtained BM-MNCs were usually planted into culture flasks for cultivation at a density of 5 x 105/cm2. It has been reported that after density-gradient centrifugation, 2-3 x 107 BM-MNCs can be obtained for every 5 mL of porcine bone marrow23. So, in this protocol, we recommend to plant total BM-MNCs isolated from each 20 mL porcine bone marrow into a 175 cm2 culture flask for a suitable density. Thirdly, impurities should be avoided during the isolation and culture of pBM-MSCs. When drawing the mononuclear cell phase, the pipette should not be inserted into the Percoll phase so as not to mix with the separating liquid. Moreover, after 24 h of cell planting, the culture flask should be gently shaken to reduce the adherence of red blood cells.
During the process of ultracentrifugation, high levels of protein aggregate and lipoprotein contamination through this method inevitably compromise the quantification and functional analysis of EVs14. In order to reduce contamination as much as possible in the process, 5 mm depth of liquid in the bottom should be retained every time when transferring the supernatant before the ultracentrifugation step. Meanwhile, after the first ultracentrifugation, resuspending the pellet in sterile PBS and then performing ultracentrifugation again can effectively reduce lipoprotein contamination.
Although density-gradient centrifugation and ultracentrifugation have been widely used in isolating BM-MSCs and their derived EVs, respectively, these two techniques also have their own limitations. On the one hand, the Percoll technique is lengthy and cumbersome, and yielding BM concentrate specimen via a bedside cell concentration device has been reported as an alternative method to isolate MSCs24. On the other hand, the ultracentrifugation method requires not only highly trained technicians but also expensive equipment; therefore, the combined application of two or more techniques may present a reasonable strategy for more efficient isolation of EVs25. Besides, the identification of pBM-MSCs and their derived EVs also needs improvement. For example, according to the international criteria for defining MSCs26, the expression of some positive or negative markers, such as CD73, CD34, and HLA-DR, is still missing from the identification results of BM-MSC phenotypes by flow cytometry in this study. In addition, although measures have been taken to avoid contamination during the process of isolating EVs, due to the limitations of our laboratory, we are unable to assess the purity of the EVs sample to help improve the follow-up work.
This study combines methods for the isolation of pBM-MSCs and their derived EVs sequentially, proved by the subsequent identification results systematically. In particular, we have highlighted the key operations in this series of steps, explaining some specific experimental conditions which can solve the problem of heterogeneity existing in different laboratories during this process to a certain extent. This methodic work might be widely used in pre-clinical studies on the transplantation efficacy of BM-MSCs and their derived EVs, which could provide an experimental basis with a sufficient level for clinical research.
Disclosures
All authors have no conflicts of interest to declare.
Acknowledgements
We thank Yang Jianzhong and Wang Xuemin for their contributions to the operation of bone marrow extraction. This work was supported by grants from CAMS Innovation Fund for Medical Sciences (CIFMS) [grant number 2016-I2M-1-009], National Natural Science Foundation of China (no: 82070307; no: 81874461).
Materials
| Name | Company | Catalog Number | Comments |
| 175 cm2 cell culture flask | Thermo Fisher | 159910 | used for cell culture |
| 0.25% Trypsin/EDTA | Thermo Fisher | 25200056 | used to digest cells |
| Adipogenic Differentiation Kit for Bone Marrow Mesenchymal Stem Cell | OriCell | GUXMX-90031 | used for adipogenic differentiation assay |
| Alix Monoclonal Antibody | Thermo Fisher | MA1-83977 | used to identify extracellular vesicles(Evs) by western blotting |
| APC Mouse IgG1 kappa Isotype Control | Thermo Fisher | 17-4714-42 | used to eliminate the effects of non-specific staining in flow cytometry |
| CD105 (Endoglin) Monoclonal Antibody | Thermo Fisher | 17-1057-42 | used to identify pBM-MSCs by flow cytometry |
| CD14 Monoclonal Antibody | Thermo Fisher | MA1-82074 | used to identify pBM-MSCs by flow cytometry |
| CD29/IGTB1 Monoclonal Antibody | Thermo Fisher | MA1-19458 | used to identify pBM-MSCs by flow cytometry |
| CD45 Monoclonal Antibody | Thermo Fisher | MA5-28383 | used to identify pBM-MSCs by flow cytometry |
| CD63 Monoclonal Antibody | Thermo Fisher | 10628D | used to identify EVs by western blotting |
| CD81 Monoclonal Antibody | Thermo Fisher | MA5-13548 | used to identify EVs by western blotting |
| CD90 Monoclonal Antibody | Thermo Fisher | A15794 | used to identify pBM-MSCs by flow cytometry |
| Chondrogenic Differentiation Kit for Bone Marrow Mesenchymal Stem Cell | OriCell | GUXMX-90041 | used for chondrogenic differentiation assay |
| Fetal Bovine Serum | Gibco | 10099141C | used to prepare complete medium |
| FITC Mouse IgG1 kappa Isotype Control | Thermo Fisher | 11-4714-42 | used to eliminate the effects of non-specific staining in flow cytometry |
| Flow cytometry | BD | Accuri C6 | used for identification of cell phenotype |
| FlowJo software | BD | V10 | used to analyze data from flow cytometry |
| High-speed centrifuge tube (50 mL) | Beckman | 357003 | used for high-speed centrifugation |
| Iscove's Modified Dulbecco's Medium | Gibco | C12440500BT | used for cell culture |
| Low-temperature high-speed floor centrifuge | Avanti | J-26XPI | used for high-speed centrifugation to obtain clean conditioned medium |
| Nonyl phenoxypolyethoxylethanol (NP-40) | Sigma-Aldrich | NP40S | used for the composition of RIPA lysis buffer |
| Osteogenic Differentiation Kit for Bone Marrow Mesenchymal Stem Cell | OriCell | GUXMX-90021 | used for osteogenic differentiation assay |
| PE Mouse IgG1 kappa Isotype Control | Thermo Fisher | 12-4714-42 | used to eliminate the effects of non-specific staining in flow cytometry |
| Percoll | Cytiva | 17089102 | used to isolate porcine bone marrow mesenchymal stem cells(pBM-MSCs) |
| Phenylmethanesulfonyl fluoride (PMSF) | Thermo Scientific | 36978 | used for the composition of RIPA lysis buffer |
| Phosphate Buffered Saline(10x) | Beyotime | ST476 | used to prepare isotonic Percoll solution |
| Phosphate Buffered Saline(1x) | Cytiva | AF29561133 | used to dilute Percoll and wash cells |
| Protease inhibitor (1x) | Thermo Scientific | A32955 | used for the composition of RIPA lysis buffer |
| sodium chloride | Sigma-Aldrich | S9888 | used for the composition of RIPA lysis buffer |
| sodium deoxycholate | Sigma-Aldrich | D6750 | used for the composition of RIPA lysis buffer |
| sodium dodecyl sulfate (SDS) | Sigma-Aldrich | L3771 | used for the composition of RIPA lysis buffer |
| Transmission electron microscopy | Hitachi | HT7700 | used for electron microscopy imaging |
| Tris·HCl | Sigma-Aldrich | 93363 | used for the composition of RIPA lysis buffer |
| TSG101 Monoclonal Antibody | Thermo Fisher | MA1-23296 | used to identify EVs by western blotting |
| Ultracentrifuge (Type 50.2 Ti Rotor) | Beckman | optima L-100XP | used for ultracentrifugation to isolate exosomes |
| Ultracentrifuge tube (26.3 mL) | Beckman | 355654 | used for ultracentrifugation |
| ZetaVIEW | Particle Metrix | S/N 17-310 | used for Nanoparticle Tracking Analysis |
References
- Heslop, J. A., et al. Concise review: workshop review: understanding and assessing the risks of stem cell-based therapies. Stem Cells Translational Medicine. 4 (4), 389-400 (2015).
- Berebichez-Fridman, R., Montero-Olvera, P. R. Sources and clinical applications of mesenchymal stem cells: State-of-the-art review. Sultan Qaboos University Medical Journal. 18 (3), 264-277 (2018).
- Antebi, B., Mohammadipoor, A., Batchinsky, A. I., Cancio, L. C. The promise of mesenchymal stem cell therapy for acute respiratory distress syndrome. The Journal of Trauma and Acute Care Surgery. 84 (1), 183-191 (2018).
- Théry, C., et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. Journal of Extracellular Vesicles. 7 (1), 1535750 (2018).
- Valadi, H., et al. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nature Cell Biology. 9 (6), 654-659 (2007).
- Farooqi, A. A., et al. Exosome biogenesis, bioactivities and functions as new delivery systems of natural compounds. Biotechnology Advances. 36 (1), 328-334 (2018).
- Lener, T., et al. Applying extracellular vesicles based therapeutics in clinical trials - an ISEV position paper. Journal of Extracellular Vesicles. 4 (1), 30087 (2015).
- Castro-Malaspina, H., et al. Characterization of human bone marrow fibroblast colony-forming cells (CFU-F) and their progeny. Blood. 56 (2), 289-301 (1980).
- vander Spoel, T. I. G., et al. Human relevance of pre-clinical studies in stem cell therapy: systematic review and meta-analysis of large animal models of ischaemic heart disease. Cardiovascular Research. 91 (4), 649-658 (2011).
- Gupta, D., Zickler, A. M., El Andaloussi, S. Dosing extracellular vesicles. Advanced Drug Delivery Reviews. 178, 113961 (2021).
- Mareschi, K., et al. Multipotent mesenchymal stromal stem cell expansion by plating whole bone marrow at a low cellular density: a more advantageous method for clinical use. Stem Cells International. , 920581 (2012).
- Van Vlasselaer, P., Falla, N., Snoeck, H., Mathieu, E. Characterization and purification of osteogenic cells from murine bone marrow by two-color cell sorting using anti-Sca-1 monoclonal antibody and wheat germ agglutinin. Blood. 84 (3), 753-763 (1994).
- Yang, D., et al. and perspective on exosome isolation - efforts for efficient exosome-based theranostics. Theranostics. 10 (8), 3684-3707 (2020).
- Li, P., Kaslan, M., Lee, S. H., Yao, J., Gao, Z. Progress in exosome isolation techniques. Theranostics. 7 (3), 789-804 (2017).
- Willms, E., Cabañas, C., Mäger, I., Wood, M. J. A., Vader, P. Extracellular vesicle heterogeneity: subpopulations, isolation techniques, and diverse functions in cancer progression. Frontiers In Immunology. 9, 738 (2018).
- Huang, P., et al. Combinatorial treatment of acute myocardial infarction using stem cells and their derived exosomes resulted in improved heart performance. Stem Cell Research & Therapy. 10 (1), 300 (2019).
- Akbar, N., Pinnick, K. E., Paget, D., Choudhury, R. P. Isolation and characterization of human adipocyte-derived extracellular vesicles using filtration and ultracentrifugation. Journal of Visualized Experiments: JoVE. (170), e61979 (2021).
- Jung, M. K., Mun, J. Y. Sample preparation and imaging of exosomes by transmission electron microscopy. Journal of Visualized Experiments: JoVE. (131), e56482 (2018).
- Menck, K., Bleckmann, A., Schulz, M., Ries, L., Binder, C. Isolation and characterization of microvesicles from peripheral blood. Journal of Visualized Experiments: JoVE. (119), e55057 (2017).
- Yang, Y. J., et al. Atorvastatin treatment improves survival and effects of implanted mesenchymal stem cells in post-infarct swine hearts. European Heart Journal. 29 (12), 1578-1590 (2008).
- Lee, W. -. J., et al. Isolation and cellular phenotyping of mesenchymal stem cells derived from synovial fluid and bone marrow of minipigs. Journal of Visualized Experiments: JoVE. (113), e54077 (2016).
- Lindqvist, R., Wilson, I. D. Food Microorganisms: Buoyant Density Centrifugation. Encyclopedia of Separation Science. , 2843-2849 (2000).
- Yao, Y. L., Zhang, H., Gong, D. J., Song, Z. G., Xu, Z. Y. Transfecting hyperpolarization-activated cyclic nucleotide-gated channel 2 gene into porcine bone marrow mesenchymal stem cells. Journal of Clinical Rehabilitative Tissue Engineering Research. 13 (49), 9673-9676 (2009).
- Hegde, V., et al. A prospective comparison of 3 approved systems for autologous bone marrow concentration demonstrated nonequivalency in progenitor cell number and concentration. Journal of Orthopaedic Trauma. 28 (10), 591-598 (2014).
- Gallet, R., et al. Exosomes secreted by cardiosphere-derived cells reduce scarring, attenuate adverse remodelling, and improve function in acute and chronic porcine myocardial infarction. European Heart Journal. 38 (3), 201-211 (2017).
- Dominici, M., et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 8 (4), 315-317 (2006).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved