
Method Article
Vergleicht man die Affinität der GTPase-bindende Proteine mit Competition-Assays
In diesem Artikel
Zusammenfassung
This protocol compares the relative affinities of binding partners for Rho-family GTPases, including Rac1. In vivo, Rac1-binding proteins compete for a single binding interface, the conformation of which is dictated by a bound nucleotide. The nucleotide is both important and difficult to control experimentally, due to the high hydrolysis rate.
Zusammenfassung
In this protocol we demonstrate a method for comparing the competition between GTPase-binding proteins. Such an approach is important for determining the binding capabilities of GTPases for two reasons: The fact that all interactions involve the same face of the GTPases means that binding events must be considered in the context of competitors, and the fact that the bound nucleotide must also be controlled means that conventional approaches such as immunoprecipitation are unsuitable for GTPase biochemistry. The assay relies on the use of purified proteins. Purified Rac1 immobilized on beads is used as the bait protein, and can be loaded with GDP, a non-hydrolyzable version of GTP or left nucleotide free, so that the signaling stage to be investigated can be controlled. The binding proteins to be investigated are purified from mammalian cells, to allow correct folding, by means of a GFP tag. Use of the same tag on both proteins is important because not only does it allow rapid purification and elution, but also allows detection of both competitors with the same antibody during elution. This means that the relative amounts of the two bound proteins can be determined accurately.
Einleitung
The actin cytoskeleton that determines the shape, polarity and migratory properties of mammalian cells is regulated by the Rho-family of small GTPases. The Rho-family GTPases include RhoA that stimulates cytoskeletal contraction, Rac1 that stimulates actin branching and membrane protrusion, and Cdc42 that has similar effects on actin polymerization to Rac1 and causes the formation of filopodia 1,2. GTPase signaling activity is determined by binding of a nucleotide, which controls the contraction and relaxation of the switch I and switch II loops that mediate the protein-protein interactions with both regulators and effectors. Guanosine 5’-triphosphate (GTP)-bound GTPases activate downstream effectors, whereas the Guanosine 5’-diphosphate (GDP)-bound form is inactive. In the cell, cycles of GTP hydrolysis and nucleotide exchange allow rapid turnover of GTPase signals that are necessary for cytoskeletal dynamics. Nucleotide turnover is regulated by three mechanisms. Guanine nucleotide exchange factors (GEFs) stabilize the nucleotide-free GTPase, catalyzing exchange of GDP for GTP, and thereby stimulating GTPase signaling activity 3,4. GTPase-activating proteins (GAPs) catalyze hydrolysis of GTP to GDP, thereby inhibiting GTPase signaling activity 5. Sequestering molecules such as regulator of chromatin condensation 2 (RCC2) and guanine nucleotide dissociation inhibitors (GDIs) obscure the switch loops and in the case of GDIs remove the GTPase from the membrane by interaction with the prenyl tail 6,7. Each of the three classes of regulatory molecule interact with the switch loops, as do the downstream effectors and some trafficking regulators such as coronin-1C 7. The purpose of this protocol is to measure competition for the switch I/II binding site between putative regulators and downstream signaling molecules. It should be noted that competition assays test binding to a shared binding site, so that this protocol is not suitable for testing interactions with other sites, such as binding of GDIs to the prenyl tail.
The subtlety of the conformation differences between active and inactive forms, combined with the labile nature of the bound nucleotide, has made study of GTPase-binding events difficult. The role of the bound nucleotide means that conventional binding assays such as immunoprecipitation or surface plasmon resonance are not well suited to investigation, as the nucleotide cannot be controlled. This obstacle is compounded by the overlap in the binding sites of GEFs, GAPs, effectors, sequestering molecules and trafficking molecules, which make binding data for a single interaction difficult to interpret in the context of the competition that will occur in the cell. Immunoprecipitation, in particular, is compromised by competition between binding partners, as under certain cellular conditions, one binding partner might be identified at the expense of all others, while under other conditions, another partner might dominate. The dynamic nature of GTPase signaling is essential to GTPase function and must be considered when analyzing the relationships between the binding interactions of different regulators. Indeed, we recently described a pathway that relied heavily on competitive binding. We identified coronin-1C as a trafficking molecule that bound to the switch loops of GDP-Rac1 7. In areas of low GEF activity, trafficking would dominate, removing Rac1 from those regions. However, when Rac1 is delivered to regions of the cell where GEF activity is high, the GEF would outcompete coronin-1C, thereby both activating Rac1 and preventing coronin-1C-mediated removal of Rac1 from that area. The model goes further, because the action of the GEF exchanges bound GDP for GTP, shifting the equilibrium still further from coronin-1C. Consequently, Rac1 activity could be explained entirely in terms of competition and relative affinity.
In this protocol, we describe a method for comparing the relative affinities of different binding partners for small GTPases, using Rac1 as an example. By using a purified protein approach, it is possible to piece together a chain of signaling events by pair wise comparison, in an experiment where the bound nucleotide can be closely controlled.
Protokoll
1. Reinigung von GST-tagged GTPase
- Kultur ein E. coli-Stamm, wie beispielsweise BL21 mit pGEX-Rac1 O / N transformierten bei 37 ° C, bei 220 Upm Schütteln in 500 ml Autoinduktion Medium (25 mM Na 2 HPO 4, 25 mM KH 2 PO 4, 50 mM NH 4 Cl, 5 mM Na 2 SO 4, 2 mM MgSO 4, 2 mM CaCl 2, 0,5% Glycerin, 0,05% Glucose, 0,2% Lactose, 5 g Trypton, 2,5 g Hefeextrakt, 100 ug / ml Ampicillin).
- Ernte Bakterien durch Zentrifugation für 10 min bei 10.000 × g, 4 ° C.
- Resuspendieren Bakterienpellet in 20 ml Protein Extraktionsreagens, 1x Proteaseinhibitor und Inkubation für 20 min bei RT mit Inversion.
- Klärung des Lysats durch Zentrifugation bei 40.000 xg für 30 min.
- 2 ml Glutathion-magnetische Kügelchen, mit Phosphat-gepufferter Salzlösung (PBS: 10 mM Na 2 HPO 4, 1,8 mM KH 2 PO 4, 137 mM NaCl, 2,7 mM KCl).
- Inkubation für 2 h, das Mischen durch Inversion bei 4 ° C.
- Viermal Waschen Kügelchen proteinbeladene mit 10 ml PBS unter Verwendung eines magnetischen Teilchens Sortierer, um die Perlen in jedem Schritt auszufällen.
- Resuspendieren Protein beladenen Kügelchen in 2 ml PBS und bei -80 ° C in 100 ul Aliquots bis sie benötigt.
2. Expression von GTPase-bindenden Proteinen
- Am Tag vor dem Experiment Transfektion Plasmide grün fluoreszierende Protein (GFP) kodiert -markierte Versionen jedes GTPase-bindenden Proteins in einem separaten 75-cm 2 -Kolben von HEK293T wie folgt. Zur Validierung von Nukleotid-Laden, transfizieren GFP-markierten TrioD1 in ein drittes 75-cm 2-Kolben von HEK293T.
- Verdünnter polyethylamine bis 1 mg / ml in 100 & mgr; l sterilem 150 mM NaCl.
- In 27 ul verdünnt polyethylamine bis 223 & mgr; l reduziert Serum Medien.
- In 12 & mgr; g Plasmid-DNA zu 250 & mgr; l reduziert Serum Medien.
- Inkubieren jedes Röhrchen für 2 min bei RT.
- Kombinieren Sie die polyethylamine und DNA vermischt sich in einem einzigen Rohr und Vortex für 2 min.
- Inkubieren Sie für 15-20 min bei RT.
- Ersetzen Sie das Wachstumsmedium (Dulbeccos modifiziertem Eagle Medium, das 10% fötales Rinderserum, 2 mM L-Glutamin, keine Antibiotika), die auf 90% konfluent HEK293T mit 5 ml frisches Wachstumsmedium.
- In der kombinierten polyethylamine / DNA-Gemisch in den Kolben und Inkubation O / N bei 37 ° C, 5% CO 2.
3. Reinigung der GTPase-bindenden Proteinen
- Spülen Sie die Flasche aus transfizierten Zellen in PBS und Drain-Kolben für 5 min, Absaugen freie Flüssigkeit.
- Abzukratzen Zellen in 500 ul Lysepuffer (50 mM Tris-HCl (pH 7,8), 1% Nonidet P-40, 1x Proteaseinhibitor) in Mikrozentrifugenröhrchen.
- Zellen lysieren durch Mischen durch Inversion bei 4 ° C für 30 min.
- Während der Lyse, waschen zwei Lose von 40 ul GFP-Trap-Kügelchen dreimal mit frischem Lysepuffer, sedimentierenden Perlen bei 2700 × g für 2 Minuten zwischen den Waschvorgängen.
- Klärung Lysate durch Zentrifugation bei 21.000 × g für 10 min.
- Übertragungs geklärte Lysat eines jeden der Wettbewerber Proteine gewaschen GFP-trap Perlen abzutrennen und zu ermöglichen GFP-Fusionsproteine, für 2 h binden, Mischen durch Umdrehen bei 4 ° C. Halten Lysat aus GFP-TrioD1 Zellen auf Eis.
- Zweimal waschen geladen GFP-Trap-Perlen in 50 mM Tris-HCl (pH 7,8), 50 mM NaCl, 0,7% (w / v) Nonidet P-40 und zweimal in 50 mM Tris-HCl (pH 7,6), 20 mM MgCl 2 , sedimentierenden Perlen bei 2700 × g für 2 Minuten zwischen den Waschgängen.
- Eluieren GFP-Fusionsproteinen durch Zugabe von 40 ul 0,2 M Glycin (pH 2,5) und Auf- und Abpipettieren für 30 sec. Sofort Sediment Kügelchen bei 21.000 × g für 60 s und Transferflüssigkeit auf ein neues Mikrozentrifugenröhrchen, enthaltend 4 & mgr; l 1 M Tris-HCl (pH 10,4). Tun dies schnell zur Beschädigung des gereinigten Proteins zu begrenzen.
- Analyse 1 ul jedes gereinigten Proteins durch Western-Blot und die Sonde mit einem anti-GFP-Antikörper, um die relative Ausbeute herzustellen unter Verwendung eines quantitativen Blotting System gemäß dem Protokoll des Herstellers. Alternativ bestimmen Proteinkonzentrationen von Bicinchoninsäure (BCA) Assay aber dies führt Fehler, wenn die Proteine nicht mit dem Assay in einer identischen Weise reagiert oder es kontaminierende Proteine.
- Leichen molaren Proteinkonzentration durch die Zugabe von 50 mM Tris-HCl (pH 7,6), 20 mM MgCl 2.
4. Nucleotide Belastung der GTPase
- Tauwetter ein Aliquot von GST-Rac1 magnetische Kügelchen, die in Schritt 1 hergestellt.
- Nehmen 90 ul von GST-Rac1 Perlen und dreimal mit 20 mM Tris-HCl (pH 7,6), 25 mM NaCl, 0,1 mM DTT, 4 mM EDTA, unter Verwendung eines magnetischen Partikelsortierer, um die Perlen in jedem Schritt auszufällen.
- Saugen Sie Puffer aus Perlen und fügen Sie 100 ul 20 mM Tris-HCl (pH 7,6), 25 mM NaCl, 0,1 mM DTT, 4 mM EDTA.
- Je nachdem, ob das BIP, GTP oder keine Nukleotid-Belastung für den Wettbewerb Experiment erforderlich, fügen Sie 12 ul 100 mM BIP, 12 & mgr; l 10 mM guanosine 5 '- [γ-thio] Triphosphat (GTP & ggr;) oder keine Nukleotid bis 60 & mgr; l GST-Rac1 Perlen.
- Für die Nukleotid-Ladesteuerungen, teilen Sie die restlichen Perlen in drei 10-ul-Aliquots und fügen Sie 2 ul 100 mM GDP, 2 & mgr; l 10 mM GTPyS oder keine Nukleotid in jedes Röhrchen.
- Inkubieren Wulst Mischungen für 30 Minuten bei 30 ° C unter Rühren.
- Stabilisierung Nucleotid gebundene Rac1 durch Zugabe von 1 M MgCl 2: 3 & mgr; l auf die Versuchsmischung (Schritt 4.4), 0,5 & mgr; l zu jeder der Kontrollmischungen (Schritt 4.5).
5. Wettbewerb bindend.
- Einrichten 6 Mikrozentrifugenröhrchen, die jeweils:
200 ul 50 mM Tris-HCl (pH 7,6), 20 mM MgCl 2
10 & mgr; experimentelle Nukleotid-beladenen Rac1-Perlen (ab Schritt 4.7)
5 ul Rac1-bindenden Protein A (Konstante Bindungsprotein) - Zu jedem Röhrchen hinzu 0, 1, 2,5, 5, 10 oder 20 ul Rac1-bindendes Protein B (variable Bindungsprotein). Diese Bände davon ausgehen, approximately gleich stock Konzentrationen der konstanten und variablen Bindungsproteine und muss unter Umständen angepasst werden.
- Einstellen Volumina bindende Proteine A und B, wenn es große Unterschiede in den Bindungsaffinitäten der beiden Proteine und dies sollte empirisch durch die Versuchswiederholungen bestimmt werden. Bilden das Gesamtvolumen des Bindungsgemisch auf 235 & mgr; l durch Zugabe von 50 mM Tris-HCl (pH 7,6), 20 mM MgCl 2.
- Einrichtung eines Mikrozentrifugenröhrchen enthält:
200 ul 50 mM Tris-HCl (pH 7,6), 20 mM MgCl 2
10 & mgr; experimentelle Nukleotid-beladenen Rac1-Perlen (ab Schritt 4.7)
10 ul Rac1-bindenden Protein A (Konstante Bindungsprotein) - Einrichten des BIP, GTPyS und keine Nukleotid-Kontrollröhrchen:
200 ul 50 mM Tris-HCl (pH 7,6), 20 mM MgCl 2
10 ul Steuer Rac1 Perlen in Schritt 4.5 mit dem BIP, GTPyS oder kein Nukleotid geladen und in Schritt 4.7 stabilisiert.
180 ul HEK293T GFP-TrioD1 Lysat, wie in Schritt 3.6 hergestellten
4 & mgr; l 1 M MgCl 2 - Inkubieren der Mischung für 2 h, das Mischen durch Inversion bei 4 ° C.
- Dreimal mit 50 mM Tris-HCl (pH 7,6), 20 mM MgCl 2 Waschen der Kügelchen.
- Eluieren die gebundenen Proteine werden in 20 ul reduzierendem Probenpuffer (50 mM Tris-HCl (pH 7), 5% SDS, 20% Glycerin, 0,02 mg / ml Bromphenolblau, 5% β-Mercaptoethanol).
6. Analyse der Wettbewerbs
- Beheben 10 ul des gebundenen Proteins (Schritt 5.6), bestimmt durch Natriumdodecylsulfat-Polyacrylamidgelelektrophorese (SDS-PAGE) und Western-Blot.
- Inkubieren der Membran bei 4 ° CO / N im anti-GFP-Antikörper, verdünnt 1/1000 in Blockierungspuffer verdünnt, in PBS 1X, 0,1% Tween-20 auf die beiden markierten GTPase-bindende Proteine erkennen.
- Dreimal 10 min mit PBS, 0,1% Tween-20 waschen der Membran.
- Inkubieren der Membran für 30 min bei RT in 800 DyLight konjugiertem Anti-Kaninchen-secondary Antikörper, verdünnt 1 / 10.000 in Blockierungspuffer verdünnt, in PBS, 0,1% Tween-20 1 x auf.
- Dreimal 10 min mit PBS, 0,1% Tween-20 waschen der Membran.
- Scannen der Membran unter Verwendung eines Infrarot-Abbildungssystem mit der Software, um die Bandintensität nach dem Protokoll des Herstellers gemessen.
- Zur Erstellung der Bandintensität jedes Proteins vor der Verstellpumpe Konkurrent (Protein B).
- Teilen das Volumen der variable Konkurrent an dem Punkt, an dem die Linien sich durch das Volumen konstant Konkurrent (Protein A, 5 & mgr; l), um den Konkurrenzverhältnis, bei dem ein Gleichgewicht erreicht bestimmen.
- Zur Validierung der Nukleotid-Ladezustand, Sondenmembranen für p21-activated kinase 1 (PAK1) (ein Effektor) und GFP-TrioD1 (a GEF), wie in den Schritten 6.1-6.6 beschrieben.
Ergebnisse
Dieses Protokoll wurde entwickelt, um die relativen Affinitäten von Bindungspartnern für Rac1 zu berechnen, ohne die Notwendigkeit, die genaue Konzentration der Wettbewerber zu geben (Abbildung 1). Bestimmung der Proteinkonzentration führt Fehler und bei der Betrachtung der Wettbewerb zwischen Molekülen in einem Signalweg, ist nicht erforderlich. Jedoch ist es wichtig zu wissen, dass die beiden Wettbewerber haben die gleiche molare Konzentration in der Stammlösungen für einfache, Verhältnisse berechnet werden, wenn die Zugabe verschiedener Volumina auf den Test. 40 ul von GFP-Trap-Perlen eine Bindungskapazität von ~ 300 pmol so eine konfluente 75 cm 2-Kolben hoch exprimierenden Zellen werden die Perlen zu sättigen, mit dem Ergebnis, dass die Vorbereitungen der beiden unterschiedlichen Bindungsproteine ähnlich vor Anpassung (Abbildung 2A). Wenn eines der Proteine schlecht exprimiert, kann dieses Problem durch Reinigen dieses Proteins aus mehr als einem Kolben von Zellen überwunden werden können.
Die Bindung der meisten GTPase Effektoren und Regler abhängig von der Nukleotid-Beladung des Köder GTPase, so ist es wichtig zu untersuchen, ob Laden erfolgreich gewesen ist. Belastung kann durch Ausfällen bekannten Bindungsproteinen aus Zellysaten überprüft werden. Effektor-Proteine, wie PAK1 binden an GTP-Rac1 und kann leicht aus Lysaten präzipitiert und durch Western Blotting 8 (2B) erfasst werden. GEFs Substanzen binden Nukleotid freien GTPase den Übergangszustand zu stabilisieren. Wie GEFs sind von geringer Menge, in der Regel nicht aktiv und häufig schlecht auslöschen, ist es besser, ein GEF oder GEF-Fragment zu Test Nukleotid-freien GTPase überexprimieren. Wir verwenden häufig die erste Dbl Homologie von Trio, ausgedrückt als GFP-Fusions (GFP-TrioD1 9) (2B) aber jede GEF funktionieren würde. Proteine, die an der BIP-geladen GTPase binden, sind seltener. Vor kurzem berichteten wir RCC2 als eine solche Protein 7 oder BIP-Belastung kann einfach als bindi validiert werdenng bis weder GEF noch Effektorzellen.
Der Ausgang aus dem Experiment wird ein Western-Blot-Darstellung die beiden GFP-markierten Bindungspartner für die GTPase gebunden sein. Durch die Verwendung eines einzelnen Antikörpers, beide Proteine zu erkennen, können die Konzentrationen, bei denen gleiche Mengen der beiden Wettbewerber binden bestimmt und daher die relativen Affinitäten abgeleitet. In diesem Beispiel ist der Wettbewerb zwischen dem Propeller-Domäne des Rac1-Trafficking-Protein Coronin-1C (Rac1-bindenden Protein A) und die Rac1 sequestrierenden Protein RCC2 (Rac1-bindendes Protein B) wird nachgewiesen (3A). Unter Verwendung eines konstanten Volumens Coronin 1C Propeller (5 ul), und das Hinzufügen von steigenden Mengen von RCC2, wir von der GFP sehen auslöschen, dass ein Gleichgewicht bei 1,25-2,5 ul RCC2 (Sternchen) erreicht ist, was zeigt, dass RCC2 hat eine stärkere Affinität für Rac1 als Coronin-1C. Durch Messen der Intensität der Banden mittels quantitativer Western Blot und Aufzeichnen der Mittelwerte für jede competitor kann der Gleichgewichtspunkt genau durch Ermittlung der Mengen an denen die Kurven schneiden (3B) berechnet werden.
Eine der möglichen Hindernisse für eine erfolgreiche Konkurrenz-Assay ist, wenn die Bindungspartner miteinander sowie die Bindung an Rac1 binden. In 3A + B zeigen wir den Wettbewerb zwischen RCC2 und Propeller-Domäne von Coronin-1C, anstatt voller Länge Coronin-1C. Der Grund für die Verwendung des trunkierten Coronin dass Coronin 1C bindet ebenfalls RCC2 durch die Schwanzdomäne. Bei voller Länge Coronin-1C gegen RCC2 titriert, Bindung beider Proteine erkannt wird, aufgrund von ternären Komplexbildung, anstatt den Wettbewerb (3C). Wenn Konkurrenz auftritt, wird Bindung eines Proteins, während das andere abnimmt erhöhen und gesamte gebundene GFP-Fusions konstant. In Fällen, in denen ein ternärer Komplex bildet es notwendig ist, eine der GTPase-bindendes Protein abzuschneiden, so dass der competitors nicht mehr interagieren.

Abbildung 1. Arbeitsablauf. Schematische Darstellung der Arbeitsablauf zur Bestimmung der Affinität der GTPase-bindende Proteine mit Konkurrenz-Assays. Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
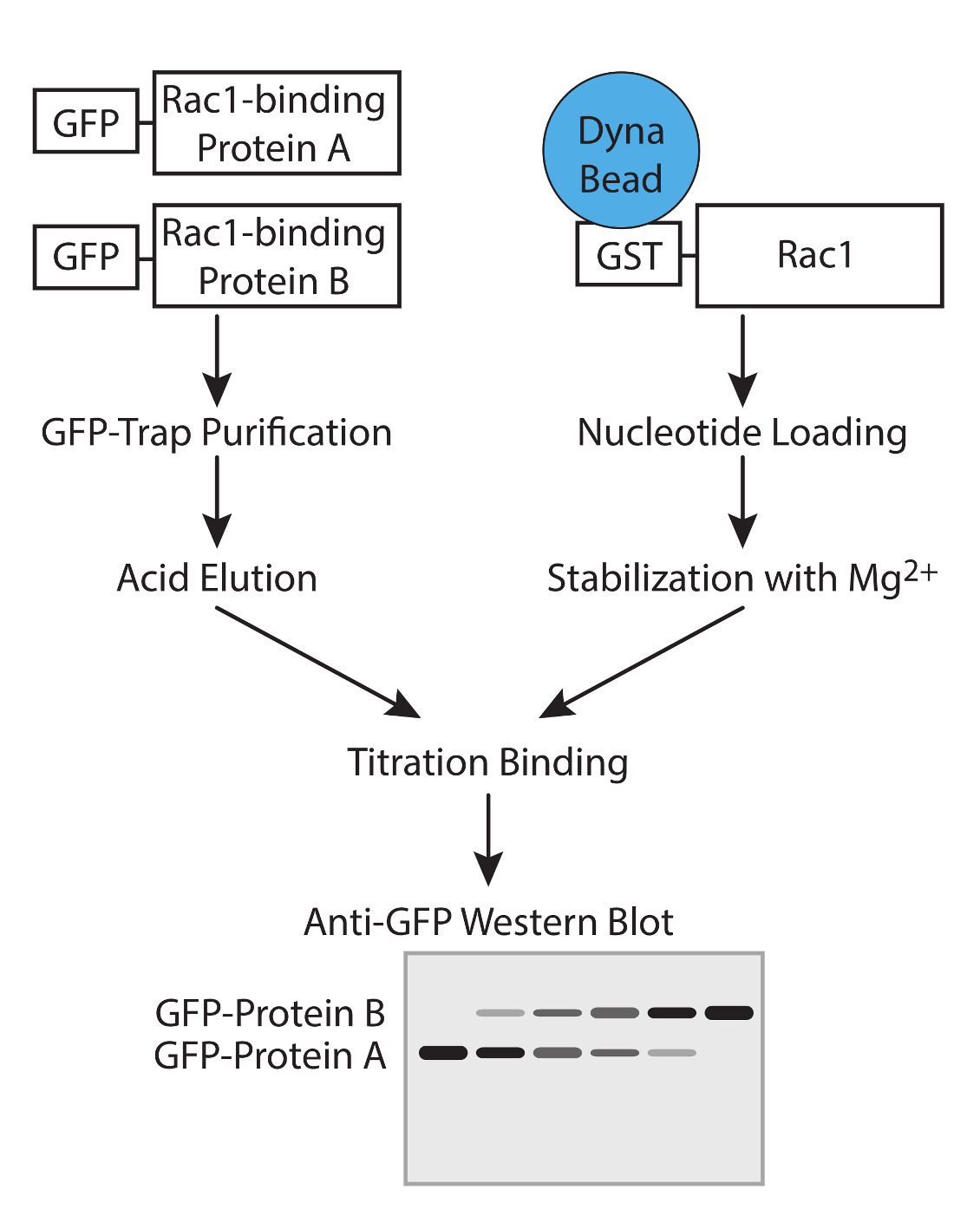{kind=link}

Abbildung 2. Validierung von gereinigten Proteinen. (A) gereinigtes GFP-markierten Rac1 bindenden Proteinen durch Western-Blot analysiert, Sondierung mit Anti-GFP, um die relative Ausbeute der beiden Proteine zu bestimmen. Diese Art von Entzerrung während des Experiments kann die Konzentration der beiden Proteine angepasst werden, so dass sie in dem Bindungsexperiment übereinstimmen. (B) GDP, GTPyS und keine Nukleotid-beladenen GST-Rac1 mit Lysat aus HEK293T, die GFP-TrioD1 Proteine durch Auftragen einer endogenen PAK1 oder überexprimiert GFP-TrioD1 erkannt inkubiert und gebunden. Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
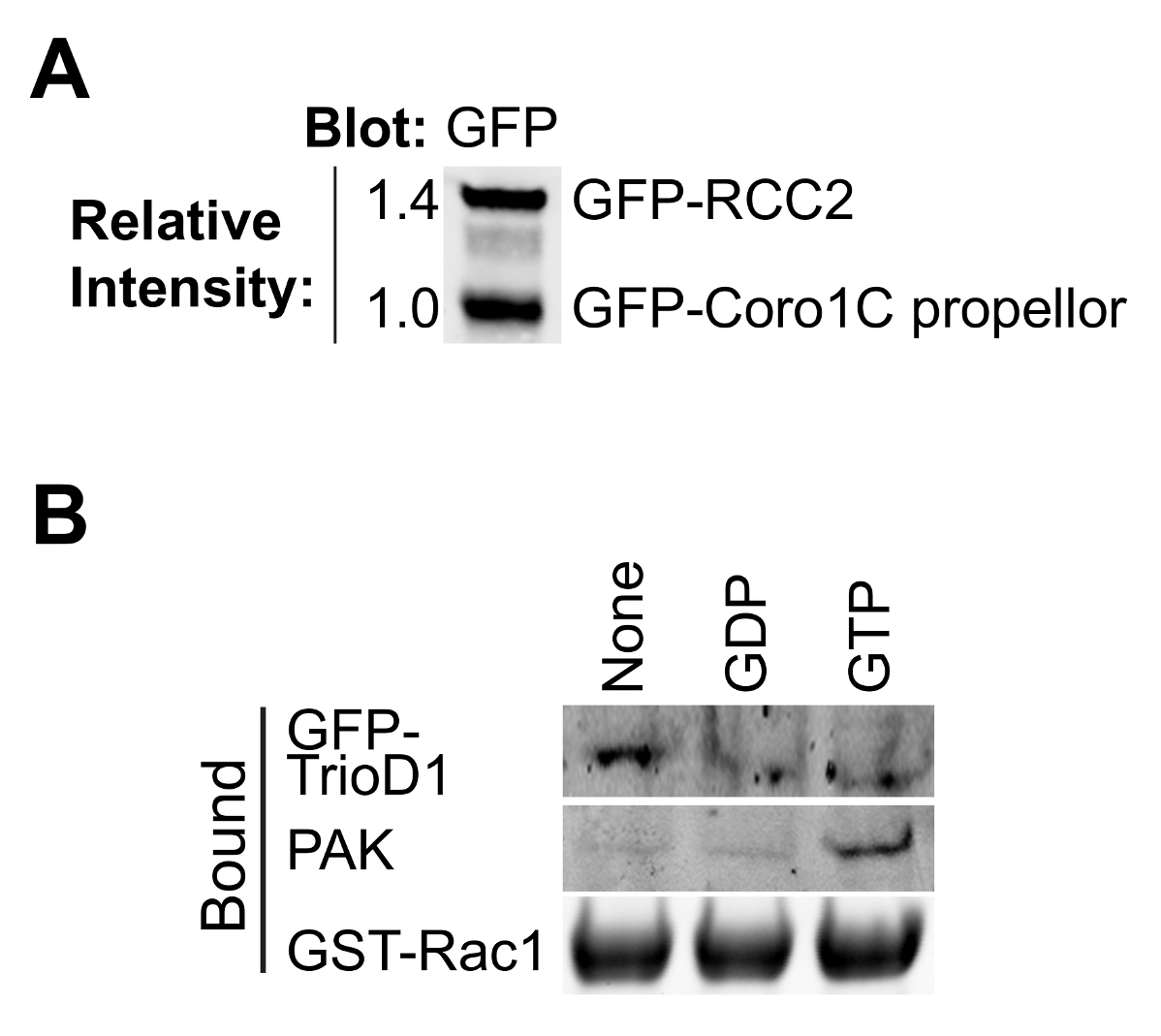{kind=link}

Abbildung 3. Western-Blot-Analyse der relativen Proteinbindung. Beispiel Ausgaben von Konkurrenz-Bindungs-Assays. (A) BIP belasteten Rac1 wurde mit 5 ul GFP-Coronin-1C-Propellerdomäne gemischt und steigenden Mengen von GFP-RCC2 wurden titriert. Durch Westernblot gebundenen Proteine GFP, sind Probleme mit der Differenzerfassung der beiden Proteine vermieden und das GFP-Signal meldet das Molverhältnis zwischen den beiden Fusionsproteinen. Sternchen zeigen die Wettbewerbsverhältnisse auf either Seite des Gleichgewichtspunkt. (B) Bandenintensitäten von gebundenem GFP-Fusionsproteinen aus drei unabhängigen Experimenten wurden durch quantitative Western-Blotting gemessen, wobei Fluorophor-konjugierten sekundären Antikörpern und Mittelwerte aufgetragen, um die Menge an RCC2 Berechnung erforderlich, um das Gleichgewicht zu erreichen. (C ) Ausgabe aus einem Experiment, wo Rac1-bindende Proteine binden aneinander und bilden einen ternären Komplex, anstatt konkurrierende. BIP belasteten Rac1 wurde mit 5 ul GFP-RCC2 gemischt und steigenden Mengen von GFP-Coronin-1C in voller Länge wurden ohne Verlust an gebundenem GFP-RCC2 titriert. Der Anstieg der gebundenen GFP-Coronin-1C zeigt ternären Komplexbildung. Bitte Klicken Sie hier, um eine größere Version dieser Figur zu sehen.
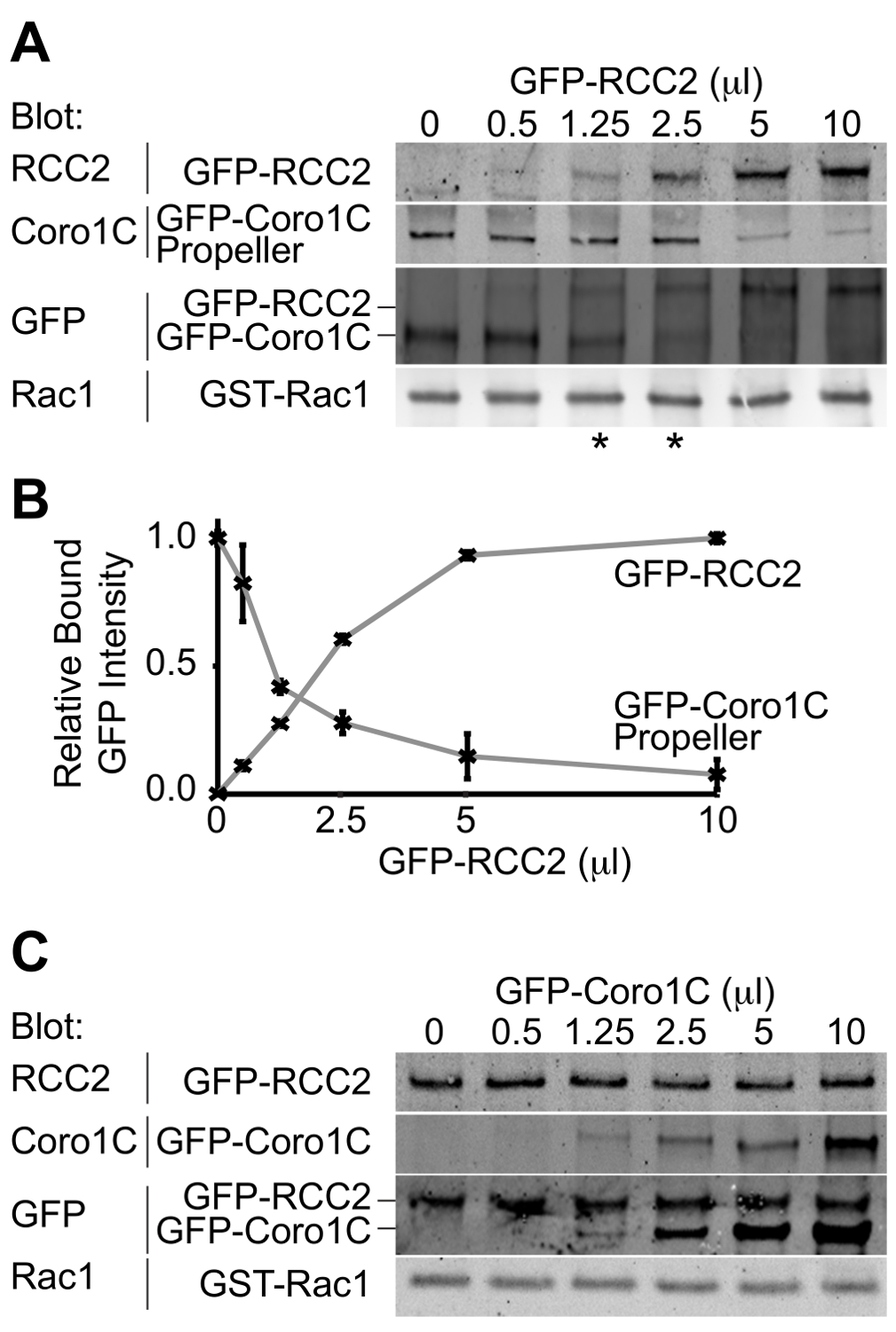{kind=link}
Diskussion
This protocol describes a method for comparing the relative affinities of pairs of small GTPase-binding proteins. The key steps are the preparation of purified GTPase-binding proteins and the nucleotide loading of the GTPase. The use of GTPase-binding proteins with the same GFP tag, allows the concentrations at which similar amounts of each competitor binds to be accurately determined. The use of recombinant nucleotide-loaded GTPase allows interrogation of the binding properties of the GTPase under specific activity conditions. This step is also the most sensitive as nucleotides will both hydrolyze and detach from the GTPase if the magnesium conditions are not maintained precisely.
In the cell, the large number of GTPase-binding proteins combined with the rapid nucleotide turnover makes such pathways difficult to interpret. The simplicity of this method in comparing only pairs of binding proteins and using carefully controlled nucleotide-loading conditions allows signaling pathways to be elucidated. However, the greatest strength of the protocol is also the greatest weakness as it is a simplification of the in vivo situation. Competition assays can be used to build a robust hypothesis, but this should then be tested in cells by knockdown experiments.
There are three features that must be considered when selecting the GFP-tagged GTPase-binding proteins to be used in the experiment. First, the fusion proteins must express well in mammalian cells, such as HEK293T, as competition assays require a reasonable amount of protein. Second, it must be possible to purify the recombinant protein without significant degradation, and where this is not possible, cloning of a GTPase-binding fragment should be considered. Third, the two GTPase-binding proteins must resolve from one another on SDS-PAGE to allow analysis in section 6.
There are a number of potential caveats to the experiment that need to be considered, and possibly addressed:
Possible denaturation of purified GTPase-binding proteins during the acid elution step or steric hindrance by the GFP tag. In our hands, these have not been a problem, but must be tested. The purified proteins can be tested in functional assays 10. Commercial kits now exist for testing the activity of GEFs or GAPs without the need for isotope-labeled nucleotides. Sequestering proteins, by their nature protect GTPases from GEF or GAP activity, so can be used as competitive inhibitors in the commercial GEF or GAP assays, as we did in our recent publication 7. The relevant feature of proteins that traffic GTPase are the capacity to bind the GTPase, and this can be tested easily in a pull down assay. An alternative approach to testing protein integrity that is applicable to all binding proteins is to titrate protein eluted from GFP-trap beads with glycine with the same protein removed from GFP-trap beads by enzymatic cleavage. The experiment would be analyzed by probing both the GFP-tagged and cleaved protein with an antibody against the protein itself. If the protein is undamaged by elution, equilibrium should be achieved at a 1:1 ratio. This approach would also indicate whether the presence of the GFP tag itself compromises the binding properties of the candidate protein, though this does require the production of a construct with an enzymatic cleavage site between the tag and the binding protein. Whether the protein is compromised by the tag or the elution step, the problem could be addressed by modifying the protocol to use an alternative purification method. Rather than GFP, binding proteins could be His-tagged, purified using Ni-NTA and analyzed using an antibody against the His-tag. The important feature is that both binding proteins must share a common tag although, if necessary, two tags could be added to a protein, one for purification and the other for detection.
The protocol is designed to investigate competition between interactions with the switch I/II domains. Although the majority of GTPase interactions are mediated by this motif, there are some exceptions, most notably the interactions of GDIs that bind to the prenyl tail, as well as obscuring the switch domains. In principle, the protocol could be adapted to use GTPase purified from mammalian cells, so that the GTPase is prenylated, however, the presence of multiple binding sites or allosteric effects complicate the interpretation of competition-binding data. Further problems associated with such a modification are that GDIs co-purify with GTPase from mammalian cells, compromising the purity of the isolated proteins and the hydrophobic nature of the prenyl groups means that prenylated GTPases are associated with either GDI or lipid membrane and such factors would need to be considered in the experiment.
The amount of GST-Rac1 being used in the assay. The constant GTPase binding protein must be at a greater concentration than the Rac1, or when the competitor is added, it will simply bind to free Rac1. It will be immediately obvious if this has happened as binding of the competitor, without a loss of the constant protein, will be detected in much the same way as when the two competing proteins bind to one another as shown in Figure 3B. As an additional control (Step 5.3), a binding reaction containing double the amount of constant binding protein and no variable binding protein should be included (Step 5.3). If the Rac1 in the titration experiment is saturated, doubling the amount of constant binding protein will have no effect on the output. The volumes suggested in the protocol should be appropriate, but the amount of Rac1 can be easily reduced. If binding of the competitor without loss of the constant binding partner is observed, reducing the amount of Rac1 should be attempted before trying to map binding sites to avoid ternary complex formation.
Non-specific interaction of GTPase-binding proteins with the GST or bead, as well as specifically with Rac1. This problem would be manifested by residual binding of the constant GTPase-binding protein, even when the variable GTPase-binding protein has reached a plateau at high concentration. Identification of this issue will be aided by conducting reciprocal experiments where the constant and variable GTPase-binding proteins are swapped. Reciprocal experiments will also greatly improve the accuracy of the estimate of equilibrium point, so should always be included. In cases of non-specific binding, the relative concentrations at which equilibrium is achieved can still be calculated by comparing band intensity between the maxima and minima for each protein, or by measuring the extent of non-specific binding by using GST beads as bait, rather than GST-Rac1.
Pull down assays using different nucleotide-loading conditions should be used to complement the competition assay described in this protocol. Determining the nucleotide preference of partners is important for both understanding the competition events and understanding the signaling pathway that the GTPase-binding protein is involved in. In Figure 2B we analyze binding of proteins with established preference for GTP-loaded or nucleotide-free GTPase as a means to validate nucleotide loading. However, it is sensible to investigate the effect of nucleotide loading on each of the competitors as well. If the hypothetical competitors show different preferences, competition will make less of a contribution to the signaling pathway, and indeed nucleotide turnover is likely to be the mechanism that directs exchange of the binding proteins.
Offenlegungen
Die Autoren haben nichts zu offenbaren.
Danksagungen
This work was supported by Wellcome Trust grant 088419 to MDB.
Materialien
| Name | Company | Catalog Number | Comments |
| Bugbuster | Novagen | 70584-3 | |
| COMPLETE protease inhibitor | Roche | 05 056 489 001 | |
| Glutathione magnetic beads | Pierce | 88821 | |
| Polyethylenimine, branched, average Mw ~25,000 | Sigma Aldrich | 408727-100ML | |
| OPIMEM | Life Technologies | 31985-047 | |
| Dulbecco's Modified Eagle Media | Sigma Aldrich | D5796 | |
| Fetal Bovine Serum | Life Technologies | 10270-1-6 | |
| L-Glutamine | Life Technologies | 25030-024 | |
| GFP-Trap_A | Chromotec | gta-20 | |
| GDP | Sigma Aldrich | G7127 | Highly unstable. Aliquot and store at -80 immediately upon reconstritution |
| GTPγS | Sigma Aldrich | G8634 | Highly unstable. Aliquot and store at -80 immediately upon reconstritution |
| Blocking Buffer | Sigma Aldrich | B6429 | |
| Tween-20 | Sigma Aldrich | P9416 | |
| Anti-GFP antibody | Living Colors | 632592 | Use at 1/1000 dilution |
| DyLight 800 conjugated goat anti-rabbit secondary antibody | Fisher Scientific | 10733944 | |
| Anti-PAK1 antibody | Cell Signaling | 2602S | Use at 1/1000 dilution |
| Odyssey SA Infrared Imaging System | Li-cor | 9260-11PC |
Referenzen
- Burridge, K., Rho Wennerberg, K. and Rac take center stage. Cell. 116 (2), 167-179 (2004).
- Raftopoulou, M., Hall, A. Cell migration: Rho GTPases lead the way. Dev Biol. 265 (1), 23-32 (2004).
- Rossman, K. L., Der, C. J., Sondek, J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol. 6 (2), 167-180 (2005).
- Worthylake, D. K., Rossman, K. L., Crystal Sondek, J. structure of Rac1 in complex with the guanine nucleotide exchange region of Tiam1. Nature. 408 (6813), 682-688 (2000).
- Scheffzek, K., Ahmadian, M. R. GTPase activating proteins: structural and functional insights 18 years after discovery. Cell Mol Life Sci. 62 (24), 3014-3038 (2005).
- Del Pozo,, A, M., et al. Integrins regulate GTP-Rac localized effector interactions through dissociation of Rho-GDI. Nat Cell Biol. 4 (3), 232-239 (2002).
- Williamson, R. C., et al. Coronin-1C and RCC2 guide mesenchymal migration by trafficking Rac1 and controlling GEF exposure. J Cell Sci. 127 (Pt 19), 4292-4307 (2014).
- Del Pozo, M. A., Price, L. S., Alderson, N. B., Ren, X. D., Schwartz, M. A. Adhesion to the extracellular matrix regulates the coupling of the small GTPase Rac to its effector PAK. Embo J. 19 (9), 2008-2014 (2000).
- Van Rijssel, J., Hoogenboezem, M., Wester, L., Hordijk, P. L., Van Buul, J. D. The N-terminal DH-PH domain of Trio induces cell spreading and migration by regulating lamellipodia dynamics in a Rac1-dependent fashion. PLoS. 7 (1), e29912(2012).
- Self, A. J., Hall, A. Measurement of intrinsic nucleotide exchange and GTP hydrolysis rates. Methods Enzymol. 256, 67-76 (1995).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten