
Method Article
具有更严格特异性的限制内核内核酶的体外定向进化
摘要
具有新序列特异性的限制内联酶可以从识别部分退化序列的酶中开发。在这里,我们提供了一个详细的协议,我们成功地用于改变NlaIV酶的序列特异性。该协议的关键成分是转录/翻译反应的体外分块和具有新序列特性的变体的选择。
摘要
限制内核酶(REase)特异性工程极其困难。在这里,我们描述了一个多步骤协议,帮助产生比父酶更严格的特性的REase变异。该协议要求为REase的变体创建表达选择盒库(ESCs),理想情况下,在可能影响DNA结合的位置有可变性。ESC 的一侧是限制站点活动所需的序列和生物素标记,另一侧是用于意外活动和引物退火位的限制站点。ESCs被转录并转化为油中水乳液,在使每个液滴存在多个DNA分子的情况下。因此,每个盒式分子中的DNA只受翻译的编码酶的活性的影响。所需特异性的 REase 变体可去除生物素标记,但不会去除引素退火位位。破坏乳液后,DNA分子受到生物蛋白下拉,只有那些在上清液保留。此步骤可确保仅保留未丢失所需活动的变体的 ESC。这些DNA分子然后受到第一次PCR反应。不需要序列中的裂解会切断其中一个引漆的引底器结合位点。因此,PCR 仅从液滴中放大 ESC,而不需要进行所需的活动。然后进行第二次PCR反应,以重新引入所需特异性和生物素标签的限制位点,以便重新选择步骤。选定的开放式阅读帧可以在细菌细胞中过度表达,这些细胞也表达出父母REase的可亲甲基转移酶,因为新进化的REase只针对甲基转移酶靶点的一个子集。
引言
序列特异性工程对 II 类 REase 极具挑战性。在此类内生酶中,序列识别和催化紧密地交织在一起,很可能是作为进化保护,防止产生比其同质甲基转移酶具有更广泛特异性的内联酶,后者会损害宿主DNA。由于需要保护宿主DNA免受新设计的内丘酶活性的影响,细胞中新特异性的定向进化变得更加复杂。因此,只有几个成功的REase工程报告,他们都利用特定酶11,2,3,4,5,6,72,3,4,5,6,7的独特特性。
在这里,我们为特异性工程提供了详细的协议,可用于生成比父母酶更窄的内生酶变异,这种酶基于我们成功的NlaIV内核酶8工程。对于任何具有任意识别序列的此类酶,可以为齿面中的碱基引入额外的特异性。对于识别部分退化序列(如NlaIV及其GGNNCC靶点)的亲酶,还可以在识别序列中引入额外的特异性。由于额外的特异性可能需要蛋白质-DNA接触,新认识的碱基应位于DNA上父母内核酸酶的足迹内。原则上,可以为识别序列的任何所需专门化设置选择方案。然而,大多数识别蛋白酶体和几乎蛋白酶靶序列的REase是功能昏暗器,只识别半位处的苍白。因此,选择违反蛋白质核相互作用对称性的新特性不太可能奏效。例如,对于小NlaIV,GGNNCC序列理论上可以缩小到GGATCC,但将特异性缩小到GGAACC预计更加困难。我们的方案包括正面和消极选择。
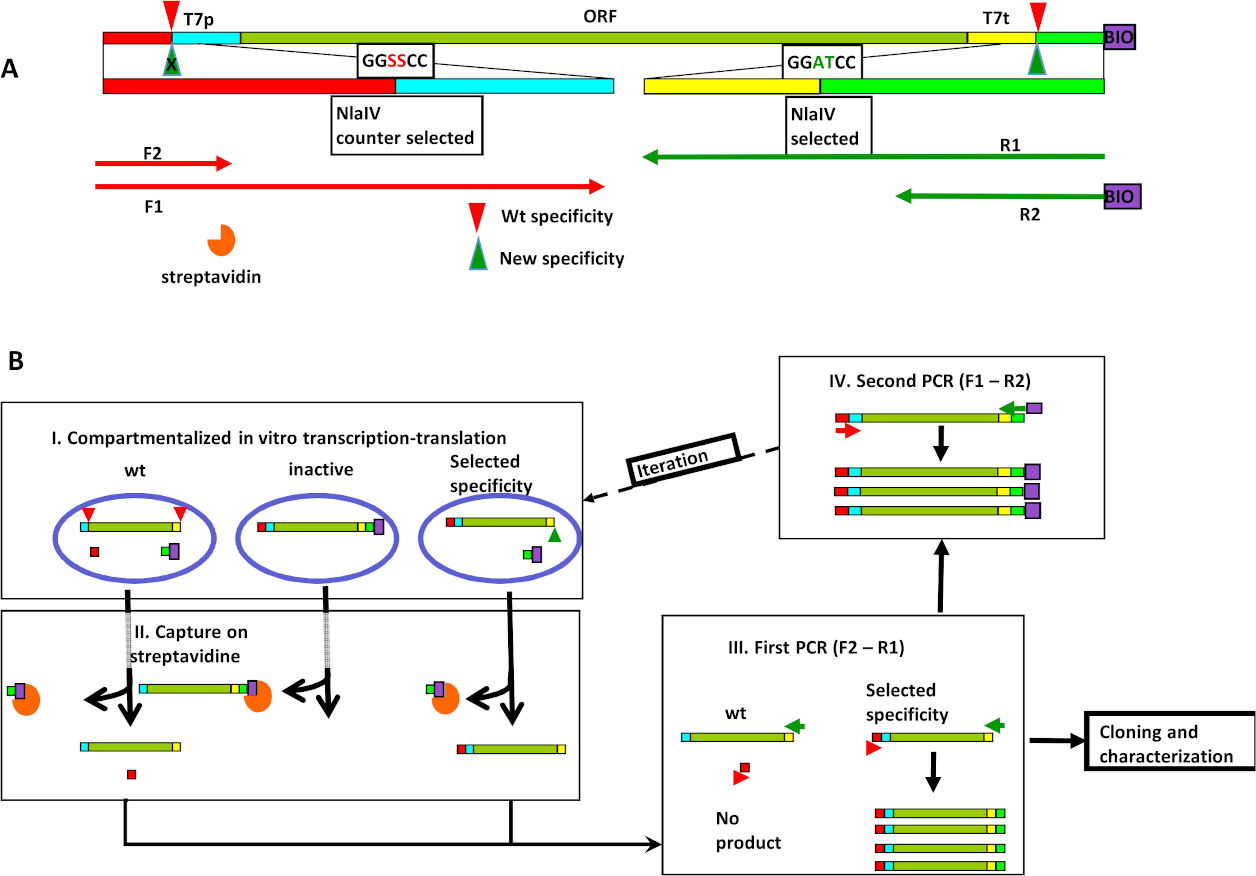
当还使用负选择来删除能够切除首选较窄特异性之外的所有序列的特异性时,该过程会更有效。例如,GGATCC 的选择可以与针对 GGBVCC 的反选择相结合(其中 B 是 A 以外的任何基,V 是 T 以外的任何基点)。当一些可能的目标序列未涵盖时,选择实验的结果取决于正和负选择的有效性。在我们的NlaIV工作中,我们选择了GGATCC,针对GGSSCC(其中S是G或C),并获得了一个特异性,忽略对称突破目标,可以描述为GGWWCC(其中W是A或T),这表明在这种情况下,负选择更多比积极选择重要。
我们的方法从创建表达式选择盒 (ESC) 开始。ESC 按部分进行构建。在内部核心部分,在 T7 启动器控制下,REase 的开放读取帧 (ORF) 有变体。ESC 的此核心部分不能包含任何工程 REase 的认知站点。核心夹在两个野生类型 REase 的认知站点之间:用于意外活动的裂解站点(计数器选定序列,本例中的 GGSSCC)和所需活动的裂解站点(所选序列,示例中的 GGATCC)。PCR 中 ESC 制备的最后一步在 5' 末端添加接近所需活性的生物素,并创建各种计数器选择序列(示例中为 GGSSCC)。选择策略依赖于在体外转录/翻译/选择协议(图1A)之后,在ESC重新放大协议中使用精心设计的引物。ESC库以体外分录转录9、10、1110,11表示。9在每个滴中,表达酶的特异性影响ESC的状态(图1B,步骤I)。对于描述的排列,翻译的蛋白质所需的裂解活性会去除DNA的生物素标签,但不影响其他ESC末端与计数器选择的序列。当乳液破裂时,通过链球菌亲和力下拉下去除生物微化碎片,以便仅保留具有所需活性的液滴碎片(图1B,步骤II)。此步骤删除非活动 REase 变体。然后,PCR 放大了下拉步骤的上清分数。在第一个PCR反应引漆F2和R1中使用(图1A,B,步骤III)。引物 F2 与计数器选定序列和分子端之间的 ESC 部分结合。因此,表达能够分割计数器选定序列的变体(因此,将引物 F2 和 R1 的结合位点分离为两个不同的 DNA 分子)的 ESC 不会放大,因此从库中移除。底漆 R1 在所选站点和 ESC 核心之间绑定,使其不受所选站点的裂解状态的影响,并还原所需活动的裂解位点 (GGATCC)。循环由第二个 PCR(引物 F1 和 R2)关闭,该 PCR 在靠近选定站点的 5' 端添加生物素,并在靠近 ESC 另一端的计数器选定地点恢复设计变异(图 1B,步骤 IV)。由此产生的DNA混合物准备进行另一轮选择。
选择协议的成功在很大程度上取决于正确选择新的、更严格的目标识别序列,以及仔细设计诱变策略及其有效实施。因为与克服它们相比,在REase的轻微预先存在的偏好上改进要容易得多,因此我们建议从任何预先存在的偏好进行动力学研究。仔细的突变设计的必要性来自突变库的有限大小,该库可以通过提交的协议处理(在单个实验中有109个克隆)。因此,所有20种可能的氨基酸替代品只能在几个位置进行有效测试(参见讨论)。随机突变,如作为替代方法呈现的容易出错的PCR(EP-PCR),将导致现有复杂性的严重低估。如果有任何有关与DNA接触涉及的潜在氨基酸位置的信息(甚至位于与同源序列中退化核苷酸的接近处),它肯定应该用于选择一些氨基酸,用于寡核苷酸引导饱和突变(协议步骤1.6-3.10)。
研究方案
1. ESC的准备
- 克隆限制-改性系统的甲基转移酶,以低拷贝数质粒(例如,pACYC184 或 pACYC174 或其衍生物)进行设计。
注:细菌宿主菌株必须能够耐受克隆酶引入的甲基化,并提供T7RNA聚合酶的诱导表达。建议使用 ER2566 菌株(携带 McrA、McrBC 和 Mrr 突变)。 - 确认重组质粒DNA通过干酪内联酶保护,在2小时2小时的缓冲和温度下用10单位的凝酶RE处理0.5μg的质粒DNA,防止裂解。
- 准备这种菌株的合格细胞。
注:可以使用任何方法。NlaIV工程项目采用了简单的氯化钙法12。 - 在T7促进子体的控制下,用ORF构造重组质粒,其选择标记与步骤1.1中含有甲基曲酶基因的粒度标记不同。可以使用矢量 pET28 和 pET30。
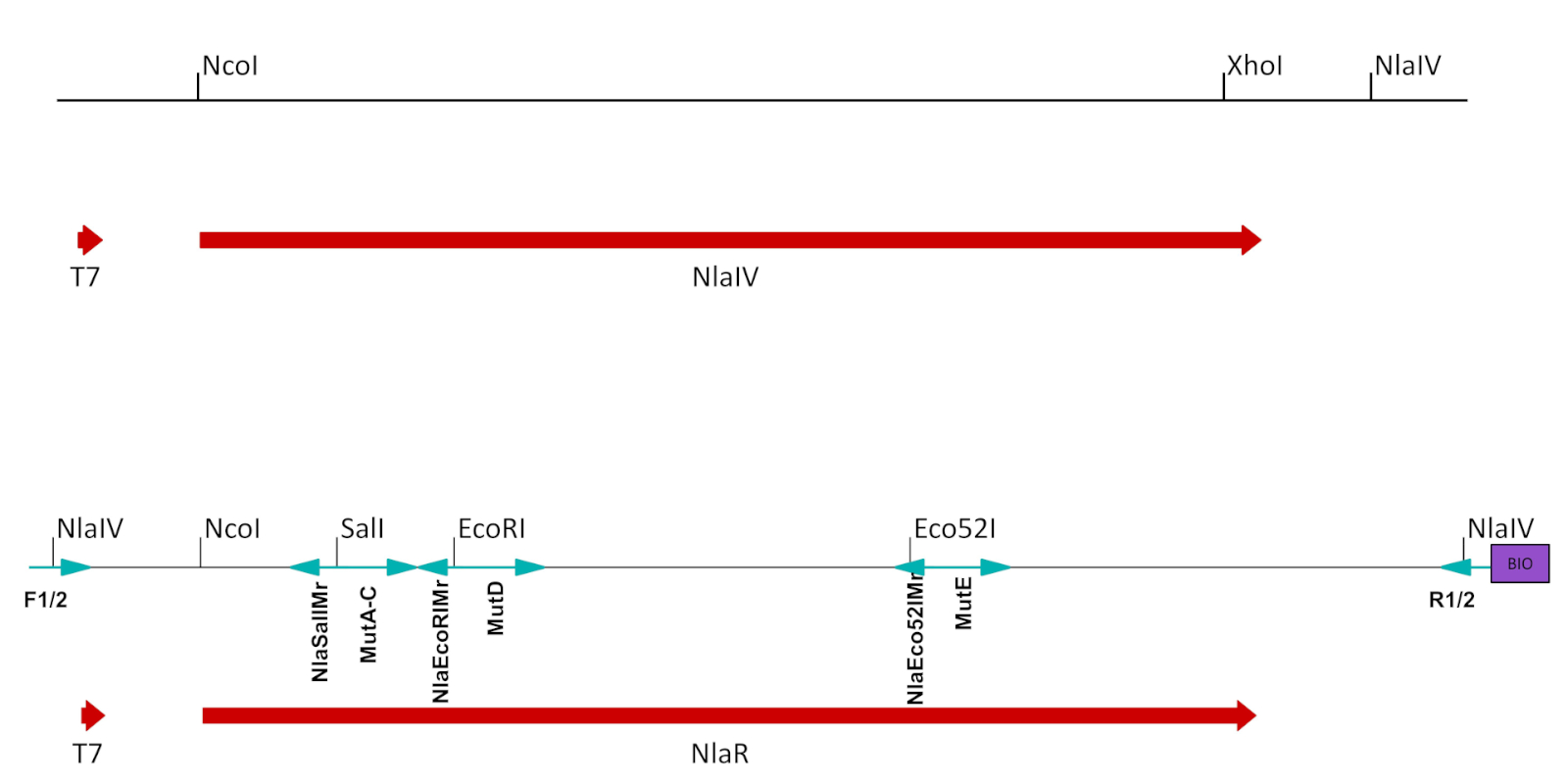
- 通过引入静默突变,从T7促进剂和酶ORF停止的停止子体之间的重组质粒部分移除工程酶的所有识别位点(图2,表1A)。
注:如果必须移除多个此类位点,则需要多个突变轮(步骤 1.5.1_1.5.7)。- 使用内向的PCR反应,用底漆5端引入的设计变异来放大全长质粒(表2A)。
- 通过在PCR反应的50μL中加入10U DpnI内核酸,在37°C下孵育2小时,去除模板DNA。
- 通过琼脂胶电泳解决产品。切掉与全长质粒对应的带子,用商业套件纯化。
- 加入10倍结扎缓冲液(浓度为1倍),并补充ATP(至1 mM)。加入10U的T4多核苷酸激酶,并在37°C孵育20分钟。在70°C加热10分钟,使酶失去活性。
- 加入PEG 4000至5%,再次补充ATP(至1 mM),并加入5U的T4DNA连接。在室温 (RT) 下孵育 2 小时。
- 转化为携带同二苯甲醚甲基转移酶的合格细菌菌株(步骤1.1)。
- 小规模分离质粒DNA,确认通过多氧测序引入序列变化。
- 引入靠近寡核苷酸引导诱变所针对的序列的唯一限制位点(图2,表1B)。按照每个站点的步骤 1.5.1_1.5.7 操作。
注:仅当使用目标突变时,才会执行此步骤。如果执行随机突变,跳过步骤 2-3,然后转到第 3 部分。在所述示例中,所有站点都引入到目标区域的上游,但它们也可以引入下游。 - 为 ESC 放大设计底漆(表 1C)。
- 设计一个反向引底结合下游的内核酶ORF,将引入选定的识别位点(R1)及其较短的版本(R2),这些识别位点(R2)在选定的NlaIV序列之外结合,并在5'末端包含生物素(参见图1)。
- 设计一个向前底漆 (F1) 绑定到 T7 启动器上游的 ESC。此引漆还应引入原始识别序列的反选择变体(即原始酶识别的序列变化的最大,所选反向序列除外)。
注:稍后在选择性 PCR(步骤 5.9)中,将使用此底漆 (F2) 的较短版本(涵盖与反选择序列的远端序列)。
2. 诱生引物的分裂和混合合成
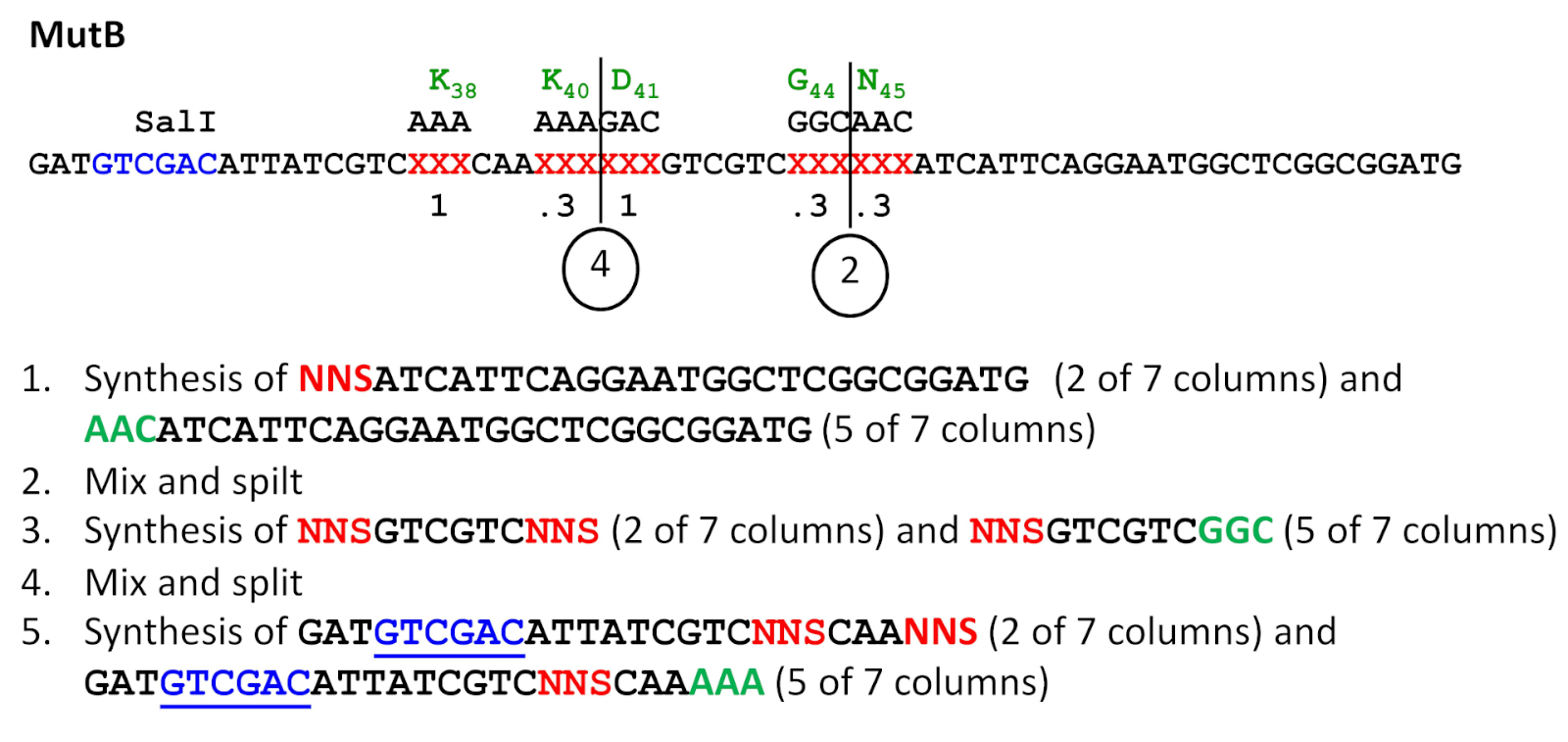
注:此步骤仅用于需要在多个站点上需要子饱和突变的项目。需要具有多个合成列的合成器。根据诱变频率分配用于合成随机NNS子体三胞胎和野生型三胞胎的列。例如,如果有 7 个相等的卷合成列,并且给定站点的突变率为 0.3 是可取的,则在 ±0.3 x 7 或两列中添加随机 NNS 协成,在 ±0.7 x 7 或 5 列中添加野生类型 codons(图 3)。
- 确定子饱和突变的站点。根据站点的假设重要性(即站点越重要,频率越高),选择突变频率,同时牢记整个库的复杂性限制(请参阅讨论)。
- 合成所有列中的寡核苷酸,在3'端的第二次次饱和突变位计数之前,达到三重。不要在最后一个合成周期移除 5'-trityl 保护组(使用合成器上的三元打开选项)。保护组将在下一个合成周期开始时删除(图 3中的步骤 1)。
- 打开合成列。将受控孔隙玻璃 (CPG) 合成支持收集到干燥的 1.5 mL 管中,并通过涡旋混合。将混合 CPG 树脂重新分区为新的合成柱。避免引入湿度,因为它会降低总体产量(图 3中的步骤 2 和 4)。
- 继续合成,从亚饱和突变位三合三开始。根据所需的诱变频率将列分配给随机的 NNS 三胞胎或野生类型三胞胎(见上文注释)。如果存在其他子饱和点,则仅继续在下一个子饱和突变位之前的三重位。同样,在合成器上保留一个 5'-trityl 组(合成器上的 5'-trityl 上选项)(图 3 中的步骤3)。然后继续步骤 2.3。
- 如果下游不再存在子饱和点,则完成合成,在合成器上留下一个 5'-trityl 组(合成器上为 5'-trityl 上选项)(图 3中的步骤 5)。
- 根据净化盒制造商的说明,对寡核苷酸库进行保护并净化。
注:通过从CPG中解保护释放的寡核苷酸也可以在反向相高性能液相色谱(HPLC)中用三乙基进行纯化,然后进行手动三聚基去除(RT用80%醋酸进行1小时治疗)和第二次HPLC纯化。 - 检查尿素-PAGE凝胶中的寡核苷酸库质量。
3. 生成变体库
注:使用步骤 1.6 中的重组质粒。
-
通过寡核苷酸定向诱变生成库。
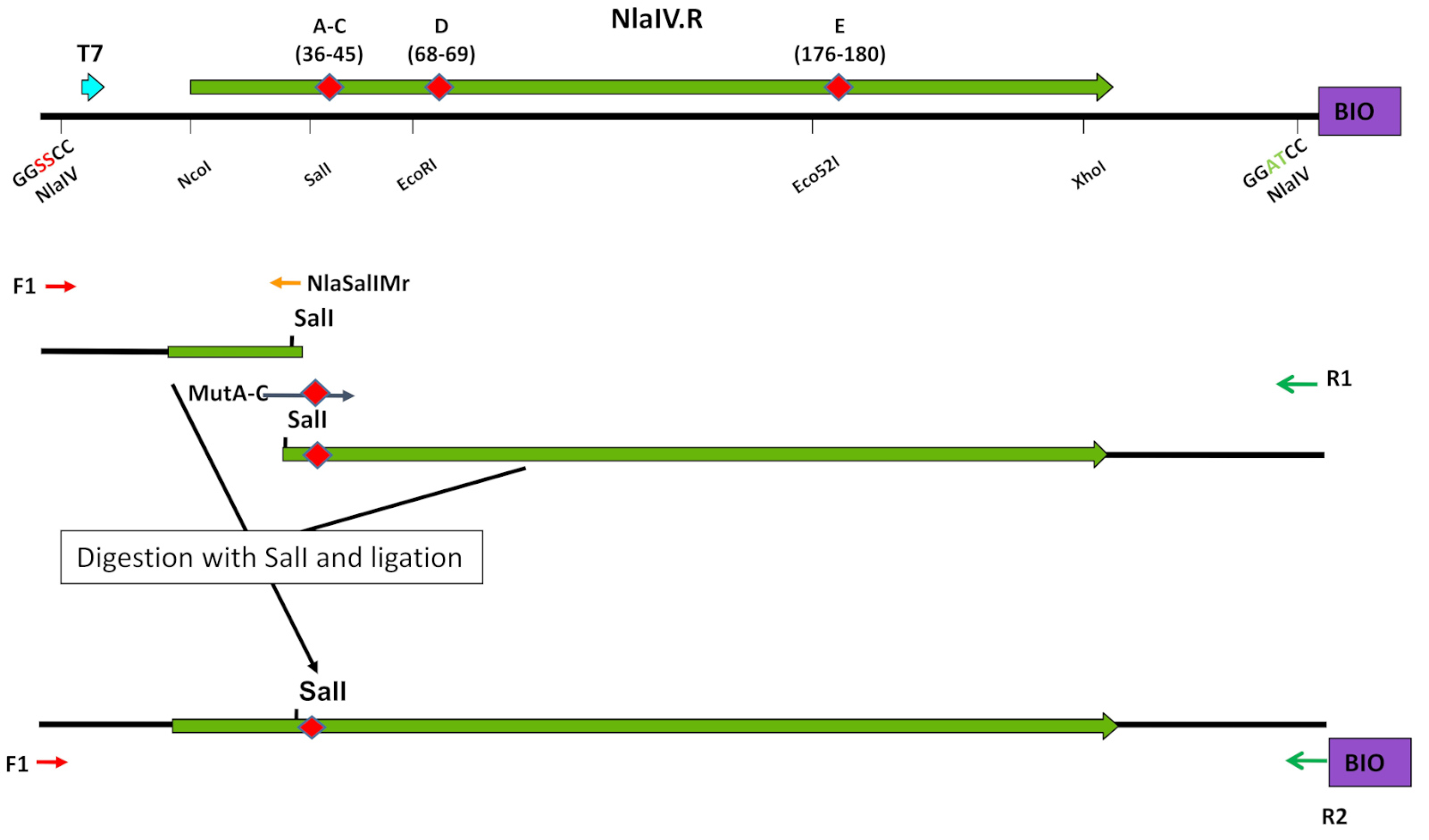
注:或者,使用 EP-PCR 协议(步骤 3.2)。- 将 T7 促进器的一个部分放大到具有诱变靶向的序列两侧的唯一限制性酶位点(在 NlaIV:SalI、EcoRI 或 Eco52I 的情况下)(表 1B-C、表 2B、图 4)。将第二部分从独特的限制性酶位点放大到ESC的3'末端。
- 将 PCR 反应的 5 μL(从步骤 3.1.1 起)与 8 μL 水、1.5 μL 的 10 倍限制酶缓冲液和 5 个相应限制性酶(SalI、EcoRI 或 Eco52I)单元分别混合,并在适当的温度下孵育 2 小时。
- 使用琼脂糖凝胶电泳解决两种反应的产品。切掉预期尺寸的带子,用商业套件进行净化。
- 在琼脂胶中运行多达 1/3 的纯化产品,并通过密度测定测量每个纯化带的浓度。
- 将ESCs的两个部分的结扎在1:1摩尔比与1x连接缓冲液和1UT4DNA连接和孵育2小时RT。
- 解决琼脂糖凝胶中的反应产物。切掉预期尺寸的产品,用商业套件进行净化。
- 使用引物 F1 和 R2(表 1C和表 2A)在 PCR 反应中放大纯化连接产品。不要运行超过 20 个放大周期。
- 分馏琼胶凝胶中的PCR反应。切掉产品,用商业套件进行净化。
- 从琼脂胶凝胶的前一步运行纯化库的5μL等位,并通过密度测量测量浓度。
- 克隆库的小样本(最多5μL)和序列>15克隆,以检查突变频率和分布(表3)。继续步骤 4。
注:或者,可以使用 ESC 小样本的高吞吐量排序。
-
执行 EP-PCR。
- 使用引物 F1 和 R1 从步骤 1.5.7 中获得的质粒放大 ESC。使用 Taq I 聚合酶 (表 1B) 运行 20 个循环。
- 凝胶净化PCR产品。
- 使用上一步 2 纳的纯化 PCR 产品设置 EP-PCR,使用 F1 和 R1 引漆运行 15 个 EP-PCR 周期(表 1C)。
- 凝胶净化产品,并通过凝胶密度测定来量化。
注:由于纯化EP-PCR产物浓度低,使用约1/3进行定量。 - 克隆库的小样本(最多1/5)和序列>15克隆,以检查突变频率和分布(表4)。
注:或者,对 ESC 的小样本执行高通量排序。
4. 进行分段体转录-翻译反应
-
在体外转录翻译中测试内核酶表达和酶活性。
- 准备一个短(200~500 bp)基板,并带有一个识别位点,用于靠近分子中心的内核酸酶,以便容易地检测到裂解反应。
注:制备基质的最简单方法是通过PCR放大任何DNA分子的适当片段。基板可以贴上放射性标签或荧光标记,以简化裂解检测。 - 根据制造商的建议,用0.5μg的野生ESC型的转录-转化反应设置50μL。将镁盐(MgCl2、MgSO4和醋酸镁可测试)添加到 1.5 mM 和上一步中适当数量的基质(如果 DNA 未标记时至少 0.5 μg)。
注:可以使用任何不含镁激活的核酸核酸核酸的转录/翻译套件。一些试剂盒供应商在生产过程中使用核酸酶去除DNA污染,然后添加包质剂作为核酶抑制剂。此类套件与此方法不兼容。 - 根据制造商的说明孵化转录-翻译反应。然后将反应混合物转移到限制酶的最佳温度2小时。
- 分析琼脂凝胶基质的裂解,然后进行适当的检测(例如DNA染色、荧光可视化或自放射成像)(图5)。
注:在进行分块化之前,至少需要部分裂解基板。如果无法实现,需要进一步优化镁的化学形式或其浓度。
- 准备一个短(200~500 bp)基板,并带有一个识别位点,用于靠近分子中心的内核酸酶,以便容易地检测到裂解反应。
- 通过在15 mL锥形管中加入225μL的跨度80和25μL的特威80至5mL矿物油,制备油表面活性剂混合物。通过轻轻反转管 15 倍彻底混合。
- 每个库将950 μL的油表面活性剂混合物转移到2 mL圆形底部低温小瓶,标签带有库名,并转移到冰上。将一个小圆柱形搅拌棒(5 x 2 mm2)放入每个小瓶中。
- 根据制造商的建议,制备体外转录-翻译反应混合物(每个库50μL)。用氯化镁补充混合物,最终浓度为1.5 mM(参见步骤4.1.4)。
- 将 50 μL 等分点分配到冰上 1.5 mL 管中。
- 在冰上的反应混合物中加入1.7个库(第3节)。
注:不要使用较多的表达式库来选择效率。尽量减少含有多个DNA分子的水滴的频率至关重要。 -
为每个库连续准备油中水乳液。
- 将装满冰块的小烧杯(或大瓶杯)放在磁性搅拌器上,搅拌速度设置为 1,150 rpm。
- 将含有 950 μL 油表面活性剂混合物的低温小瓶和从步骤 4.3 到磁性搅拌器上的冷烧杯的小搅拌棒转移。检查搅拌杆是否旋转。
- 在30秒的间隔内加入体外库转录-翻译混合物的5个10μL等位,然后继续搅拌一分钟。将装有乳液的小瓶转移到冰容器。从步骤 4.7.2 开始,继续下一个库。
- 处理完所有库后,根据套件制造商的建议,开始孵化所有库。
- 将小瓶转移到工程内核酸的最佳温度上,再延长2小时,然后将其放入冰上至少10分钟。
5. 继续处理图书馆和选择
- 将低温小瓶中的乳液转移到冷1.5 mL管中,加入1 μL 0.5 M EDTA,并在室温下以13,000 x g离心5分钟。
- 使用移液器拆下上油相。如果油水相位不可见,在-20°C孵育管至少5分钟以冻结水相,然后立即移出液油相。
- 加入100 μL的10mM Tris HCl pH 8.0,立即进行150μL酚:氯仿(1:1 v/v)的萃取,通过短涡旋,然后相分离,以30s离心在13,000 x g。收集上水相。
- 加入醋酸钠(pH = 5.2)的0.1伏(15μL),2.5×5μg的糖原和2.5伏(375μL)乙醇,沉淀DNA。在-20°C下孵育1小时,在13,000 x g,4°C下15分钟。 g丢弃上清液,用 1 mL 冷 70% 乙醇短暂清洗颗粒。
- 在速度真空或空气干燥中干燥 DNA/糖原颗粒,进行 >5 分钟。
- 将颗粒溶解在 50 μL 的 10 mM Tris-HCl (pH = 7.5)。加入5 μL链球菌磁珠,根据制造商的说明制备,在RT时混合1小时,最好在转盘搅拌机或轻柔涡旋。
- 将磁架上的珠子分开,收集不含生物tin的DNA中丰富的液体。
- 通过乙醇沉淀浓缩DNA(步骤5.4~5.5)。
- 将上一步中的浓缩DNA溶解在5μL水中,并用作F2和R1底漆的PCR反应的模板(表1A)。
注:为了避免模板污染问题,尽量减少PCR伪酶使用Taq聚合酶(不是Pfu或Phusion),并运行18-20个周期与模板大小成比例的扩展时间(1 kb = 1 分钟)(参见表2B)。 - 将 PCR 产品分馏为琼脂胶凝胶,并切出预期尺寸的产品。一些涂抹表明有不同尺寸的产品(参见图 6)。用商业套件从凝胶板中净化DNA。
- 使用步骤 5.10 和引引剂 F1 和 R2 的 DNA 进行第二次 PCR 反应,使用步骤 5.9 中相同的协议。按照 5.10 中所述的产品纯化操作。通过琼脂胶密度测定(不是紫外线光谱)定量后纯化DNA可用于下一轮体外选择(步骤4.6)。
6. 更改序列特异性的屏幕变体
- 克隆选定的变体。
- 从步骤 5.10 消化产品 2 小时与 10 U 的限制酶适合克隆到表达载体 (NlaIV: NcoI 和 XhoI) 在酶供应商推荐的温度和缓冲.用琼脂凝胶电泳解决产品,并分离预期尺寸片段。
- 使用与步骤 6.1.1 相同的酶通过双裂解制备质粒载体(例如 pET28),并通过商用 DNA 凝胶纯化试剂盒进行凝胶纯化。
- 用琼脂糖凝胶电泳测定出病媒的浓度和插入。
- 设置一个连接与1+5 U的T4DNA连接和载体:插入摩尔比1:3-1:5在1x加糖缓冲酶供应商推荐。在RT孵育2小时,并通过转化或电穿孔12引入适当的宿主细菌(从第1.3步起)。
- 在含有适当抗生素(pET28 或 pET30 载体的 50 μg/mL 卡那霉素)和 1% 葡萄糖的 LB 板上选择转化剂。
- 快速蛋白质变异。
- 从转化的单菌落点(在一次运行中可以很容易地处理24个克隆)成2 mL的LB与卡那霉素(50μg/mL)和1%葡萄糖,并在37°C摇抖下生长过夜。
- 接种15 mL的温暖(37°C)LB含有100μg卡那霉素,无葡萄糖与0.75 mL的过夜培养,并在37°C孵育剧烈的震动。
注:可以使用 50 mL 离心机管或 100 mL Erlenmayer 烧瓶。 - 将176μL的甘油加入1 mL的过夜培养(甘油的最终浓度 = 15%)彻底混合,在-70°C下冷冻。
- 在 2⁄3 h 后,使用 IPTG 补充 15 mL 的培养(从步骤 6.2.1)到 1 mM 和培养,额外 5 小时。
- 通过离心(10,000 x g,4°C,10 分钟)收集细菌颗粒,并在 -70 °C 下冷冻。
- 净化蛋白质变异。
- 将 20 μL 镍亲和树脂悬浮液转移到 200 μL B1 缓冲液管中,在具有宽孔移液器尖端的 1.5 mLg管中,轻轻混合,并离心机(5,000 x g,30 s,4 °C)。通过移液去除上清液,并将管子留在冰上。
- 通过剧烈涡旋将细菌颗粒从B1300μL的步进6.2.5中重新悬浮。将悬架转移到 1.5 mL 管中。
- 在B1中加入3μL的100x蛋白酶抑制剂鸡尾酒和淋索素溶液(最终浓度为1mg/mL)。通过用配备尖端的探头声波分解细胞。每个样品使用六个 10 s 突发,中间的冰中有 >15 s 尖端冷却时间。始终保持细胞悬浮在冰上。
- 通过离心(2 分钟、12,000 x g、4°C)将 250 μL 的上清液从步骤 6.3.1 转移到树脂等位物的颗粒细胞碎片。
- 在寒冷的房间里混合15分钟,最好在旋转木马搅拌机中或通过温和的涡旋。
- 离心机(5,000 gx g,30 s,4 °C),用移液器吸气上清器。
- 加入 500 μL 的 W 缓冲液,轻轻重新悬浮树脂。离心机(5,000 gx g,30 s,4 °C),用移液器吸气上清器。
- 重复步骤 6.3.7。
- 加入20 μL的缓冲液E,轻轻重新悬浮树脂,将样品留在冰上2~5分钟。离心(5,000 x gg,30 s,4°C),然后收集上清液。
- 重复步骤 6.3.9。池超子。
- 通过 SDS-PAGE (5~10 μL) 分析蛋白质样本(图 7)。
- 筛选具有更改特异性的变体。
- 在噬菌体lambdaDNA上检测裂解活性。蛋白质样本可构成最终反应量的10%。每0.5μgDNA中总共2μL的蛋白质样本和2小时反应时间是一个很好的起点。
- 分析由阿加丝凝胶电泳反应产物以及野生型酶产生的产品。选择产生裂解模式的克隆人与野生类型酶产生的模式明显不同,以便进一步分析(图8)。
结果
该协议只是一种工具,通过消耗(但不消除)两个不需要的类来增加工程REase所需变体的频率:非活性酶和具有不变野生类型序列特异性的内联酶。另一方面,由于改变REase特异性是极其困难的,因此,在24个克隆的单个筛选中,即使发现一种产生与野生酶不同的裂解模式的变异也被认为是成功的。在我们手中,最好的屏幕可以识别多达20%的有前途的变种(图8A)。
积极的结果在很大程度上取决于图书馆的质量(即替换频率有限及其随机分布)和图书馆成员生物微量的有效捕获(步骤3.6-3.7)。可以检测到这两个问题。应在选择之前通过对尽可能多的克隆进行排序(>15)或通过高吞吐量排序直接排序(步骤 3.10,表 3)来检查库质量。如果大多数选定的克隆未处于活动状态,则此明显表示链球菌捕获选择失败。对于经历许多选择周期的库,也观察到类似的效果,因为此类库很可能由逃避链球菌捕获选择步骤的非活动变体主导(图 8B)。因此,建议在每个选择周期后运行筛选,并进一步开发手动选择的有前途的变体,而不是依赖于选择迭代。

图1:基于NlaIV工程的新序列特异性的体外选择。(A) 表达式/选择盒 (ESC) 的组织包括 REase 的两个识别站点,1) 靠近右端的选定序列 (GGATCC) 和 2) 靠近左端的计数器选定序列 (GGSSCC),以及 T7p 和 T7t-T7 启动器和 T7 终结器。底漆绑定位如下所示。按野生类型和选定的 NlaIV 变体的裂解分别显示为红色和绿色三角形。(B) 选择周期步骤:一) 转录-翻译-裂解反应与ESC库混合的乳化;II) 所有生物微化DNA都捕获在涂有链球菌的磁性颗粒上,并去除,从而去除编码非活性变异;III) 使用野生类型活动(即能够切割 GGSSCC 序列)编码 REase 的 ESC 将被取消,因为序列的裂解将正向和反向引物的绑定位点分开。因此,不放大这些ESC;IV) 下一个选择回合的输入是通过在右端添加生物曲子和在左端重新引入计数器选定序列的变体来创建的。转载从查平斯卡等人8号,经埃尔塞维尔许可。请点击此处查看此图形的较大版本。
{kind=link}

图2:ESC的准备。在T7分子控制下的包含NlaIV ORF的表达式向量中从原始构造派生的片段被修改为适合表达式/选择。NlaIV ORF 下游的 NlaIV 站点被移除,用于改变选定位置的独特站点(SalI、EcoRI 和 Eco52I)在 NlaIV ORF 中作为静默突变引入。最后构造被放大了侧翼引漆,引入了两个侧翼NlaIV位点:左侧的计数器选择序列(GGSSCC),右侧的选定序列(GGATCC)。反向引漆还引入了生物素。用于创建突变 ECS 的引物显示为蓝色箭头,并在下面标记(参见表 1B,C)。请点击此处查看此图形的较大版本。
{kind=link}

图3:分裂和混合合成方案。该示例指 MutB 底漆合成,其中在四个位置以 0.8 频率引入 NNS 序列(另请参阅表 3)。请注意,化学合成从 3' 到 5'进行,但所有序列都以规范的 5'-3' 方向显示(即,在此方案中,从右到左进行)。突变位置处的野生类型序列以绿色显示,而 NNS 突变序列显示为红色。SalI 识别站点,后来用于引入 ESC 突变被加下划线。指示混合和分割步骤点(2 和 4)。请点击此处查看此图形的较大版本。
{kind=link}

图4:在寡核苷酸靶向诱变中使用独特的限制性酶位点。突变引入策略显示在库 A-C 构造的示例(参见步骤 3.1_3.7)。转载从查平斯卡等人8号,经埃尔塞维尔许可。请点击此处查看此图形的较大版本。
{kind=link}

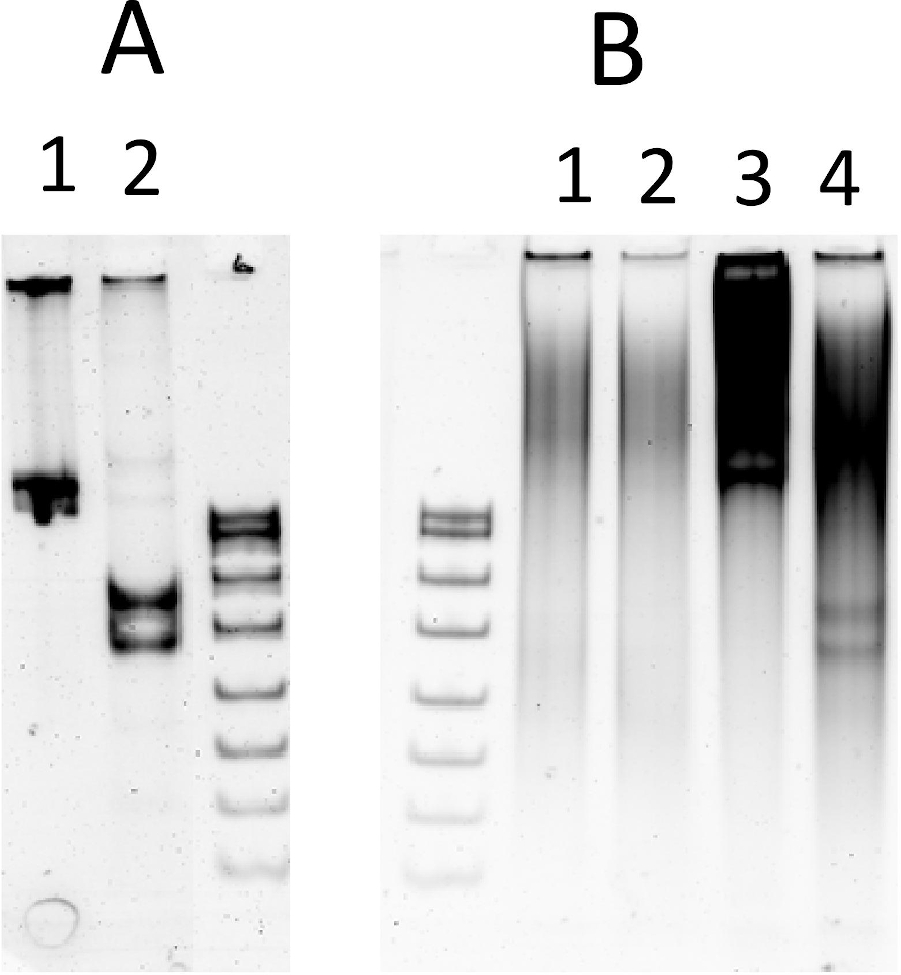
图5:体外转录翻译中的内核解裂解。(A) 最佳 REase 缓冲液中测试基板的裂解:1) 基板,612 bp PCR 产物,具有单个 NlaIV 识别位点;2) 裂解产物,355 bp和257bp.(B)体外转录-转化反应中的裂解(含有0.5微克ESC):1⁄2)15 μL无基板体转录的分录转算(第2行:反应补充1.5mM MgCl2);3⁄4) 15μL体外转录转算与1μg的测试基板;(第4行:反应补充1.5 mM MgCl2)。S-DNA 尺寸标记(pBR322 随 MspI 消化)。样本在 6% 本机 PAGE 中得到解决。脱氧核糖核酸被溴化钠污染。请点击此处查看此图形的较大版本。
{kind=link}

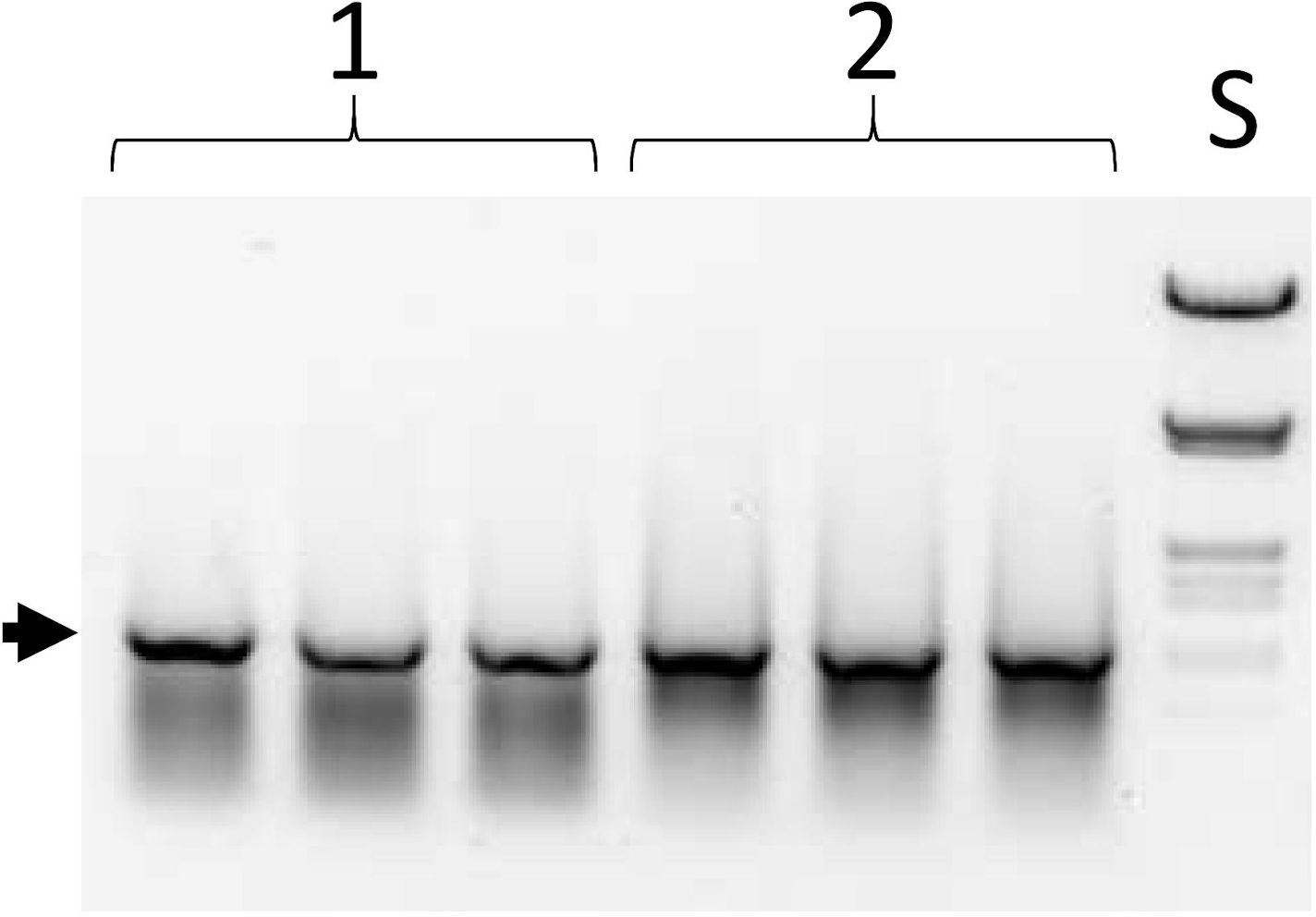
图 6:选择周期中第一个 PCR 的产品。参见图 1B,步骤 III;协议步骤 5.10。列集 1 和 2 是三联加载的两个不同库的等号。S-DNA尺寸标准(用HindIII和EcoRI消化的lambda DNA)。箭头指示全长 ESC (1,050 bp) 的位置。请点击此处查看此图形的较大版本。
{kind=link}

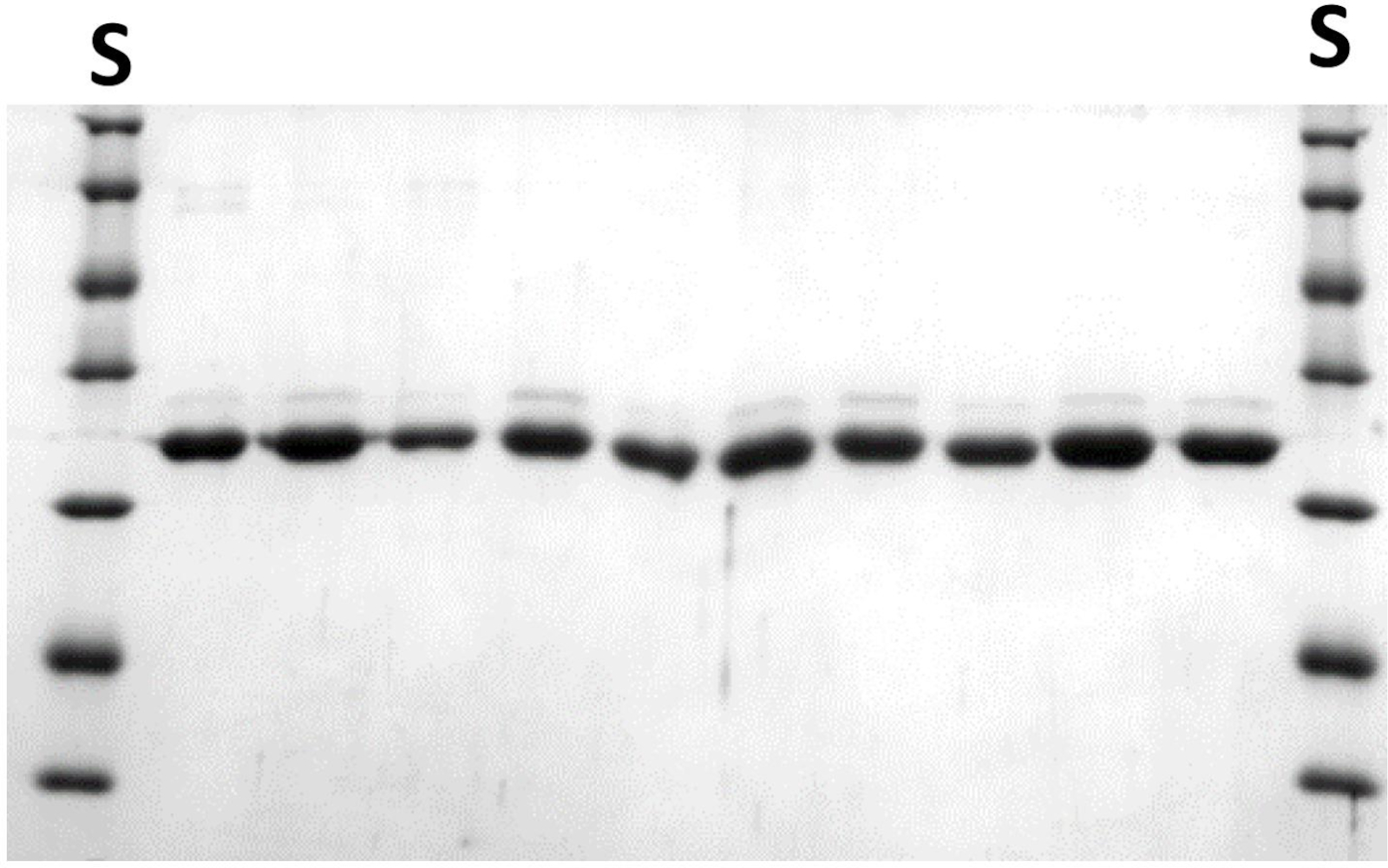
图 7:NlaIV 变型经过纯化,用于小型比例的进一步筛选。请参阅步骤 6.3.11。每条线包含不同变体的 10 μL 等位。S-蛋白质分子量标准。NaIV REase子单位的分子量为29.9 kDa。请点击此处查看此图形的较大版本。
{kind=link}

图 8:筛选 NlaIV 变体以进行序列特异性更改的示例。请参阅步骤 6.4.2。(A) 成功筛选,具有高频率的有前途的变体。S = DNA 大小标记,用 HindIII 和 EcoRI 的 lambda DNA 裂块;野生类型 (wt) = lambda DNA 与野生类型 NlaIV 的分块;* 羔羊DNA基质,未被刻;其他列 = 活动非常低的变体。变体被标记!• 有希望的变异,产生不同于野生类型酶的裂解模式;?• 可能更改序列首选项的变体。(B) 不成功的筛选,大多数变种不活动,一个变种明显没有改变的裂解模式。请点击此处查看此图形的较大版本。
{kind=link}

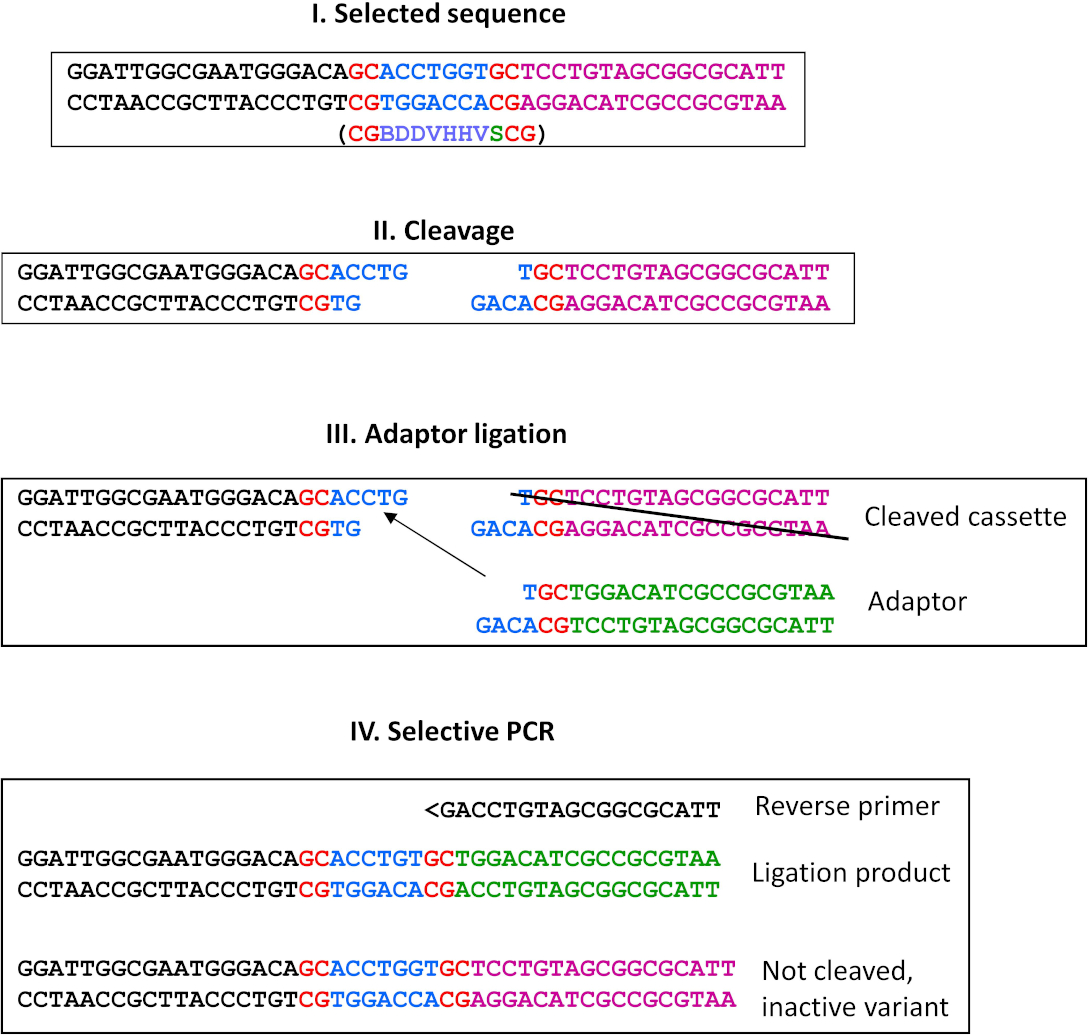
图 9:通过连接进行替代选择。此替代方法可用于生成粘性端端的所有 REase。在这里,我们提出了MwoI酶选择方案(未发布)的示例协议。I) 选定序列(位于 ESC 的右端),其定义的残留物以红色显示,并且以蓝色显示的认知序列的选定变体显示。在位于 ESC 左端的计数器所选序列下方的括号中显示;在内括号中,将放置在 ESC 的左端。II) MwoI 裂解产物;III) 终止体外转录/翻译后,对产品进行纯化,使用多余的适配器进行连接。只有在所选序列中切割的裂解产物才能参与结扎。因此,消除了非活动变体,并且不需要下拉步骤。计数器选定序列中的裂解产物(在 ESC 的左端,未显示)不能参与此连接,因为适配器的突出端与计数器所选序列不互补;IV) 选择性 PCR 使用与主协议中相同的策略来消除具有野生类型退化序列特异性的变体(F1 引漆与计数器选定站点的远端结合),而非活动变体则被无法绑定到未解(因此未通过适配器连接)端绑定的选择性反向引底器消除。在下一个周期中,可以使用与前一步骤的裂解产物(即面板 III 中的"切开盒")和适当的选择性反向引底器相同的适配器来迭代该过程。请点击此处查看此图形的较大版本。
{kind=link}
表1:NlaIV工程中使用的引素。注释中提到的限制站点的顺序带有下划线。小字母表示 DNA 模板中没有补数的序列。请点击此处查看此表(右键单击以下载)。
表2:协议中使用的PCR反应条件。Tm = 底漆熔融温度(如果底漆的 Tm不同,则应使用较低的 Tm)。请点击此处查看此表(右键单击以下载)。
表3:两种用分裂和混合策略合成的诱变引物的质量检查结果。用 [XXX] 表示的诱变性协和器。较低的索引数表示编码氨基酸的位置。改编自查平斯卡等人8号,经埃尔塞维尔许可。请点击此处查看此表(右键单击以下载)。
表4:EP-PCR的结果。从22个ECS克隆的序列分析中得出的主要参数。请点击此处查看此表(右键单击以下载)。
讨论
此处描述的选择协议对 NlaIV 8 进行了测试,NlaIV8是一种二进制 PD-(D/E)XK 折叠识别序列,用于识别具有中央 NN 基座的异位靶点,并催化 NN 基之间的钝端切口。之所以选择NlaIV,是因为NN碱基之间的裂痕表明这些碱基接近复合体中的蛋白质。原则上,该协议可用于任何折叠组的任何序列特定限制内核酶、单体或小链,催化任何交错器的双股断裂,无论催化和特异性域是否重合(如 NlaIV 示例中所示)还是单独(例如 FokI)。此外,该协议原则上不仅有助于产生新的、更窄的酶特异性,还可用于消除恒星活动或创造高保真内核酶。然而,这一切尚未测试。特别是,有针对性地消除恒星活性可能很复杂,因为同样的氨基酸残留物可能参与与期望和不需要的碱基结合。该协议中描述的体外步骤并不限于选择缩小的特异性,还可用于选择其他更改的特异性。然而,变异内联酶存在一个问题:如果基质谱包含非由父母内核酶切割的新目标,则一般没有保护细胞免受这种活性有害影响的好方法。相反,如果内核酶特异性被缩小,目标是野生类型目标的子集,因此现有的同应体甲基转移酶应完全保护。
我们的协议在许多方面不同于许多定向进化协议。开放式阅读帧多样性在实验开始时生成一次,而不是在每个迭代中生成。此外,它是通过拆分和混合合成,而不是由EP-PCR创建的。对于用于此工作中的 NNS 替换,有 (4 x 4 x 2)6 = 1.07 x 109组合的六个位置。因此,任何给定的变体平均存在一次,在 1.7 fmoles 的 ESC.这种能力可以通过与格伦研究公司提供的20个三核苷酸前体混合物的合成,或通过通过分裂和混合寡核苷酸合成在不太有前途的位置降低突变频率,增加到7个位置。如果可能,建议将变化范围限制在六个位置。显然,这种诱变靶向需要一些预先存在的知识,至少涉及基底结合的R易区域。与 EP-PCR 相比,产生多样性的拆分和混合协议具有明显的优势。使用EP-PCR,我们获得了不变的变体和序列,在同一EP-PCR中携带了8个NlaIVESC的替代(表4)。EP-PCR 的库包含应避免的克隆的很大一部分(野生类型序列、多个替换、帧移位和无意义突变,以及不太可能影响序列特异性的地方的突变)。
我们的协议也不同于许多其他定向进化协议,通过存在两个顺序选择步骤。正选择可确保保留所需的活动,否则生物素标记不会被删除,并且可以通过下拉来删除编码序列。从技术上讲,如果靠近所需的裂解部位,但在其他地方存在合适的裂解部位,则偶然出现新颖、非重叠的特异性(例如GCATGC)也可能导致生物素标签的切断。但是,这应该不太可能。负选择会删除为仍然具有不需要活性的酶编码的开放读取帧。此步骤并非严格强制性的,因为该协议仍将使用能够切合选择序列但无法在 ESC 的其他位置切合的变体来丰富输出库,因此不适合 PCR 放大。然而,选择效果预期较低,因为具有原始序列特异性的酶不会从输出中去除,并且会超越具有改变特异性的有前途的变体,但酶活性也会降低。请注意,在总体级别,所需的目标序列和不需要的目标序列都可以(但不必)退化。在 NlaIV 示例中,反目标退化,目标非退化。即使在人口一级有退化,在单个液滴中只有一个(非退化的)目标或反目标存在。在我们的协议中,每次重复选择步骤时都会重新引入目标和反目标序列。因此,开放式阅读帧必须编码能够切开所有可能目标的酶,并且无法切开任何反靶点,才能在多个选择轮中存活。请注意,在协议的每个迭代中重新引入反选择目标的需要都会强制实施两个顺序 PCR。第一个 PCR 使用底漆在反目标外进行内侧,以便反目标的裂解可防止 PCR 反应。第二个 PCR 需要一个底漆,该底漆超出反目标,并重新引入反目标,以确保在多轮选择期间,每个打开的读取帧都针对反目标的所有变体进行测试。
对于产生粘性末端的酶,可以使用基于先前描述的 REase ORF10分离方法的相关替代协议。在我们的实验中使用的生物素捕获的非活性变体的损耗在替代协议中被连接成一个序列,该序列在选择性 PCR 中用作底漆结合位点(图 9)。只有产生具有选定特异性的酶的ESC才会产生具有结扎能力的末端,因此将被选择。计数器 e选定序列的粘滞端序列的设计方式必须使其不能参与与适配器的连接。通过在两个不同的适配器之间切换,从而在选择性 PCR 中切换两个不同的反向引引器,可以轻松实现选择过程的迭代。
即使有了新的协议,在体外工程新特性的任务仍然非常具有挑战性。对于典型的 II 型 REase,序列特异性和内丘解解活性取决于相同的蛋白质区域。因此,在不影响另一种的情况下改变一个是困难的。成功更可能通过一种策略,考虑到酶的足迹,尊重蛋白质-DNA相互作用的对称性,并建立在预先存在的酶偏好之上,这应该预先确定在生化实验中,如对NlaIV的例子8所做的那样。
披露声明
作者没有什么可透露的。
致谢
这项工作得到了波兰国家科学中心(UMO-2011/02/A/NZ1/00052)提供的波兰国家科学中心(UMO-2011/02/A/NZ1/00052)的赠款(0295/B/PO1/2008/34至MB和N301 100 31/3043至KS)的资助。 UMO-2014/13/B/NZ1/03991 和 UMO-2014/14/M/NZ5/00558 到 MB)和短期 EMBO 奖学金到 KS (ATSF 277.00-05)。
材料
| Name | Company | Catalog Number | Comments |
| 1000Å CPG Support (dA, dT, dC, dG) | Biosset | 45-1000-050 | Other vendors can be used as well |
| ASM-800 DNA/RNA | Biosset | 800-001-000 | |
| GeneJET Gel Extraction Kit | Thermo Scientific | K0691 | Any other kit can be used |
| Glen-Pak DNA purification cartridge | Glen Research | 60-5200 | |
| HIS-Select Nickel Affinity Gel | Sigma | P6611 | |
| pET 28a vector | Any other vector with T7 promoter upstream of plycloning site can be used instead | ||
| Phusion High-Fidelity DNA Polymerase | Thermo Scientific | F530S | Any other high fidelity and highly processive thermophilic polymearse can be used instead |
| Porous steel foil | Biosset | 40-063 | |
| Rapid Translation System RTS 100, E.coli HY Kit | Roche | 3 186 148 | |
| Restriction endonucleases | Thermo Scientific | Obviously other vendors, enzymes can be used | |
| Streptavidin Magnetic Beads | New England Biolabs | S1420S | Other vendors can be used as well. We have positively tested beds form Sigma |
| Synthesis chemicals including phosphoramidities | Carl Roth | Other vendors can be used as well | |
| Synthesis columns (different sizes) | Biosset | ||
| T4 DNA ligase | Thermo Scientific | EL0011 | Any other ligase can be used |
参考文献
- Schöttler, S., Wenz, C., Lanio, T., Jeltsch, A., Pingoud, A. Protein engineering of the restriction endonuclease EcoRV--structure-guided design of enzyme variants that recognize the base pairs flanking the recognition site. European Journal of Biochemistry. 258 (1), 184-191 (1998).
- Wenz, C., Hahn, M., Pingoud, A. Engineering of variants of the restriction endonuclease EcoRV that depend in their cleavage activity on the flexibility of sequences flanking the recognition site. Biochemistry. 37 (8), 2234-2242 (1998).
- Samuelson, J. C., Xu, S. Y. Directed evolution of restriction endonuclease BstYI to achieve increased substrate specificity. Journal of Molecular Biology. 319 (3), 673-683 (2002).
- Samuelson, J. C., et al. Engineering a rare-cutting restriction enzyme: genetic screening and selection of NotI variants. Nucleic Acids Research. 34 (3), 796-805 (2006).
- Rimseliene, R., Maneliene, Z., Lubys, A., Janulaitis, A. Engineering of restriction endonucleases: using methylation activity of the bifunctional endonuclease Eco57I to select the mutant with a novel sequence specificity. Journal of Molecular Biology. 327 (2), 383-391 (2003).
- Morgan, R. D., Luyten, Y. A. Rational engineering of type II restriction endonuclease DNA binding and cleavage specificity. Nucleic Acids Research. 37 (15), 5222-5233 (2009).
- Skowronek, K., Boniecki, M. J., Kluge, B., Bujnicki, J. M. Rational engineering of sequence specificity in R.MwoI restriction endonuclease. Nucleic Acids Research. 40 (17), 8579-8592 (2012).
- Czapinska, H., et al. Crystal Structure and Directed Evolution of Specificity of NlaIV Restriction Endonuclease. Journal of Molecular Biology. 431 (11), 2082-2094 (2019).
- Miller, O. J., et al. Directed evolution by in vitro compartmentalization. Nature Methods. 3 (7), 561-570 (2006).
- Zheng, Y., Roberts, R. J. Selection of restriction endonucleases using artificial cells. Nucleic Acids Research. 35 (11), e83(2007).
- Takeuchi, R., Choi, M., Stoddard, B. L. Redesign of extensive protein-DNA interfaces of meganucleases using iterative cycles of in vitro compartmentalization. Proceedings of the National Academy of Science U.S.A. 111 (11), 4061-4066 (2014).
- Howland, J. L. Short Protocols in Molecular Biology. Ausubel, F., Brent, R., Kingston, R. E., Moore, D. D., Seidman, J. G., Smith, J. A., Struhl, K. , Third edition, John Wiley & Sons. New York. (1995).
- Wilson, D. S., Keefe, A. D. Chapter 8 Unit 8.3: Random mutagenesis by PCR. Current Protocols in Molecular Biology. , John Wiley & Sons. New York. (2001).
转载和许可
请求许可使用此 JoVE 文章的文本或图形
请求许可探索更多文章
This article has been published
Video Coming Soon
版权所属 © 2025 MyJoVE 公司版权所有,本公司不涉及任何医疗业务和医疗服务。