
Method Article
比较GTP酶结合蛋白的使用竞争性试验的亲和
摘要
This protocol compares the relative affinities of binding partners for Rho-family GTPases, including Rac1. In vivo, Rac1-binding proteins compete for a single binding interface, the conformation of which is dictated by a bound nucleotide. The nucleotide is both important and difficult to control experimentally, due to the high hydrolysis rate.
摘要
In this protocol we demonstrate a method for comparing the competition between GTPase-binding proteins. Such an approach is important for determining the binding capabilities of GTPases for two reasons: The fact that all interactions involve the same face of the GTPases means that binding events must be considered in the context of competitors, and the fact that the bound nucleotide must also be controlled means that conventional approaches such as immunoprecipitation are unsuitable for GTPase biochemistry. The assay relies on the use of purified proteins. Purified Rac1 immobilized on beads is used as the bait protein, and can be loaded with GDP, a non-hydrolyzable version of GTP or left nucleotide free, so that the signaling stage to be investigated can be controlled. The binding proteins to be investigated are purified from mammalian cells, to allow correct folding, by means of a GFP tag. Use of the same tag on both proteins is important because not only does it allow rapid purification and elution, but also allows detection of both competitors with the same antibody during elution. This means that the relative amounts of the two bound proteins can be determined accurately.
引言
The actin cytoskeleton that determines the shape, polarity and migratory properties of mammalian cells is regulated by the Rho-family of small GTPases. The Rho-family GTPases include RhoA that stimulates cytoskeletal contraction, Rac1 that stimulates actin branching and membrane protrusion, and Cdc42 that has similar effects on actin polymerization to Rac1 and causes the formation of filopodia 1,2. GTPase signaling activity is determined by binding of a nucleotide, which controls the contraction and relaxation of the switch I and switch II loops that mediate the protein-protein interactions with both regulators and effectors. Guanosine 5’-triphosphate (GTP)-bound GTPases activate downstream effectors, whereas the Guanosine 5’-diphosphate (GDP)-bound form is inactive. In the cell, cycles of GTP hydrolysis and nucleotide exchange allow rapid turnover of GTPase signals that are necessary for cytoskeletal dynamics. Nucleotide turnover is regulated by three mechanisms. Guanine nucleotide exchange factors (GEFs) stabilize the nucleotide-free GTPase, catalyzing exchange of GDP for GTP, and thereby stimulating GTPase signaling activity 3,4. GTPase-activating proteins (GAPs) catalyze hydrolysis of GTP to GDP, thereby inhibiting GTPase signaling activity 5. Sequestering molecules such as regulator of chromatin condensation 2 (RCC2) and guanine nucleotide dissociation inhibitors (GDIs) obscure the switch loops and in the case of GDIs remove the GTPase from the membrane by interaction with the prenyl tail 6,7. Each of the three classes of regulatory molecule interact with the switch loops, as do the downstream effectors and some trafficking regulators such as coronin-1C 7. The purpose of this protocol is to measure competition for the switch I/II binding site between putative regulators and downstream signaling molecules. It should be noted that competition assays test binding to a shared binding site, so that this protocol is not suitable for testing interactions with other sites, such as binding of GDIs to the prenyl tail.
The subtlety of the conformation differences between active and inactive forms, combined with the labile nature of the bound nucleotide, has made study of GTPase-binding events difficult. The role of the bound nucleotide means that conventional binding assays such as immunoprecipitation or surface plasmon resonance are not well suited to investigation, as the nucleotide cannot be controlled. This obstacle is compounded by the overlap in the binding sites of GEFs, GAPs, effectors, sequestering molecules and trafficking molecules, which make binding data for a single interaction difficult to interpret in the context of the competition that will occur in the cell. Immunoprecipitation, in particular, is compromised by competition between binding partners, as under certain cellular conditions, one binding partner might be identified at the expense of all others, while under other conditions, another partner might dominate. The dynamic nature of GTPase signaling is essential to GTPase function and must be considered when analyzing the relationships between the binding interactions of different regulators. Indeed, we recently described a pathway that relied heavily on competitive binding. We identified coronin-1C as a trafficking molecule that bound to the switch loops of GDP-Rac1 7. In areas of low GEF activity, trafficking would dominate, removing Rac1 from those regions. However, when Rac1 is delivered to regions of the cell where GEF activity is high, the GEF would outcompete coronin-1C, thereby both activating Rac1 and preventing coronin-1C-mediated removal of Rac1 from that area. The model goes further, because the action of the GEF exchanges bound GDP for GTP, shifting the equilibrium still further from coronin-1C. Consequently, Rac1 activity could be explained entirely in terms of competition and relative affinity.
In this protocol, we describe a method for comparing the relative affinities of different binding partners for small GTPases, using Rac1 as an example. By using a purified protein approach, it is possible to piece together a chain of signaling events by pair wise comparison, in an experiment where the bound nucleotide can be closely controlled.
研究方案
1.净化GST标记的GTP酶
- 文化的E.大肠杆菌菌株如大肠杆菌BL21转化的质粒pGEX-Rac1的O / N在37℃下,振荡以220rpm,在500ml自身诱导培养基(25mM的磷酸氢二钠,25mM的KH 2 PO 4,50mM的氯化铵 , 5mM的用 Na 2 SO 4,2mM的硫酸镁 ,2毫氯化钙 ,0.5%甘油,0.05%葡萄糖,0.2%乳糖,5克蛋白胨,2.5克酵母提取物,100微克/毫升氨苄青霉素)。
- 收获细菌通过离心10分钟以10,000×g离心,4℃。
- 重悬细菌沉淀在20ml蛋白质提取试剂,1×蛋白酶抑制剂孵育20分钟,在室温将其与倒置。
- 澄清裂解液离心以40,000×g离心30分钟。
- 加2ml谷胱甘肽磁珠,用磷酸盐缓冲盐水(PBS:10mM的磷酸氢二钠,1.8毫KH 2 PO 4,137 mM氯化钠,2.7毫米氯化钾)。
- 孵育2小时后,通过倒转在4℃下混合。
- 用10ml PBS洗涤蛋白加载珠四次,使用磁粉分拣以沉淀珠粒在每一个步骤。
- 重悬蛋白质装载于2ml PBS中珠和储存在-80℃下在100μl等份直到需要。
GTP酶结合蛋白2的表达
- 一天实验前,转染的质粒编码绿色荧光蛋白(GFP)-tagged每个GTP酶结合蛋白的版本到一个单独的75-cm 2的烧瓶HEK293T的如下。对于核苷酸装载的验证,转染GFP标记TrioD1到第三个75厘米2瓶HEK293T的。
- 稀释polyethylamine至1mg / ml,在100μl的无菌150mM的NaCl洗涤。
- 加入27微升稀释polyethylamine至223微升减少血清的培养基。
- 加12微克质粒DNA到250微升减少血清的培养基。
- 孵育各管在室温2分钟。
- 结合polyethylamine和在单管中,涡旋2分钟的DNA混合。
- 下室温温育15-20分钟。
- 用5毫升新鲜生长培养基更换生长培养基(Dulbecco氏改良的Eagle介质,10%牛胎儿血清,2mM的L-谷氨酰胺,不含抗生素)上90%汇合的HEK293T。
- 将合并的polyethylamine / DNA混合物添加到该烧瓶中,并培育O / N在37℃,5% 的 CO 2。
3.纯化GTP酶结合蛋白的
- 冲洗转染的细胞在PBS中的烧瓶和漏极烧瓶5分钟,吸出游离的液体。
- 刮去细胞在500μl裂解缓冲液(50mM的Tris-HCl(pH7.8),1%的Nonidet P-40,1×蛋白酶抑制剂)到微量离心管中。
- 裂解细胞通过倒置,在4℃下进行30分钟的混合。
- 在裂解,洗两批40微升GFP-陷阱珠三次新鲜的裂解液,沉淀的珠2,700 XG的洗涤之间2分钟。
- 澄清的裂解物通过离心21,000×g离心10分钟。
- 转印澄清每个参赛者的蛋白质分离洗涤GFP-陷阱珠和允许的GFP融合蛋白结合2小时的裂解物,由倒置,在4℃下混合。保持溶胞产物从GFP-TrioD1细胞在冰上。
- 在50mM的Tris-HCl(pH值7.8),50mM NaCl中洗两次加载GFP-陷阱珠,0.7%(重量/体积)的Nonidet P-40的和两次在50mM的Tris-HCl(pH值7.6),20mM的MgCl 2的,2,700 xg离心洗涤之间2分钟沉降珠。
- 通过加入40微升的0.2M甘氨酸(pH值2.5)和移液器上下吹吸30秒洗脱GFP的融合蛋白。立即沙珠在21000 XG为60秒和转让液含有4微升的1M的Tris-HCl(pH值10.4),一个新的离心管中。做到这一点很快,限制损坏纯化蛋白。
- 分析1微升通过Western印迹和探针用抗GFP抗体每种纯化蛋白的使用定量blott建立相对产量根据制造商的协议的系统上。可替代地,确定的蛋白浓度通过二辛可宁酸(BCA)测定法但这会引入错误,如果蛋白质不与化验反应中以相同的方式或有污染蛋白质。
- 通过加入50mM的Tris-HCl(pH值7.6),20mM的MgCl 2的摩尔相等蛋白质浓度。
GTP酶的4核苷酸装
- 解冻的GST-Rac1的磁珠,在步骤1中制备一个等份。
- 采取90微升的GST-Rac1的珠和洗涤三次用20mM的Tris-HCl(pH为7.6),25 mM氯化钠,0.1mM的DTT,4mM EDTA的,使用磁性粒子分选器以沉淀珠粒在每一个步骤。
- 从珠吸缓冲区,并添加100微升20毫米的Tris-HCl(pH值7.6),25毫米氯化钠,0.1毫米DTT,4毫摩尔EDTA。
- 根据国内生产总值,GTP或没有加载核苷酸是否需要竞争的实验中,加入12微升100毫米的GDP,12微升10毫区anosine 5' - [γ-硫代]三磷酸(GTPγS)或无核苷酸到60微升的GST-Rac1的珠。
- 对于核苷酸负荷控制,分割剩余的珠串成三个10微升等份,并添加2微升100毫米GDP,2微升10毫米GTPγS或无核苷酸,每管。
- 孵育珠混合物30分钟,在30℃下搅拌。
- 稳定通过加入1M MgCl 2的核苷酸结合的Rac1:3μl到实验混合物(步骤4.4),0.5微升至每个控制混合(步骤4.5)的。
5.竞争结合。
- 设立了6个离心管,每片含:
200微升的50mM的Tris-HCl(pH值7.6),20mM的MgCl 2的
10微升试验核苷酸加载Rac1的珠(来自步骤4.7)
5微升Rac1的结合蛋白A(常结合蛋白) - 到各管中,加入0,1,2.5,5,10或20微升的Rac1结合蛋白B(可变结合蛋白)。这些卷假设pproximately等于库存浓度恒定和可变结合蛋白的,并可能需要调整。
- 调节结合蛋白A和卷乙如果在两种蛋白质的结合亲和力大的差异,这应根据经验通过实验重复来确定。补结合混合物与235微升的总体积通过加入50mM的Tris-HCl(pH值7.6),20mM的MgCl 2的。
- 建立含有微量离心管:
200微升的50mM的Tris-HCl(pH值7.6),20mM的MgCl 2的
10微升试验核苷酸加载Rac1的珠(来自步骤4.7)
10微升Rac1的结合蛋白A(常结合蛋白) - 成立了国内生产总值,GTPγS无核苷酸对照管:
200微升的50mM的Tris-HCl(pH值7.6),20mM的MgCl 2的
10微升控制Rac1的珠子装入步骤4.5与GDP,GTPγS或无核苷酸和稳定步骤4.7。
180微升水中EK293T GFP-TrioD1裂解物,制备如步骤3.6
4微升1M的氯化镁 - 2小时孵育该混合物中,通过倒转在4℃下混合。
- 用50mM的Tris-HCl(pH值7.6),20mM的MgCl 2的洗珠三次。
- 洗脱结合的蛋白于20μl还原样品缓冲液(50mM的Tris-HCl(pH 7)中,5%的SDS,20%甘油,0.02毫克/毫升溴酚蓝,5%β巯基乙醇)。
竞争6.分析
- 解决将10μl结合蛋白(步骤5.6)的十二烷基硫酸钠聚丙烯酰胺凝胶电泳(SDS-PAGE)和Western印迹。
- 孵育该膜在4℃的CO / N的抗GFP抗体在封闭缓冲液稀释1/1000稀释于PBS 1倍,0.1%Tween-20的同时检测标记的GTP酶结合蛋白。
- 10分钟,用PBS,0.1%Tween-20的洗膜三次。
- 在DyLight孵育膜在室温30分钟800缀合的抗兔秒ondary抗体,稀释1 / 10,000中的封闭缓冲液稀释在PBS,0.1%Tween-20的1倍。
- 10分钟,用PBS,0.1%Tween-20的洗膜三次。
- 扫描使用红外成像系统的膜,使用该软件根据制造商的协议来测量带强度。
- 图中的每个蛋白的带强度对变量的竞争对手(蛋白B)的量。
- 除以变量竞争者的体积在的点上的线相交由恒定竞争者(蛋白A,5微升)的容积,以确定在哪些达到平衡的竞争者比率。
- 为了验证核苷酸负荷状态,探测膜的按照步骤6.1-6.6中描述的p21激活激酶1(PAK1)(效应)和GFP-TrioD1(全球环境基金)。
结果
这个协议设计为计算Rac1的结合伴侣的相对亲和力,而不需要知道的竞争者的精确浓度( 图1)。蛋白浓度的测定将错误引入并考虑分子之间竞争时在一个信号通路是不需要的。但是,重要的是要知道,两个竞争者具有相同摩尔浓度的储备溶液,以允许简单的比率来计算的添加不同量的测定时。 40微升的GFP-陷阱珠具有的〜300皮摩尔如此汇合75厘米2烧瓶高度表达细胞将饱和的珠子,其结果是在两个不同的结合蛋白的制剂将是调整前相似的结合能力(图2A)。如果蛋白质之一表示较差,这个问题可以通过纯化蛋白质从细胞中的一个以上的烧瓶被克服。
最GTP酶的效应和调节器的结合依赖于核苷酸 - 装载的诱饵GTP酶的,因此,以测试负载是否成功是非常重要的。装载可以通过从细胞裂解物中沉淀已知结合蛋白进行验证。效应蛋白,如PAK1绑定到的GTP-Rac1和可从裂解物容易沉淀和Western印迹8(图2B)进行检测。 GEFS优先结合到核苷酸的无GTP酶稳定过渡状态。由于全环基金是丰度低,通常不活跃的,经常涂抹不好,最好是过表达或全球环境基金GEF片段的核苷酸检测无GTP酶。我们经常使用第一张双人同源重奏的,表示为GFP融合(GFP-TrioD1 9)(图2B),但任何环基金会工作。结合对GDP加载GTP酶蛋白质是罕见的。我们最近报道RCC2作为一个这样的蛋白7,占GDP加载可以简单地为宾迪验证纳克既不全球环境基金,也不效应。
从实验的输出将是一个Western印迹描绘绑定到GTP酶两个GFP标记的结合伴侣。通过使用单一抗体来检测两种蛋白质,在该类似量既是竞争对手的结合浓度可被确定,因此,相对亲和力推断。在Rac1的拐蛋白的螺旋桨结构域之间的这种实施例的竞争,coronin-1C(Rac1的结合蛋白A)中,和Rac1的-截存蛋白,RCC2(Rac1的结合蛋白B)证明(图3A)。通过使用coronin-1C螺旋桨(5微升)的恒定体积,并加入RCC2体积增加,我们可以从GFP看到涂抹达到平衡在1.25-2.5微升RCC2(星号)的,这表明RCC2具有更强对于Rac1的比coronin-1C亲和力。通过测量用定量免疫印迹条带的强度,并绘制平均值为每个competitoR,平衡点可以准确地通过识别在该曲线相交(图3B)的卷来计算。
其中一个可能的障碍一个成功的竞争测定法是,如果结合伴侣结合到彼此以及结合Rac1的。在图3A + B我们证明RCC2和coronin-1C的螺旋桨域之间的竞争,而不是全长coronin-1C。之所以使用截短coronin是coronin-1C还通过尾域结合RCC2。当全长coronin-1C滴定针对RCC2,两种蛋白的结合进行检测,由于三元复合物形成,而不是竞争(图3C)。如果竞争的现象发生,一种蛋白质结合会增加,而其他的减少,总的约束GFP融合将保持不变。的情况下一个三元复合物形成,有必要截断GTP酶结合蛋白之一,使得competitors不再进行交互。

图1.工作流程。工作流程来确定使用竞争性试验GTP酶结合蛋白的亲和力的示意图。 请点击此处查看该图的放大版本。
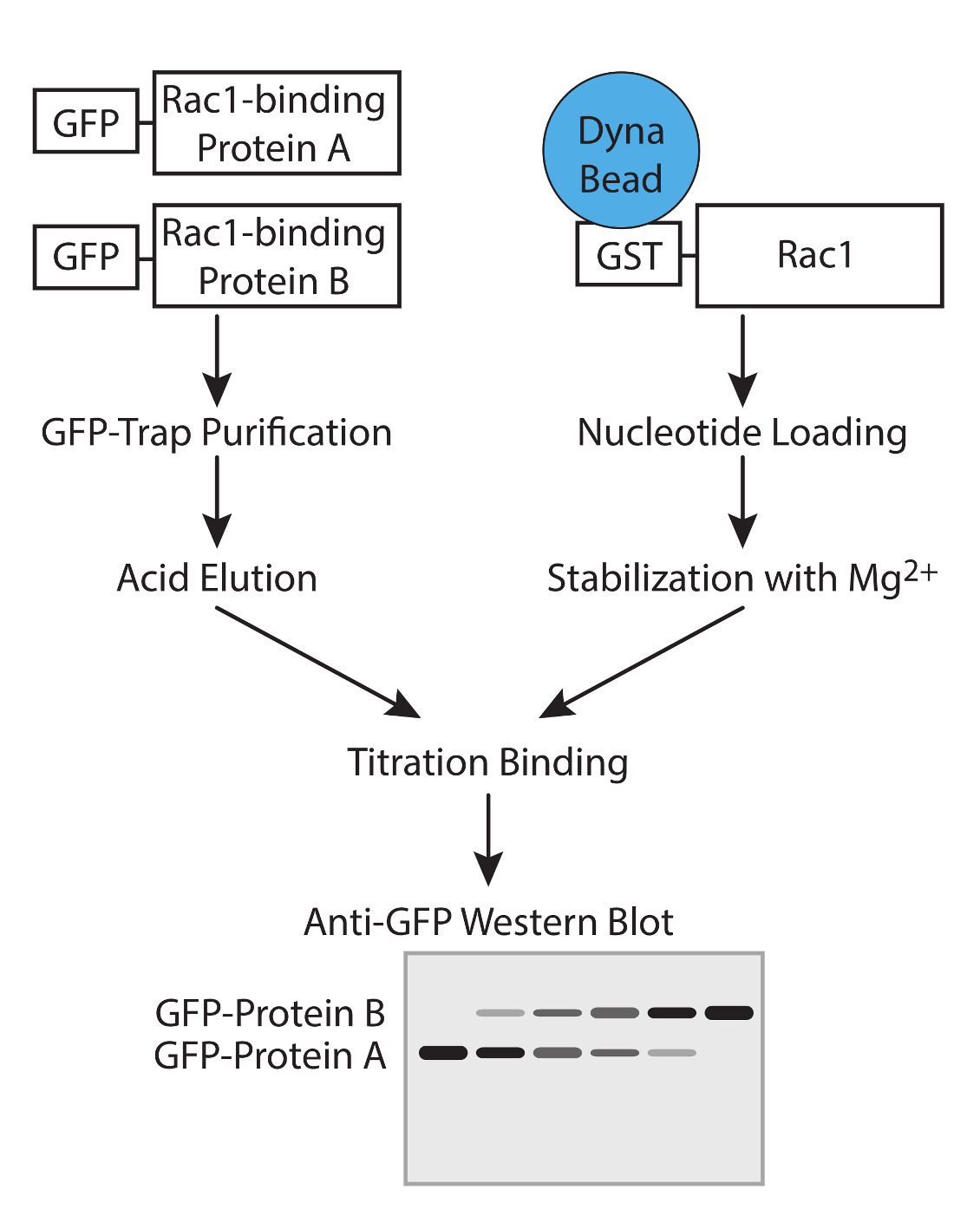{kind=link}

图2.验证纯化的蛋白质。 (A)纯化 GFP标记的Rac1结合通过蛋白质印迹分析的蛋白质,具有抗GFP探测,以确定两个蛋白的相对产率。这在实验期间类型均衡允许调整两种蛋白质的浓度,以便它们在结合实验匹配。(B)中的GD磷,GTPγS无核苷酸加载GST-Rac1的培养与HEK293T裂解表达GFP-TrioD1和约束绘制了内源性PAK1或过表达GFP-TrioD1检测蛋白质。 请点击此处查看该图的放大版本。
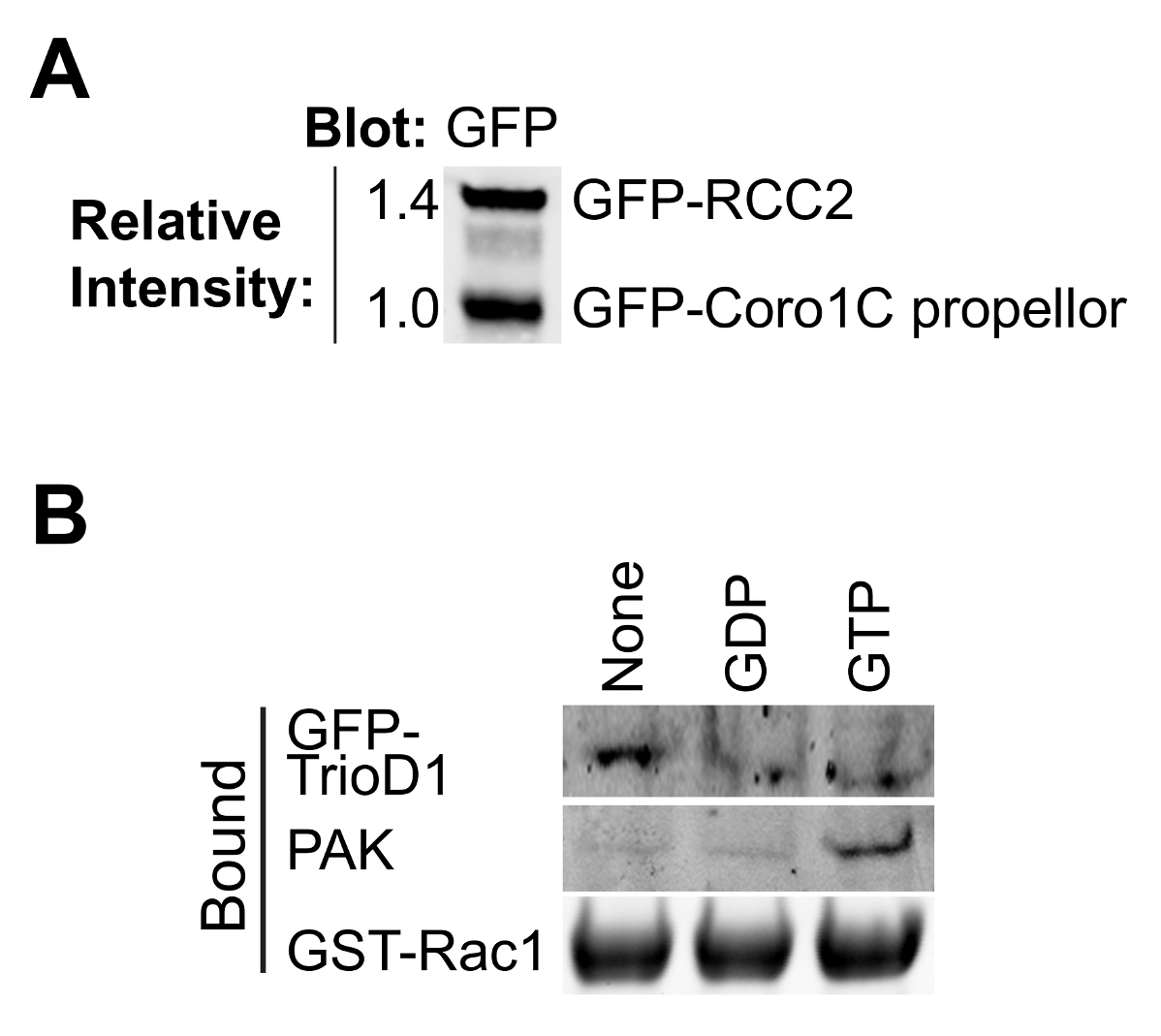{kind=link}

相对蛋白质的图3.蛋白质印迹分析 ,从竞争结合测定结合。实施例输出。(A)中的GDP加载的Rac1中加入5微升的GFP-coronin-1C螺旋桨域和增加的GFP-RCC2的体积滴定,通过Western印迹结合的蛋白质的GFP,与差分检测两种蛋白质的问题被避免和GFP信号报告两个融合蛋白之间的摩尔比。星号表示在eithe竞争比例平衡点的R侧。(B)的来自三个独立实验结合GFP融合蛋白的谱带强度通过定量蛋白质印迹法测定,使用荧光标记的二抗和平均数作图来计算RCC2的量需要以达到平衡。(℃从一个实验,其中Rac1的结合蛋白结合到彼此并形成三元复合物,而不是竞争)实施例输出。 GDP-加载 的Rac1中加入5微升的GFP-RCC2和增加的GFP-coronin-1C全长的体积滴定。在结合的GFP-coronin-1C的增加没有约束的GFP-RCC2的损耗表示三元复合物的形成。 请点击此处查看该图的放大版本。
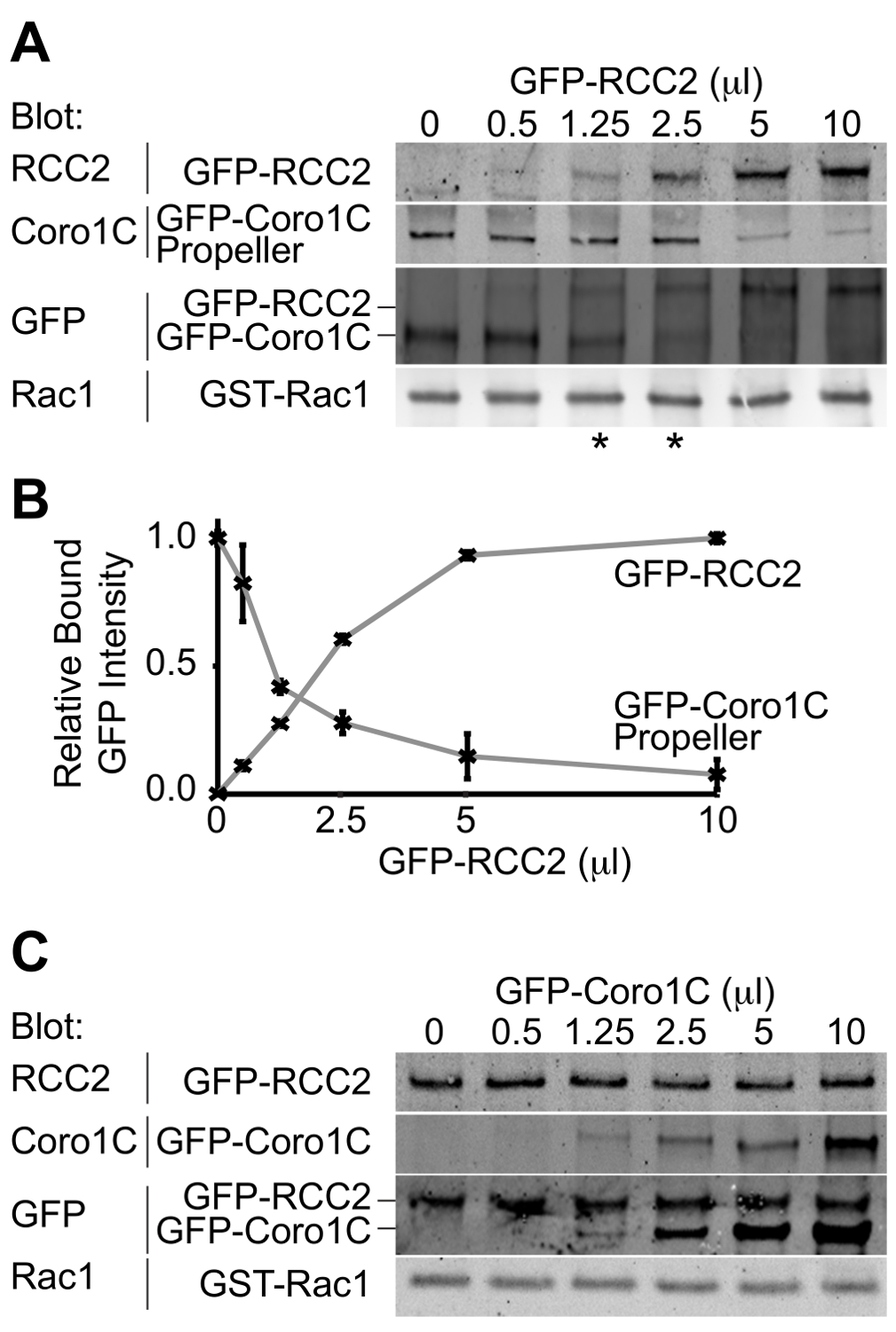{kind=link}
讨论
This protocol describes a method for comparing the relative affinities of pairs of small GTPase-binding proteins. The key steps are the preparation of purified GTPase-binding proteins and the nucleotide loading of the GTPase. The use of GTPase-binding proteins with the same GFP tag, allows the concentrations at which similar amounts of each competitor binds to be accurately determined. The use of recombinant nucleotide-loaded GTPase allows interrogation of the binding properties of the GTPase under specific activity conditions. This step is also the most sensitive as nucleotides will both hydrolyze and detach from the GTPase if the magnesium conditions are not maintained precisely.
In the cell, the large number of GTPase-binding proteins combined with the rapid nucleotide turnover makes such pathways difficult to interpret. The simplicity of this method in comparing only pairs of binding proteins and using carefully controlled nucleotide-loading conditions allows signaling pathways to be elucidated. However, the greatest strength of the protocol is also the greatest weakness as it is a simplification of the in vivo situation. Competition assays can be used to build a robust hypothesis, but this should then be tested in cells by knockdown experiments.
There are three features that must be considered when selecting the GFP-tagged GTPase-binding proteins to be used in the experiment. First, the fusion proteins must express well in mammalian cells, such as HEK293T, as competition assays require a reasonable amount of protein. Second, it must be possible to purify the recombinant protein without significant degradation, and where this is not possible, cloning of a GTPase-binding fragment should be considered. Third, the two GTPase-binding proteins must resolve from one another on SDS-PAGE to allow analysis in section 6.
There are a number of potential caveats to the experiment that need to be considered, and possibly addressed:
Possible denaturation of purified GTPase-binding proteins during the acid elution step or steric hindrance by the GFP tag. In our hands, these have not been a problem, but must be tested. The purified proteins can be tested in functional assays 10. Commercial kits now exist for testing the activity of GEFs or GAPs without the need for isotope-labeled nucleotides. Sequestering proteins, by their nature protect GTPases from GEF or GAP activity, so can be used as competitive inhibitors in the commercial GEF or GAP assays, as we did in our recent publication 7. The relevant feature of proteins that traffic GTPase are the capacity to bind the GTPase, and this can be tested easily in a pull down assay. An alternative approach to testing protein integrity that is applicable to all binding proteins is to titrate protein eluted from GFP-trap beads with glycine with the same protein removed from GFP-trap beads by enzymatic cleavage. The experiment would be analyzed by probing both the GFP-tagged and cleaved protein with an antibody against the protein itself. If the protein is undamaged by elution, equilibrium should be achieved at a 1:1 ratio. This approach would also indicate whether the presence of the GFP tag itself compromises the binding properties of the candidate protein, though this does require the production of a construct with an enzymatic cleavage site between the tag and the binding protein. Whether the protein is compromised by the tag or the elution step, the problem could be addressed by modifying the protocol to use an alternative purification method. Rather than GFP, binding proteins could be His-tagged, purified using Ni-NTA and analyzed using an antibody against the His-tag. The important feature is that both binding proteins must share a common tag although, if necessary, two tags could be added to a protein, one for purification and the other for detection.
The protocol is designed to investigate competition between interactions with the switch I/II domains. Although the majority of GTPase interactions are mediated by this motif, there are some exceptions, most notably the interactions of GDIs that bind to the prenyl tail, as well as obscuring the switch domains. In principle, the protocol could be adapted to use GTPase purified from mammalian cells, so that the GTPase is prenylated, however, the presence of multiple binding sites or allosteric effects complicate the interpretation of competition-binding data. Further problems associated with such a modification are that GDIs co-purify with GTPase from mammalian cells, compromising the purity of the isolated proteins and the hydrophobic nature of the prenyl groups means that prenylated GTPases are associated with either GDI or lipid membrane and such factors would need to be considered in the experiment.
The amount of GST-Rac1 being used in the assay. The constant GTPase binding protein must be at a greater concentration than the Rac1, or when the competitor is added, it will simply bind to free Rac1. It will be immediately obvious if this has happened as binding of the competitor, without a loss of the constant protein, will be detected in much the same way as when the two competing proteins bind to one another as shown in Figure 3B. As an additional control (Step 5.3), a binding reaction containing double the amount of constant binding protein and no variable binding protein should be included (Step 5.3). If the Rac1 in the titration experiment is saturated, doubling the amount of constant binding protein will have no effect on the output. The volumes suggested in the protocol should be appropriate, but the amount of Rac1 can be easily reduced. If binding of the competitor without loss of the constant binding partner is observed, reducing the amount of Rac1 should be attempted before trying to map binding sites to avoid ternary complex formation.
Non-specific interaction of GTPase-binding proteins with the GST or bead, as well as specifically with Rac1. This problem would be manifested by residual binding of the constant GTPase-binding protein, even when the variable GTPase-binding protein has reached a plateau at high concentration. Identification of this issue will be aided by conducting reciprocal experiments where the constant and variable GTPase-binding proteins are swapped. Reciprocal experiments will also greatly improve the accuracy of the estimate of equilibrium point, so should always be included. In cases of non-specific binding, the relative concentrations at which equilibrium is achieved can still be calculated by comparing band intensity between the maxima and minima for each protein, or by measuring the extent of non-specific binding by using GST beads as bait, rather than GST-Rac1.
Pull down assays using different nucleotide-loading conditions should be used to complement the competition assay described in this protocol. Determining the nucleotide preference of partners is important for both understanding the competition events and understanding the signaling pathway that the GTPase-binding protein is involved in. In Figure 2B we analyze binding of proteins with established preference for GTP-loaded or nucleotide-free GTPase as a means to validate nucleotide loading. However, it is sensible to investigate the effect of nucleotide loading on each of the competitors as well. If the hypothetical competitors show different preferences, competition will make less of a contribution to the signaling pathway, and indeed nucleotide turnover is likely to be the mechanism that directs exchange of the binding proteins.
披露声明
作者什么都没有透露。
致谢
This work was supported by Wellcome Trust grant 088419 to MDB.
材料
| Name | Company | Catalog Number | Comments |
| Bugbuster | Novagen | 70584-3 | |
| COMPLETE protease inhibitor | Roche | 05 056 489 001 | |
| Glutathione magnetic beads | Pierce | 88821 | |
| Polyethylenimine, branched, average Mw ~25,000 | Sigma Aldrich | 408727-100ML | |
| OPIMEM | Life Technologies | 31985-047 | |
| Dulbecco's Modified Eagle Media | Sigma Aldrich | D5796 | |
| Fetal Bovine Serum | Life Technologies | 10270-1-6 | |
| L-Glutamine | Life Technologies | 25030-024 | |
| GFP-Trap_A | Chromotec | gta-20 | |
| GDP | Sigma Aldrich | G7127 | Highly unstable. Aliquot and store at -80 immediately upon reconstritution |
| GTPγS | Sigma Aldrich | G8634 | Highly unstable. Aliquot and store at -80 immediately upon reconstritution |
| Blocking Buffer | Sigma Aldrich | B6429 | |
| Tween-20 | Sigma Aldrich | P9416 | |
| Anti-GFP antibody | Living Colors | 632592 | Use at 1/1000 dilution |
| DyLight 800 conjugated goat anti-rabbit secondary antibody | Fisher Scientific | 10733944 | |
| Anti-PAK1 antibody | Cell Signaling | 2602S | Use at 1/1000 dilution |
| Odyssey SA Infrared Imaging System | Li-cor | 9260-11PC |
参考文献
- Burridge, K., Rho Wennerberg, K. and Rac take center stage. Cell. 116 (2), 167-179 (2004).
- Raftopoulou, M., Hall, A. Cell migration: Rho GTPases lead the way. Dev Biol. 265 (1), 23-32 (2004).
- Rossman, K. L., Der, C. J., Sondek, J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol. 6 (2), 167-180 (2005).
- Worthylake, D. K., Rossman, K. L., Crystal Sondek, J. structure of Rac1 in complex with the guanine nucleotide exchange region of Tiam1. Nature. 408 (6813), 682-688 (2000).
- Scheffzek, K., Ahmadian, M. R. GTPase activating proteins: structural and functional insights 18 years after discovery. Cell Mol Life Sci. 62 (24), 3014-3038 (2005).
- Del Pozo,, A, M., et al. Integrins regulate GTP-Rac localized effector interactions through dissociation of Rho-GDI. Nat Cell Biol. 4 (3), 232-239 (2002).
- Williamson, R. C., et al. Coronin-1C and RCC2 guide mesenchymal migration by trafficking Rac1 and controlling GEF exposure. J Cell Sci. 127 (Pt 19), 4292-4307 (2014).
- Del Pozo, M. A., Price, L. S., Alderson, N. B., Ren, X. D., Schwartz, M. A. Adhesion to the extracellular matrix regulates the coupling of the small GTPase Rac to its effector PAK. Embo J. 19 (9), 2008-2014 (2000).
- Van Rijssel, J., Hoogenboezem, M., Wester, L., Hordijk, P. L., Van Buul, J. D. The N-terminal DH-PH domain of Trio induces cell spreading and migration by regulating lamellipodia dynamics in a Rac1-dependent fashion. PLoS. 7 (1), e29912(2012).
- Self, A. J., Hall, A. Measurement of intrinsic nucleotide exchange and GTP hydrolysis rates. Methods Enzymol. 256, 67-76 (1995).
转载和许可
请求许可使用此 JoVE 文章的文本或图形
请求许可探索更多文章
This article has been published
Video Coming Soon
版权所属 © 2025 MyJoVE 公司版权所有,本公司不涉及任何医疗业务和医疗服务。