
Method Article
Confrontando l'affinità delle proteine-GTPasi legame con saggi Concorso
In questo articolo
Riepilogo
This protocol compares the relative affinities of binding partners for Rho-family GTPases, including Rac1. In vivo, Rac1-binding proteins compete for a single binding interface, the conformation of which is dictated by a bound nucleotide. The nucleotide is both important and difficult to control experimentally, due to the high hydrolysis rate.
Abstract
In this protocol we demonstrate a method for comparing the competition between GTPase-binding proteins. Such an approach is important for determining the binding capabilities of GTPases for two reasons: The fact that all interactions involve the same face of the GTPases means that binding events must be considered in the context of competitors, and the fact that the bound nucleotide must also be controlled means that conventional approaches such as immunoprecipitation are unsuitable for GTPase biochemistry. The assay relies on the use of purified proteins. Purified Rac1 immobilized on beads is used as the bait protein, and can be loaded with GDP, a non-hydrolyzable version of GTP or left nucleotide free, so that the signaling stage to be investigated can be controlled. The binding proteins to be investigated are purified from mammalian cells, to allow correct folding, by means of a GFP tag. Use of the same tag on both proteins is important because not only does it allow rapid purification and elution, but also allows detection of both competitors with the same antibody during elution. This means that the relative amounts of the two bound proteins can be determined accurately.
Introduzione
The actin cytoskeleton that determines the shape, polarity and migratory properties of mammalian cells is regulated by the Rho-family of small GTPases. The Rho-family GTPases include RhoA that stimulates cytoskeletal contraction, Rac1 that stimulates actin branching and membrane protrusion, and Cdc42 that has similar effects on actin polymerization to Rac1 and causes the formation of filopodia 1,2. GTPase signaling activity is determined by binding of a nucleotide, which controls the contraction and relaxation of the switch I and switch II loops that mediate the protein-protein interactions with both regulators and effectors. Guanosine 5’-triphosphate (GTP)-bound GTPases activate downstream effectors, whereas the Guanosine 5’-diphosphate (GDP)-bound form is inactive. In the cell, cycles of GTP hydrolysis and nucleotide exchange allow rapid turnover of GTPase signals that are necessary for cytoskeletal dynamics. Nucleotide turnover is regulated by three mechanisms. Guanine nucleotide exchange factors (GEFs) stabilize the nucleotide-free GTPase, catalyzing exchange of GDP for GTP, and thereby stimulating GTPase signaling activity 3,4. GTPase-activating proteins (GAPs) catalyze hydrolysis of GTP to GDP, thereby inhibiting GTPase signaling activity 5. Sequestering molecules such as regulator of chromatin condensation 2 (RCC2) and guanine nucleotide dissociation inhibitors (GDIs) obscure the switch loops and in the case of GDIs remove the GTPase from the membrane by interaction with the prenyl tail 6,7. Each of the three classes of regulatory molecule interact with the switch loops, as do the downstream effectors and some trafficking regulators such as coronin-1C 7. The purpose of this protocol is to measure competition for the switch I/II binding site between putative regulators and downstream signaling molecules. It should be noted that competition assays test binding to a shared binding site, so that this protocol is not suitable for testing interactions with other sites, such as binding of GDIs to the prenyl tail.
The subtlety of the conformation differences between active and inactive forms, combined with the labile nature of the bound nucleotide, has made study of GTPase-binding events difficult. The role of the bound nucleotide means that conventional binding assays such as immunoprecipitation or surface plasmon resonance are not well suited to investigation, as the nucleotide cannot be controlled. This obstacle is compounded by the overlap in the binding sites of GEFs, GAPs, effectors, sequestering molecules and trafficking molecules, which make binding data for a single interaction difficult to interpret in the context of the competition that will occur in the cell. Immunoprecipitation, in particular, is compromised by competition between binding partners, as under certain cellular conditions, one binding partner might be identified at the expense of all others, while under other conditions, another partner might dominate. The dynamic nature of GTPase signaling is essential to GTPase function and must be considered when analyzing the relationships between the binding interactions of different regulators. Indeed, we recently described a pathway that relied heavily on competitive binding. We identified coronin-1C as a trafficking molecule that bound to the switch loops of GDP-Rac1 7. In areas of low GEF activity, trafficking would dominate, removing Rac1 from those regions. However, when Rac1 is delivered to regions of the cell where GEF activity is high, the GEF would outcompete coronin-1C, thereby both activating Rac1 and preventing coronin-1C-mediated removal of Rac1 from that area. The model goes further, because the action of the GEF exchanges bound GDP for GTP, shifting the equilibrium still further from coronin-1C. Consequently, Rac1 activity could be explained entirely in terms of competition and relative affinity.
In this protocol, we describe a method for comparing the relative affinities of different binding partners for small GTPases, using Rac1 as an example. By using a purified protein approach, it is possible to piece together a chain of signaling events by pair wise comparison, in an experiment where the bound nucleotide can be closely controlled.
Protocollo
1. Purificazione di GTPase GST-tagged
- Cultura una E. coli come BL21 trasformato con pGEX-Rac1 O / N a 37 ° C, agitando a 220 rpm, in 500 ml di media autoinduzione (25 mM Na 2 HPO 4, 25 mM KH 2 PO 4, 50 mM NH 4 Cl, 5 Na mM 2 SO 4, 2 MgSO mm 4, 2 mM CaCl 2, 0,5% glicerolo, 0,05% di glucosio, lattosio 0,2%, 5 g Triptone, 2,5 g estratto di lievito, 100 mg / ml ampicillina).
- Batteri Harvest per centrifugazione per 10 minuti a 10.000 xg, 4 ° C.
- Risospendere pellet batterico in 20 ml di reagente di estrazione di proteine, inibitore della proteasi 1x e incubare per 20 min a temperatura ambiente con inversione.
- Chiarire il lisato mediante centrifugazione a 40.000 xg per 30 min.
- Aggiungere 2 ml di glutatione biglie magnetiche, lavati con PBS (PBS: Na 10 mm 2 HPO 4, 1,8 mm KH 2 PO 4, 137 mM NaCl, 2,7 mM KCl).
- Incubare per 2 ore, mescolando per inversione a 4 ° C.
- Lavare perline proteina-caricato quattro volte con 10 ml di PBS, utilizzando un sorter particella magnetica per precipitare le perle ad ogni passo.
- Perline in 2 ml di PBS e conservare risospendere proteina-caricato a -80 ° C in aliquote di 100 microlitri fino a quando necessario.
2. Espressione di proteine GTPasi binding
- Il giorno prima dell'esperimento, plasmidi transfettare che codificano la proteina fluorescente verde (GFP) -tagged versioni di ogni proteina-GTPasi legame in un pallone da 75 cm 2 separata di HEK293T come segue. Per la convalida di nucleotide carico, transfect GFP-tagged TrioD1 in un terzo di 75 cm 2 fiasco di HEK293T.
- Diluire polyethylamine a 1 mg / ml in 100 ml di NaCl 150 mM sterile.
- Aggiungere 27 ml polyethylamine diluito a 223 ml ridotti multimediali siero.
- Aggiungere 12 mg plasmide DNA per 250 microlitri ridotti multimediali siero.
- Incubare ogni provetta per 2 minuti a temperatura ambiente.
- Unire la polyethylamine e DNA mescola in una singola provetta e agitare per 2 min.
- Incubare per 15-20 minuti a temperatura ambiente.
- Sostituire i mezzi di crescita (Dulbecco Modified Eagle media, il 10% di siero bovino fetale, 2 mM di L-glutammina, antibiotici), sul 90% confluenti HEK293T con 5 ml di terreni di coltura fresco.
- Aggiungere la miscela polyethylamine / DNA combinato al pallone e incubare O / N a 37 ° C, 5% CO 2.
3. Purificazione di proteine GTPasi binding
- Sciacquare fiaschi di cellule trasfettate in PBS e scarico pallone per 5 minuti, aspirando liquido libero.
- Raschiare le cellule in 500 microlitri di buffer di lisi (50 mM Tris-HCl (pH7.8), 1% Nonidet P-40, 1x inibitore della proteasi) in provetta per microcentrifuga.
- Lisare le cellule miscelazione per inversione a 4 ° C per 30 min.
- Durante lisi, lavare due lotti di 40 microlitri GFP-Trappola perle tre volte con tampone di lisi fresco, perline sedimenta a 2.700 xg per 2 minuti tra un lavaggio.
- Chiarire lisati di centrifugazione a 21.000 xg per 10 min.
- Trasferimento chiarito lisato di ciascuna delle proteine concorrenti per separare lavate perline GFP-trappola e che consentono di proteine GFP-fusione di legarsi per 2 ore, mescolando per inversione a 4 ° C. Tenere lisato da cellule GFP-TrioD1 sul ghiaccio.
- Lavare caricate perline GFP-trap due volte in 50 mM Tris-HCl (pH 7,8), 50 mM NaCl, 0,7% (w / v) Nonidet P-40 e due volte in 50 mM Tris-HCl (pH 7,6), 20 mM MgCl 2 , sedimenta perline a 2.700 xg per 2 minuti tra lavaggi.
- Eluire proteine GFP-fusione con l'aggiunta di 40 microlitri 0,2 M glicina (pH 2,5) e pipettando su e giù per 30 sec. Immediatamente perline sedimenti a 21.000 xg per 60 s e trasferimento di liquidi in una nuova provetta da microcentrifuga contenente 4 ml 1 M Tris-HCl (pH 10,4). Fate questo in fretta per limitare i danni alla proteina purificata.
- Analizzare 1 ml di ogni proteina purificata mediante Western blot e sonda con un anticorpo anti-GFP stabilire resa relativa utilizzando un blott quantitativaSistema ing secondo il protocollo del produttore. In alternativa, la determinazione delle concentrazioni di proteine di acido bicinconinico (BCA) assay ma questo introduce errori se le proteine non reagiscono con il dosaggio in modo identico o ci sono proteine contaminanti.
- Uguagliare concentrazione proteica molare con l'aggiunta di 50 mM Tris-HCl (pH 7,6), 20 mM MgCl 2.
4. Nucleotide caricamento di GTPase
- Scongelare un'aliquota di GST-Rac1 biglie magnetiche, previste al punto 1.
- Prendere 90 ml di GST-Rac1 perline e lavare tre volte con 20 mM Tris-HCl (pH 7,6), 25 mM NaCl, 0,1 mM DTT, EDTA 4 mM, utilizzando una selezionatrice particella magnetica per precipitare le perle ad ogni passo.
- Aspirare tampone da perline e aggiungere 100 ml di 20 mM Tris-HCl (pH 7.6), 25 mM NaCl, 0,1 mM DTT, EDTA 4 mm.
- A seconda che il PIL, GTP o no nucleotide carico è necessario per l'esperimento della concorrenza, aggiungere 12 ml PIL 100 mm, 12 microlitri gu 10 mManosine 5 '- [γ-tio] trifosfato (GTPγS) o nessuna nucleotide a 60 microlitri GST-Rac1 perline.
- Per i controlli nucleotide-caricamento, dividere i restanti perle in tre aliquote di 10 microlitri e aggiungere 2 ml PIL 100 mm, 2 pl 10 GTPγS mM o no nucleotide ad ogni provetta.
- Incubare miscele tallone per 30 minuti a 30 ° C con agitazione.
- Stabilizzare nucleotide-bound Rac1 per aggiunta di 1 M MgCl 2: 3 microlitri al mix sperimentale (passo 4.4), 0,5 microlitri a ciascuna delle miscele di controllo (passo 4.5).
5. Concorrenza vincolante.
- Impostare 6 tubi microcentrifuga, ciascuna contenente:
200 microlitri 50 mM Tris-HCl (pH 7,6), 20 mM MgCl 2
10 microlitri sperimentali perline Rac1 nucleotide-caricati (dal punto 4.7)
5 microlitri proteina Rac1-binding A (proteina di legame costante) - Per ogni tubo, aggiungere 0, 1, 2,5, 5, 10 o 20 l-Rac1 binding protein B (proteina di legame variabile). Questi volumi assumono unpproximately uguali concentrazioni di magazzino delle proteine di legame costante e variabile e può essere necessario un aggiustamento.
- Regolare volumi di proteine leganti A e B se ci sono grandi differenze tra le affinità di legame delle due proteine e questo dovrebbe essere determinati empiricamente attraverso le ripetizioni sperimentali. Portare il volume totale della miscela di legame di 235 ml per aggiunta di 50 mM Tris-HCl (pH 7,6), 20 mM MgCl 2.
- Impostare una provetta da microcentrifuga contenente:
200 microlitri 50 mM Tris-HCl (pH 7,6), 20 mM MgCl 2
10 microlitri sperimentali perline Rac1 nucleotide-caricati (dal punto 4.7)
10 microlitri proteina Rac1-binding A (proteina di legame costante) - Impostare il PIL, GTPγS e senza tubi di controllo nucleotide:
200 microlitri 50 mM Tris-HCl (pH 7,6), 20 mM MgCl 2
10 microlitri perline Rac1 controllo caricati al passo 4.5 con il PIL, GTPγS o no nucleotidi e stabilizzato al punto 4.7.
180 microlitri HEK293T GFP-TrioD1 lisato, preparata come al punto 3.6
4 microlitri 1 M MgCl 2 - Incubare la miscela per 2 ore, mescolando per inversione a 4 ° C.
- Lavare le perline tre volte con 50 mM Tris-HCl (pH 7,6), 20 mM MgCl 2.
- Eluire proteine legate in 20 microlitri riducenti tampone (50 mM Tris-HCl (pH 7), 5% SDS, 20% glicerolo, 0,02 mg / ml di blu di bromofenolo, 5% β-mercaptoetanolo).
6. Analisi della concorrenza
- Risolvere 10 ml di proteina legata (passo 5,6) mediante elettroforesi dodecil solfato di sodio gel di poliacrilamide (SDS-PAGE) e Western Blot.
- Incubare la membrana a 4 ° CO / N in anticorpo anti-GFP diluito 1/1000 in tampone bloccante diluito a 1x in PBS, 0,1% di Tween-20 per rilevare entrambe le proteine GTPasi legame con tag.
- Lavare la membrana per tre volte per 10 minuti con PBS, 0,1% Tween-20.
- Incubare la membrana per 30 minuti a RT in DyLight 800 sec-coniugato anti-coniglioanticorpo daria, diluito 1 / 10.000 in tampone bloccante diluito a 1x in PBS, 0,1% di Tween-20.
- Lavare la membrana per tre volte per 10 minuti con PBS, 0,1% Tween-20.
- Scansione la membrana utilizzando un sistema di imaging a raggi infrarossi, utilizzando il software per misurare l'intensità di banda secondo il protocollo del produttore.
- Tracciare la intensità della banda di ciascuna proteina contro il volume del concorrente variabili (Protein B).
- Dividere il volume di concorrente variabile nel punto in cui le linee si intersecano dal volume di concorrente costante (Proteina A, 5 ml) per determinare il rapporto concorrente che è raggiunto l'equilibrio.
- Per la convalida dello stato di nucleotide-caricamento, membrane sonda per p21-chinasi attivata 1 (PAK1) (un'unità di effetti) e GFP-TrioD1 (un GEF), come descritto nei passaggi 6.1-6.6.
Risultati
Questo protocollo è progettato per calcolare le relative affinità di legame per i partner Rac1, senza la necessità di conoscere la precisa concentrazione dei concorrenti (Figura 1). Determinazione della concentrazione proteica introduce errori e quando si considera la concorrenza tra le molecole in un percorso di segnalazione non è necessaria. Tuttavia, è importante sapere che i due concorrenti hanno la stessa concentrazione molare nelle soluzioni madre a consentire rapporti semplici da calcolare quando si aggiungono volumi diversi per il saggio. 40 ml di perline GFP-trap hanno una capacità di legame di ~ 300 pmol così un confluente 75 centimetri 2 fiaschi di altamente cellule che esprimono saturerà le perline, con il risultato che i preparativi delle due diverse proteine di legame saranno simili prima della regolazione (figura 2A). Se una delle proteine esprime male, questo problema può essere superato purificando quella proteina da più di un pallone di cellule.
Il legame di maggior effettori GTPasi e regolatori dipende dal nucleotide-caricamento della GTPasi esca, quindi è importante verificare se il carico ha avuto successo. Caricamento in corso può essere verificata precipitando proteine di legame conosciute da lisati cellulari. Proteine effettrici, quali PAK1 legano a GTP-Rac1 e possono essere facilmente precipitati da lisati e rilevati mediante Western blotting 8 (Figura 2B). GEFs legano preferenzialmente al nucleotide-liberi GTPasi per stabilizzare lo stato di transizione. Come GEFs sono di bassa abbondanza, solitamente inattivo e spesso asciugare bene male, è meglio iperespressione di un GEF o GEF frammento di prova GTPasica senza nucleotide. Usiamo spesso la prima omologia Dbl di Trio, espresso come una fusione GFP (GFP-TrioD1 9) (Figura 2B), ma qualsiasi GEF avrebbe funzionato. Le proteine che si legano al GTPasi PIL-caricati sono più rari. Recentemente abbiamo riportato RCC2 come una di queste proteine 7, o PIL di carico può essere convalidato semplicemente come binding né alla GEF né effettrici.
L'uscita dal esperimento sarà una macchia occidentale raffigurante i due partner di legame GFP-tagged legati alla GTPasi. Utilizzando un singolo anticorpo per rilevare entrambe le proteine, le concentrazioni alle quali simili quantità di entrambi i concorrenti legano può essere determinato e quindi le relative affinità presupposta. In questa competizione esempio, tra il dominio propulsore della proteina Rac1-tratta, Coronin-1C (Rac1-binding protein A), e la proteina Rac1-sequestro, RCC2 (Rac1-binding protein B), è dimostrato (Figura 3A). Utilizzando un volume costante di Coronin-1C elica (5 ml), e l'aggiunta di crescenti volumi di RCC2, possiamo vedere dalla GFP macchia che l'equilibrio è raggiunto a 1,25-2,5 ml di RCC2 (asterisco), dimostrando che ha una forte RCC2 affinità per Rac1 di Coronin-1C. Misurando l'intensità delle bande utilizzando Western blotting quantitativa, e riportando i valori medi per ogni competitoR, il punto di equilibrio può essere calcolata con precisione identificando i volumi in cui le curve si intersecano (Figura 3B).
Uno dei possibili ostacoli ad un dosaggio competere con successo è se il partner di legame si legano l'uno all'altro così come legame Rac1. Nella Figura 3A + B dimostriamo concorrenza tra RCC2 e il dominio propulsore di Coronin-1C, piuttosto che full-length Coronin-1C. La ragione per usare la Coronin troncato è che Coronin-1C si lega anche RCC2 attraverso il dominio di coda. Quando full-length Coronin-1C è titolato con RCC2, legame di entrambe le proteine viene rilevato, a causa della formazione del complesso ternario, piuttosto che la concorrenza (Figura 3C). Se la concorrenza è in corso, legame di una proteina aumenta mentre l'altra diminuisce, e totale legato GFP-fusione rimarrà costante. Nei casi in cui un complesso ternario forma è necessario troncare una delle proteine GTPasi vincolanti, talché la competmodifiche ai contenuti interagire non più.

Figura 1. Flusso di lavoro. Rappresentazione schematica del flusso di lavoro per determinare l'affinità delle proteine GTPasi legame con saggi di concorrenza. Cliccate qui per vedere una versione più grande di questa figura.
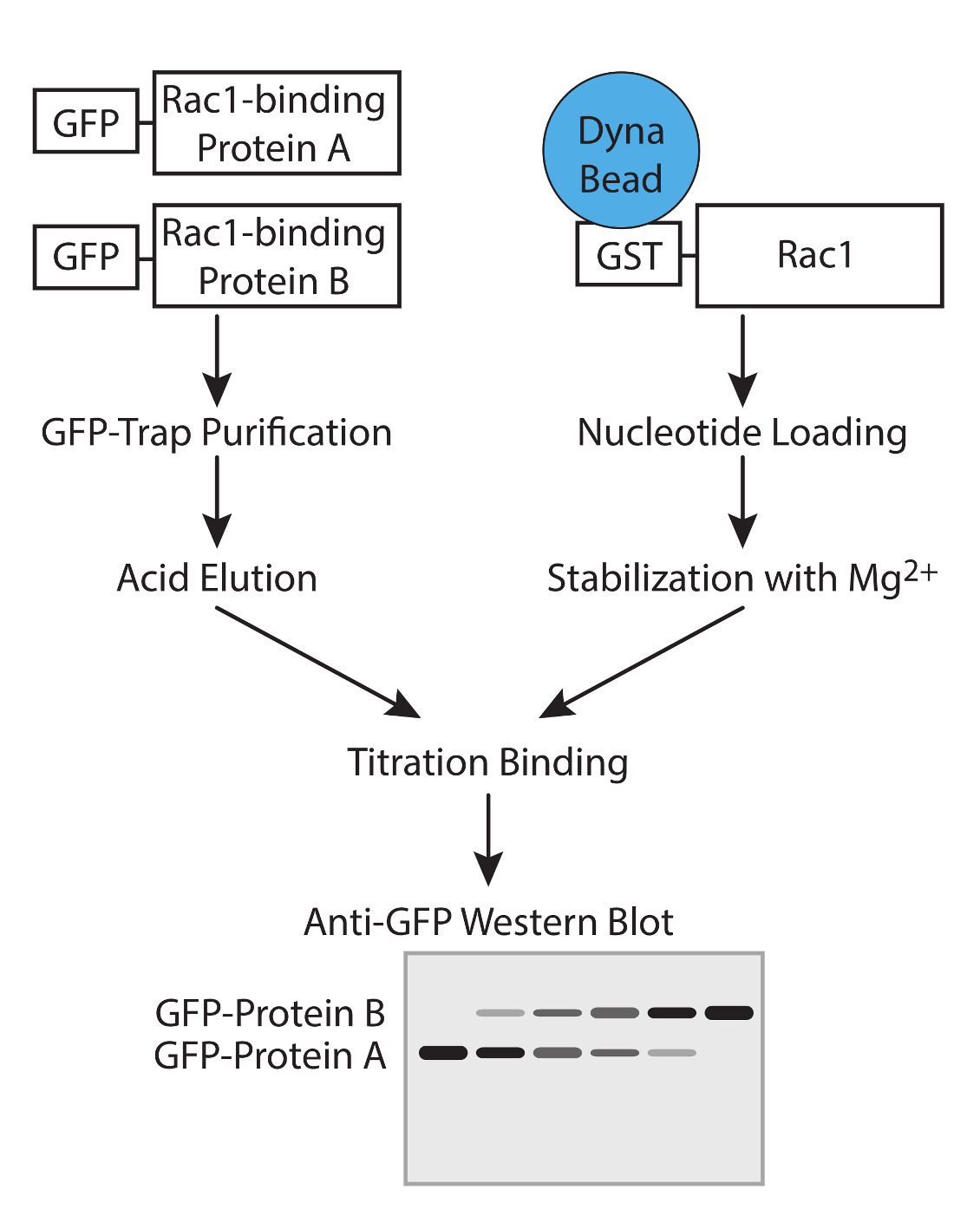{kind=link}

Figura 2. La convalida di proteine purificate. (A) purificato GFP-tagged proteine analizzate mediante Western blot vincolante, sondando con anti-GFP per determinare la resa relativa delle due proteine Rac1. Questo tipo di equalizzazione durante l'esperimento permette la concentrazione delle due proteine di essere regolata in modo che corrispondano nell'esperimento vincolante. (B) GDP, GTPγS e non nucleotide-caricato GST-Rac1 è stata incubata con lisato da HEK293T esprimono GFP-TrioD1 e proteine rilevate tracciando per PAK1 endogena o sovraespresso GFP-TrioD1 legate. Cliccate qui per vedere una versione più grande di questa figura.
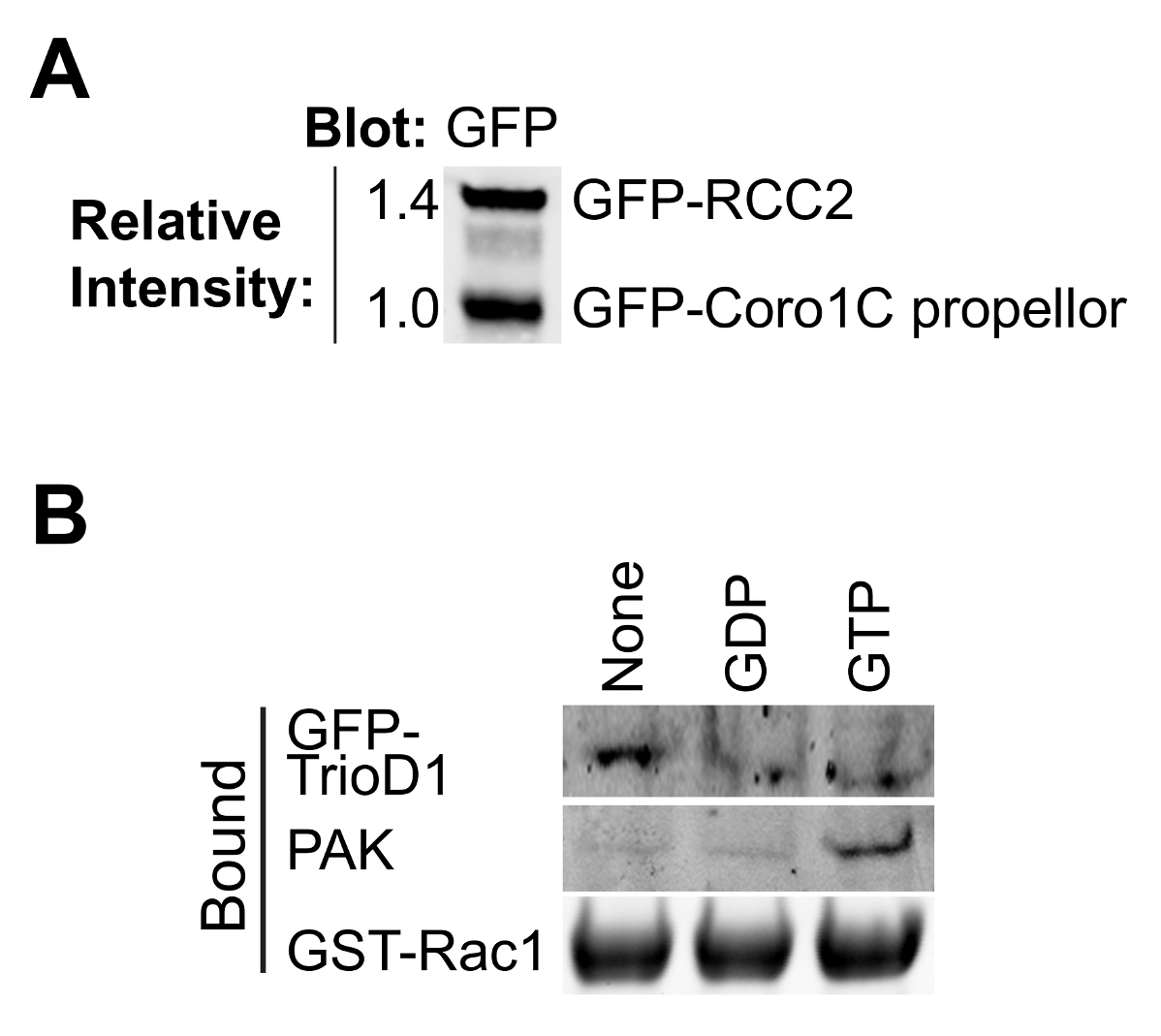{kind=link}

Figura 3. Western blot di proteine relativo vincolante. Uscite esempio da saggi di concorrenza vincolante. (A) GDP-caricato Rac1 è stato mescolato con 5 microlitri GFP-Coronin-1C dominio elica e volumi crescenti di GFP-RCC2 stati titolato in. Per Western blotting proteine legate per GFP, problemi con rilevamento differenziale delle due proteine vengono evitati e il segnale GFP riporta il rapporto molare tra i due proteine di fusione. Gli asterischi indicano i rapporti di concorrenza in eithelato r il punto di equilibrio. (B) Banda intensità rilegati proteine GFP fusione di tre esperimenti indipendenti sono stati misurati mediante Western blotting quantitativa, utilizzando fluoroforo-coniugato anticorpi secondari e le medie complottato per calcolare la quantità di RCC2 necessaria per raggiungere l'equilibrio. (C ) Esempio di output da un esperimento in cui proteine Rac1 vincolanti legano fra loro e formano un complesso ternario, invece di competere. PIL-caricato Rac1 è stato mescolato con 5 microlitri GFP-RCC2 e volumi crescenti di GFP-Coronin-1C full-length sono stati titolati in. L'aumento del limite GFP-Coronin-1C senza perdita di limite GFP-RCC2 indica ternario formazione complessa. Si prega clicca qui per vedere una versione più grande di questa figura.
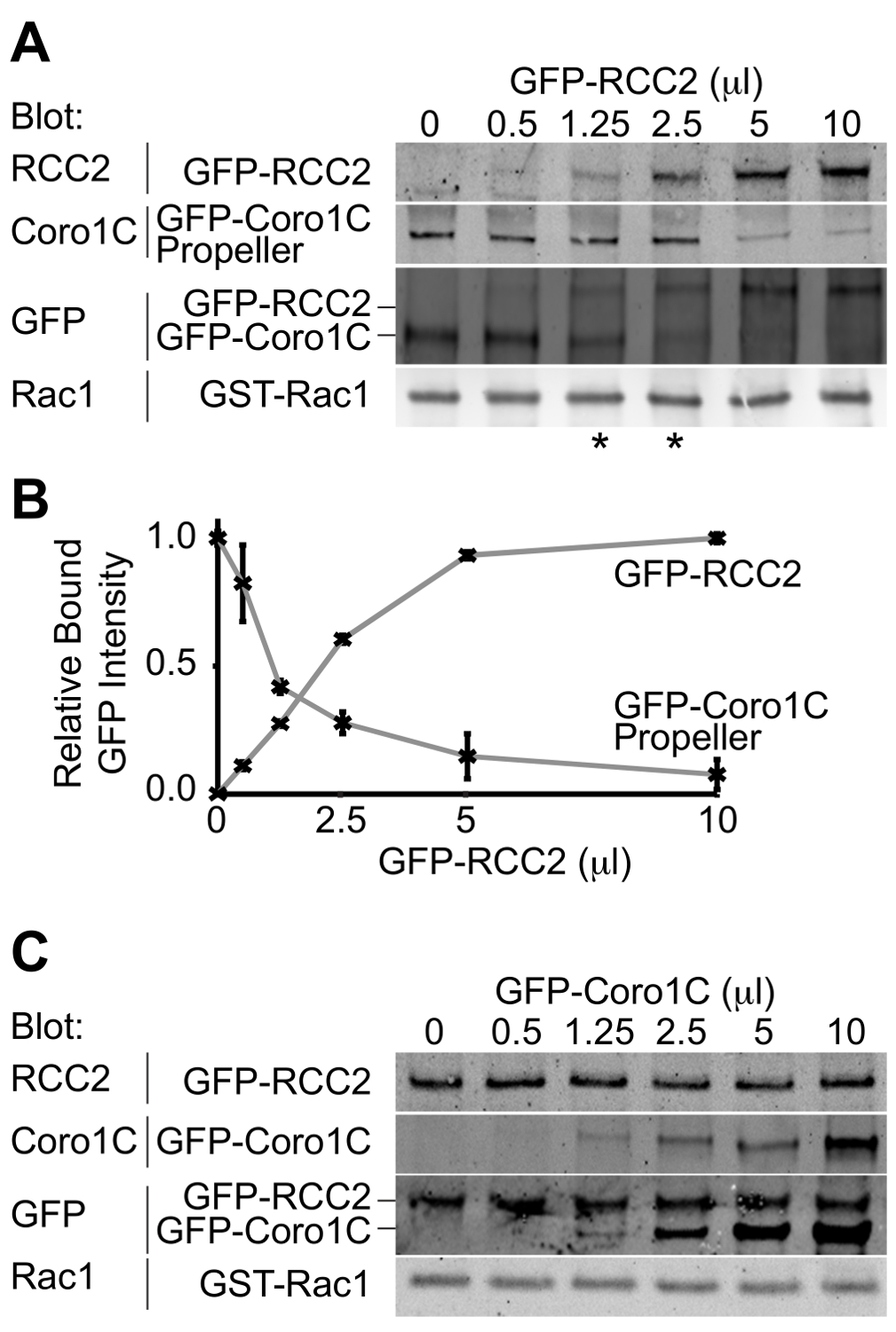{kind=link}
Discussione
This protocol describes a method for comparing the relative affinities of pairs of small GTPase-binding proteins. The key steps are the preparation of purified GTPase-binding proteins and the nucleotide loading of the GTPase. The use of GTPase-binding proteins with the same GFP tag, allows the concentrations at which similar amounts of each competitor binds to be accurately determined. The use of recombinant nucleotide-loaded GTPase allows interrogation of the binding properties of the GTPase under specific activity conditions. This step is also the most sensitive as nucleotides will both hydrolyze and detach from the GTPase if the magnesium conditions are not maintained precisely.
In the cell, the large number of GTPase-binding proteins combined with the rapid nucleotide turnover makes such pathways difficult to interpret. The simplicity of this method in comparing only pairs of binding proteins and using carefully controlled nucleotide-loading conditions allows signaling pathways to be elucidated. However, the greatest strength of the protocol is also the greatest weakness as it is a simplification of the in vivo situation. Competition assays can be used to build a robust hypothesis, but this should then be tested in cells by knockdown experiments.
There are three features that must be considered when selecting the GFP-tagged GTPase-binding proteins to be used in the experiment. First, the fusion proteins must express well in mammalian cells, such as HEK293T, as competition assays require a reasonable amount of protein. Second, it must be possible to purify the recombinant protein without significant degradation, and where this is not possible, cloning of a GTPase-binding fragment should be considered. Third, the two GTPase-binding proteins must resolve from one another on SDS-PAGE to allow analysis in section 6.
There are a number of potential caveats to the experiment that need to be considered, and possibly addressed:
Possible denaturation of purified GTPase-binding proteins during the acid elution step or steric hindrance by the GFP tag. In our hands, these have not been a problem, but must be tested. The purified proteins can be tested in functional assays 10. Commercial kits now exist for testing the activity of GEFs or GAPs without the need for isotope-labeled nucleotides. Sequestering proteins, by their nature protect GTPases from GEF or GAP activity, so can be used as competitive inhibitors in the commercial GEF or GAP assays, as we did in our recent publication 7. The relevant feature of proteins that traffic GTPase are the capacity to bind the GTPase, and this can be tested easily in a pull down assay. An alternative approach to testing protein integrity that is applicable to all binding proteins is to titrate protein eluted from GFP-trap beads with glycine with the same protein removed from GFP-trap beads by enzymatic cleavage. The experiment would be analyzed by probing both the GFP-tagged and cleaved protein with an antibody against the protein itself. If the protein is undamaged by elution, equilibrium should be achieved at a 1:1 ratio. This approach would also indicate whether the presence of the GFP tag itself compromises the binding properties of the candidate protein, though this does require the production of a construct with an enzymatic cleavage site between the tag and the binding protein. Whether the protein is compromised by the tag or the elution step, the problem could be addressed by modifying the protocol to use an alternative purification method. Rather than GFP, binding proteins could be His-tagged, purified using Ni-NTA and analyzed using an antibody against the His-tag. The important feature is that both binding proteins must share a common tag although, if necessary, two tags could be added to a protein, one for purification and the other for detection.
The protocol is designed to investigate competition between interactions with the switch I/II domains. Although the majority of GTPase interactions are mediated by this motif, there are some exceptions, most notably the interactions of GDIs that bind to the prenyl tail, as well as obscuring the switch domains. In principle, the protocol could be adapted to use GTPase purified from mammalian cells, so that the GTPase is prenylated, however, the presence of multiple binding sites or allosteric effects complicate the interpretation of competition-binding data. Further problems associated with such a modification are that GDIs co-purify with GTPase from mammalian cells, compromising the purity of the isolated proteins and the hydrophobic nature of the prenyl groups means that prenylated GTPases are associated with either GDI or lipid membrane and such factors would need to be considered in the experiment.
The amount of GST-Rac1 being used in the assay. The constant GTPase binding protein must be at a greater concentration than the Rac1, or when the competitor is added, it will simply bind to free Rac1. It will be immediately obvious if this has happened as binding of the competitor, without a loss of the constant protein, will be detected in much the same way as when the two competing proteins bind to one another as shown in Figure 3B. As an additional control (Step 5.3), a binding reaction containing double the amount of constant binding protein and no variable binding protein should be included (Step 5.3). If the Rac1 in the titration experiment is saturated, doubling the amount of constant binding protein will have no effect on the output. The volumes suggested in the protocol should be appropriate, but the amount of Rac1 can be easily reduced. If binding of the competitor without loss of the constant binding partner is observed, reducing the amount of Rac1 should be attempted before trying to map binding sites to avoid ternary complex formation.
Non-specific interaction of GTPase-binding proteins with the GST or bead, as well as specifically with Rac1. This problem would be manifested by residual binding of the constant GTPase-binding protein, even when the variable GTPase-binding protein has reached a plateau at high concentration. Identification of this issue will be aided by conducting reciprocal experiments where the constant and variable GTPase-binding proteins are swapped. Reciprocal experiments will also greatly improve the accuracy of the estimate of equilibrium point, so should always be included. In cases of non-specific binding, the relative concentrations at which equilibrium is achieved can still be calculated by comparing band intensity between the maxima and minima for each protein, or by measuring the extent of non-specific binding by using GST beads as bait, rather than GST-Rac1.
Pull down assays using different nucleotide-loading conditions should be used to complement the competition assay described in this protocol. Determining the nucleotide preference of partners is important for both understanding the competition events and understanding the signaling pathway that the GTPase-binding protein is involved in. In Figure 2B we analyze binding of proteins with established preference for GTP-loaded or nucleotide-free GTPase as a means to validate nucleotide loading. However, it is sensible to investigate the effect of nucleotide loading on each of the competitors as well. If the hypothetical competitors show different preferences, competition will make less of a contribution to the signaling pathway, and indeed nucleotide turnover is likely to be the mechanism that directs exchange of the binding proteins.
Divulgazioni
Gli autori non hanno nulla da rivelare.
Riconoscimenti
This work was supported by Wellcome Trust grant 088419 to MDB.
Materiali
| Name | Company | Catalog Number | Comments |
| Bugbuster | Novagen | 70584-3 | |
| COMPLETE protease inhibitor | Roche | 05 056 489 001 | |
| Glutathione magnetic beads | Pierce | 88821 | |
| Polyethylenimine, branched, average Mw ~25,000 | Sigma Aldrich | 408727-100ML | |
| OPIMEM | Life Technologies | 31985-047 | |
| Dulbecco's Modified Eagle Media | Sigma Aldrich | D5796 | |
| Fetal Bovine Serum | Life Technologies | 10270-1-6 | |
| L-Glutamine | Life Technologies | 25030-024 | |
| GFP-Trap_A | Chromotec | gta-20 | |
| GDP | Sigma Aldrich | G7127 | Highly unstable. Aliquot and store at -80 immediately upon reconstritution |
| GTPγS | Sigma Aldrich | G8634 | Highly unstable. Aliquot and store at -80 immediately upon reconstritution |
| Blocking Buffer | Sigma Aldrich | B6429 | |
| Tween-20 | Sigma Aldrich | P9416 | |
| Anti-GFP antibody | Living Colors | 632592 | Use at 1/1000 dilution |
| DyLight 800 conjugated goat anti-rabbit secondary antibody | Fisher Scientific | 10733944 | |
| Anti-PAK1 antibody | Cell Signaling | 2602S | Use at 1/1000 dilution |
| Odyssey SA Infrared Imaging System | Li-cor | 9260-11PC |
Riferimenti
- Burridge, K., Rho Wennerberg, K. and Rac take center stage. Cell. 116 (2), 167-179 (2004).
- Raftopoulou, M., Hall, A. Cell migration: Rho GTPases lead the way. Dev Biol. 265 (1), 23-32 (2004).
- Rossman, K. L., Der, C. J., Sondek, J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol. 6 (2), 167-180 (2005).
- Worthylake, D. K., Rossman, K. L., Crystal Sondek, J. structure of Rac1 in complex with the guanine nucleotide exchange region of Tiam1. Nature. 408 (6813), 682-688 (2000).
- Scheffzek, K., Ahmadian, M. R. GTPase activating proteins: structural and functional insights 18 years after discovery. Cell Mol Life Sci. 62 (24), 3014-3038 (2005).
- Del Pozo,, A, M., et al. Integrins regulate GTP-Rac localized effector interactions through dissociation of Rho-GDI. Nat Cell Biol. 4 (3), 232-239 (2002).
- Williamson, R. C., et al. Coronin-1C and RCC2 guide mesenchymal migration by trafficking Rac1 and controlling GEF exposure. J Cell Sci. 127 (Pt 19), 4292-4307 (2014).
- Del Pozo, M. A., Price, L. S., Alderson, N. B., Ren, X. D., Schwartz, M. A. Adhesion to the extracellular matrix regulates the coupling of the small GTPase Rac to its effector PAK. Embo J. 19 (9), 2008-2014 (2000).
- Van Rijssel, J., Hoogenboezem, M., Wester, L., Hordijk, P. L., Van Buul, J. D. The N-terminal DH-PH domain of Trio induces cell spreading and migration by regulating lamellipodia dynamics in a Rac1-dependent fashion. PLoS. 7 (1), e29912(2012).
- Self, A. J., Hall, A. Measurement of intrinsic nucleotide exchange and GTP hydrolysis rates. Methods Enzymol. 256, 67-76 (1995).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati