Method Article
Kryoschnitt-Dissektion der adulten subependymalen Zone für genaue und tiefe quantitative Proteomanalyse
In diesem Artikel
Zusammenfassung
Die Kryoschnitt-Dissektion ermöglicht die frische, gefrorene Vorbereitung der größten neurogenen Nische im murinen Gehirn für eine tiefe quantitative Proteomanalyse. Die Methode ist präzise, effizient und verursacht minimale Gewebestörungen. Daher eignet es sich ideal für die Untersuchung der molekularen Mikroumgebung dieser Nische sowie anderer Organe, Regionen und Arten.
Zusammenfassung
Die subependymale neurogene Nische besteht aus einem paraventrikulären Band der lateralen Ventrikelwand des lateralen Ventrikels. Die subependymale Zone (SEZ) ist eine dünne und ausgeprägte Region, die den Ventrikeln und der Zerebrospinalflüssigkeit ausgesetzt ist. Die Isolierung dieser Nische ermöglicht die Analyse einer neurogenen Stammzellmikroumgebung. Die Extraktion kleiner Gewebe für die Proteomanalyse ist jedoch eine Herausforderung, insbesondere für die Aufrechterhaltung einer beträchtlichen Messtiefe und das Erreichen einer zuverlässigen Robustheit. Eine neue Methode namens Kryoschnitt-Dissektion (CSD), die hohe Präzision mit minimaler Gewebestörung kombiniert, wurde entwickelt, um diese Herausforderungen zu bewältigen. Die Methode ist kompatibel mit modernsten Massenspektrometrie-Methoden (MS), die den Nachweis von Nischenregulatoren mit geringer Häufigkeit ermöglichen. Diese Studie verglich den CSD und seine Proteomdaten mit der Methode und den Daten, die durch Laser-Capture-Mikrodissektion (LCM) und eine Standard-Wholemount-Dissektion erhalten wurden. Die CSD-Methode führte zu einer doppelten Quantifizierungstiefe in weniger als der Hälfte der Vorbereitungszeit im Vergleich zum LCM und übertraf gleichzeitig die Dissektionsgenauigkeit der Wholemount-Dissektion deutlich. Daher ist CSD eine überlegene Methode zur Erfassung der SWZ für die Proteomanalyse.
Einleitung
Da die Neurogenese im erwachsenen Gehirn eingeschränkt ist, würden verschiedene Reparaturstrategien des zentralen Nervensystems stark von einem besseren Verständnis der Grundlagen des neuronalen Ersatzes bei Erwachsenen profitieren. Nagetiere haben uns geholfen, die grundlegenden Mechanismen der postnatalen Neurogenese zu verstehen, obwohl zu beachten ist, dass die adulte Neurogenese stark artenabhängig ist. Bei Mäusen gibt es drei adulte neurale Stammzellnischen (NSC). Der Hypothalamus ist eine adulte NSC-NSC-Nische mit neurogenem Potential1,2, während die kontinuierliche Neurogenese bei Erwachsenen hauptsächlich auf den Hippocampus3 und die SEZ der Seitenwände der lateralen Ventrikel beschränkt ist4,5,6. Die SEZ ist die größte Keimregion mit NSCs (Typ-B-Zellen), die sich über transitverstärkende Vorläuferzellen (Typ-C-Zellen) zu Neuroblasten (Typ-A-Zellen) entwickeln. Die SWZ enthält 20-35% der Typ-B-Zellen, 1-15% der Typ-C-Zellen, 1-30% der Typ-A-Zellen und 25-50% der ependymalen Zellen7. Die SEZ verfügt über eine komplexe Mikroarchitektur mit Endothelzellen, Mikrogliazellen und ependymalen Zellen, die sich in der Stammzellnische befinden und diese beeinflussen8,9,10. Obwohl Neuronen in der SEZ selten sind, erreichen und beeinflussen Axone, die aus entfernten Quellen wie dem Striatum, dem ventralen tegmentalen Bereich oder dem Hypothalamus stammen, Typ-B-Zellen4. Ein einzigartiges Merkmal dieser Stammzellnische ist die Trennung zwischen dem Ort der Proliferation und dem Ort der Differenzierung. Nach der Proliferation wandern die neuronalen Vorläufer mehrere Millimeter von der SEZ zum Riechkolben, wo sie sich endlos zu Neuronen differenzieren und in bereits bestehende neuronale Schaltkreise integrieren. Untersuchungen zu zellintrinsischen Programmen im Zusammenhang mit der Neurogenese haben bereits Erkenntnisse geliefert, die für experimentelle therapeutische Zellreprogrammierungs- und Transplantationsstrategien wichtig sind15,16,17,18,19,20. Zellextrinesische Signale regulieren jedoch auch die Neurogenese, und Gewebeumgebungen können das neurogene Schicksal von Stammzellen bestimmen11,12,14,21,22,23. Daher ist die Untersuchung der Mikroumgebung der neurogenen Nischen und ihrer Interaktion mit den Stammzellen von entscheidender Bedeutung.
Die extrazelluläre Matrix (ECM) und andere sezernierte Proteine sind ein großer Teil der Mikroumgebung. Für eine genaue Identifizierung und Quantifizierung ist ein proteomischer Ansatz aufgrund der geringen Korrelation zwischen Transkriptom- und Proteinspiegeln für ECM besser geeignet als ein transkriptomischer Ansatz zur Bestimmung der ECM-Zusammensetzung24,25. Darüber hinaus gibt es erhebliche Hinweise darauf, dass Nischenregulatoren in der SWZ nicht ausschließlich von Zellen produziert werden, die die Nische selbst bevölkern. Weiter entfernte Orte, wie der Plexus choroideus, sezernieren modulatorische Signale, die über die Zerebrospinalflüssigkeit an die Stammzellen übertragen werden22,23. Die Untersuchung des Nischenproteoms kann dazu beitragen, Nischenregulatoren in der Nische unabhängig von ihrem Produktionsstandort zu identifizieren, da ein erheblicher Teil der extrazellulären Mikroumgebung durch Proteine aufgebaut wird.
Um die murine ventrikuläre Zone für eine unvoreingenommene proteomische Analyse zu erfassen, ist eine Methode mit hoher Präzision erforderlich, bei der das ca. 50 μm dünne paraventrikuläre Band mit Stammzellen erfasst wird, während das Gewebe des angrenzenden Striatums ausgeschlossen wird. Darüber hinaus muss die Gewebestörung während der Dissektion für die Analyse der extrazellulären Mikroumgebung minimiert werden, da lösliche Proteine, einschließlich Wachstumsfaktoren oder Zytokine, leicht weggewaschen werden könnten. Obwohl es möglich ist, die Massenspektren von festsitzendem Gewebe zu analysieren, reduziert das erforderliche Mittel wie Paraformaldehyd die Proteinidentifikationstiefe und kann posttranslationale Modifikationen einführen. Eine übliche Wholemount-SEZ-Dissektion, z.B. zur Sammlung von Zellen für die fluoreszenzaktivierte Zellsortieranalyse, entfernt die gesamte SEZ mit einer Schere26. Diese Standarddissektion ist schnell mit minimaler Gewebestörung. Eine striatale Kontamination der Proben kann jedoch nicht vermieden werden. Umgekehrt hat LCM den herausragenden Vorteil einer überlegenen Dissektionspräzision. LCM kann jedoch Gewebestörungen verursachen, beispielsweise aufgrund von Hintergrundfärbungen oder laserbedingter Proteindenaturierung. Um die Stärken der Wholemount-Dissektion und LCM zu kombinieren, wurde eine neuartige Methode entwickelt, die mit MS kompatibel ist und als Kryo-Section-Dissektion (CSD) bezeichnet wird (Abbildung 1A-D). Der CSD ermöglicht die Extraktion der SEZ und die Dissektion der SEZ der medialen Wände der lateralen Ventrikel (MEZ), was eine ideale, meist nicht-neurogene Kontrollregion für die SEZ ist (siehe Protokoll). Das Nischenproteom, das durch die Kombination von CSD- und modernsten MS-Methoden erhalten wurde, erwies sich als nützlich für die Charakterisierung und Identifizierung neuartiger Regulatoren in dieser adulten NSC-NSC-NEK-Nische25. Daher wird diese Methode für die Bestimmung der SEZ-Gewebeproteinzusammensetzung nützlich sein.
Protokoll
Alle experimentellen Verfahren in dieser Studie wurden nach deutschen und EU-Richtlinien durchgeführt und vom institutionellen Tierpflegeausschuss und der Regierung von Oberbayern genehmigt. Nur männliche C57Bl6-Mäuse im Alter zwischen 8-10 Wochen wurden für die Experimente verwendet.
1. Vorbereitung des Mausgehirns (~ 15 min pro Maus)
- Bereiten Sie das Dissektionsmedium vor, indem Sie 5 mL 1 M HEPES (Endkonzentration 10 mM) zu 500 ml 1x Hank's Balanced Salt Solution (HBSS) hinzufügen.
HINWEIS: Die Lagerzeit des Dissektionsmediums (+4 °C) sollte 2 Wochen nicht überschreiten. - Opfern Sie die Mäuse durch zervikale Luxation und sezieren Sie vorsichtig das Gehirn.
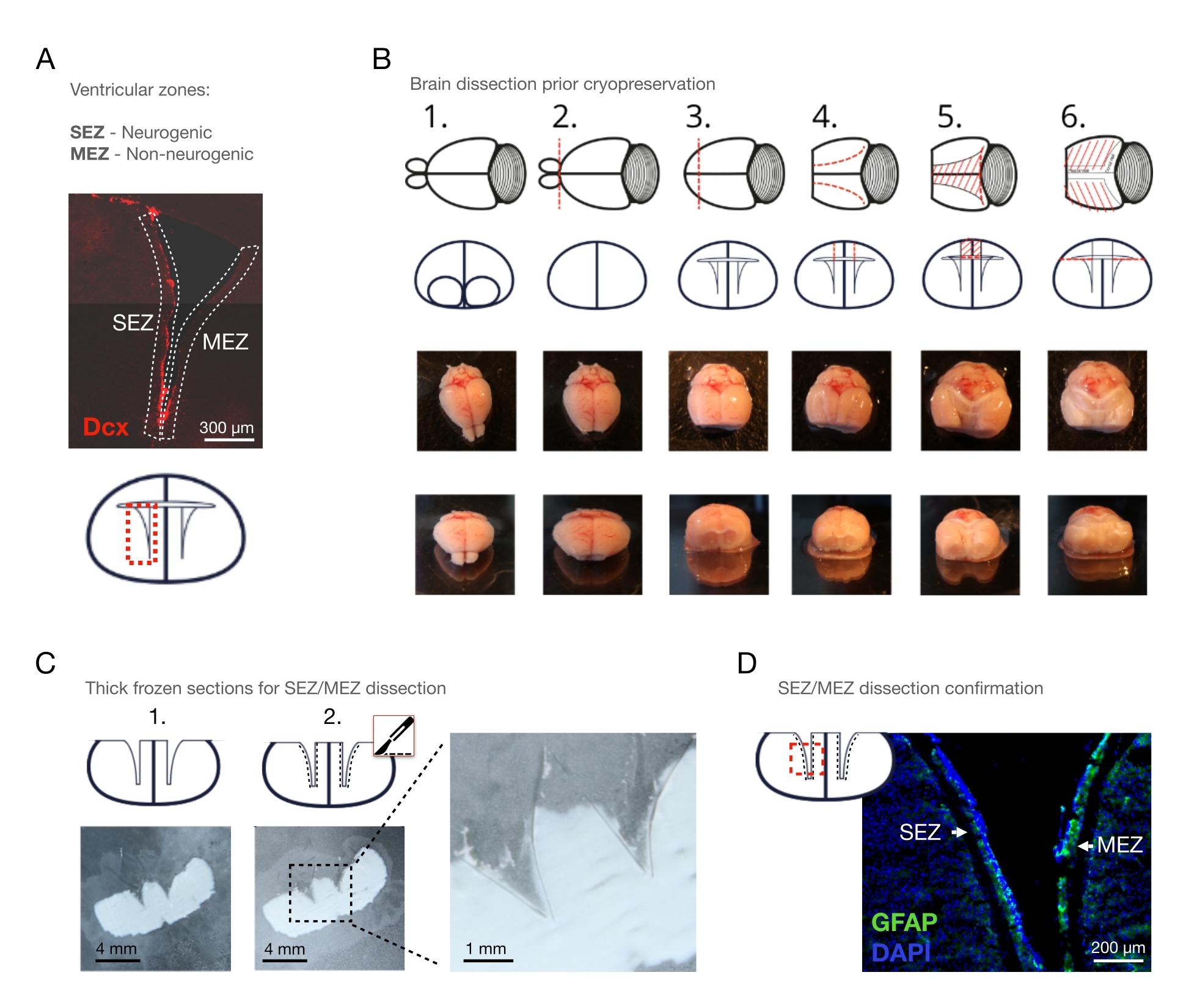
HINWEIS: Bei der Untersuchung des ECM sollte das Gewebe vorzugsweise unverändert bleiben. Die zervikale Dislokation hält die Dissektionszeit so kurz wie möglich und verhindert so die postmortale enzymatische Autodigestion so weit wie möglich. Wenn die Entfernung von Blut für die Forschungsfrage entscheidend ist, perfundieren Sie die Maus einfach transkardiell mit phosphatgepufferter Kochsalzlösung (PBS), bevor Sie das Gehirn entfernen. - Extrahieren Sie das Gehirn durch manuelle Dissektion und legen Sie es in eine Kulturschale, die eiskaltes Dissektionsmedium enthält (Abbildung 1B - 1).
HINWEIS: Halten Sie das Gehirn während der gesamten Dissektion im Dissektionsmedium auf Eis. - Entfernen Sie den Riechkolben (OB) mit einem Skalpell (Abbildung 1B - 2) durch einen geraden koronalen Schnitt zwischen dem OB und dem vorderen Pol des Kortex.
- Entfernen Sie den vorderen Pol des Kortex mit dem Skalpell mit einem koronalen Schnitt, um die lateralen Ventrikel in der koronalen Ebene sichtbar zu machen (Abbildung 1B - 3).
HINWEIS: Stellen Sie sicher, dass der koronale Schnitt ~ 5 mm Rostrally aus dem optischen Chiasma gemacht wird; Andernfalls geht der rostrale Teil der SWZ/MEZ verloren. - Öffnen Sie mit einer Schere beide seitlichen Ventrikel von oben, beginnend mit einem sagittalen Abschnitt von der kortikalen Oberfläche bis zum ventrikulären Lumen, und verlängern Sie diesen Schnitt nach der ventrikulären Flexion c-förmig (Abbildung 1B - 4).
- Verbinden Sie die kaudalen Enden des linken und rechten Sagittalschnitts mittels eines zusätzlichen koronalen Schnitts mit der Schere.
HINWEIS: Die drei Schnitte bilden nun ein Trapez und erleichtern im nächsten Schritt die Entfernung von Cortex und Corpus callosum. - Entfernen Sie den Kortex und den Corpus callosum, die die lateralen Ventrikel bedecken, mit einer Pinzette (Abbildung 1B - 5). Entfernen Sie dann den Kortex und das Corpus callosum, die die medialen ventrikulären Wände bedecken. Machen Sie hier zusätzliche Schnitte, wenn das Gewebe an den medialen Ventrikelwänden befestigt ist, oder heben Sie einfach den Kortex und den Corpus callosum mit einer Schere an, um das Gewebe zu entfernen.
- Verteilen Sie die Ventrikelwände vorsichtig mit einer Pinzette (Abbildung 1B - 6). Entfernen Sie den Aderhautplexus mit einer Pinzette.
HINWEIS: Die vollständige Entfernung des Aderhautplexus ist wichtig, um Interferenzen mit den folgenden Dissektionsschritten zu vermeiden und eine mögliche Kontamination der SEZ/MEZ-Proben zu vermeiden. - Legen Sie das Gehirn auf einen Glasobjektträger und legen Sie den Glasträger auf Trockeneis, um das Gehirn einzufrieren. Pflegen Sie die ventrikulären Wände in der offenen Konfiguration.
HINWEIS: Stellen Sie einen ausreichenden Abstand zwischen den lateralen und medialen Wänden des Ventrikels sicher, um eine präzise und exklusive Dissektion von SEZ und MEZ zu ermöglichen. Wenn sich das Gewebe wieder in eine geschlossene Konfiguration zusammenzieht, verwenden Sie die Pinzette, um die Wände während des Einfrierens in der gewünschten Position zu fixieren. Vermeiden Sie Schäden an der SEZ/MEZ. Versuchen Sie, minimale Kraft anzuwenden, hauptsächlich an der Oberkante der geöffneten Ventrikel.
2. Schnitt des präparierten Gehirns (~ 15 min pro Maus)
- Schneiden Sie 50-100 μm dicke koronale Abschnitte des Gehirns bis zum Ende des lateralen Ventrikels mit einem Kryostaten und montieren Sie die Abschnitte auf Glasobjektträgern. Stellen Sie sicher, dass das Gehirn mit OCT-Medium an der Kryostaten-Befestigungsplatte im Hinterhirn befestigt ist und dass kein OCT mit dem Vorderhirn, insbesondere an den Ventrikeln, in Kontakt kommt.
HINWEIS: OCT-Medium stört die MS-Messungen. Wenn das Gewebe jedoch für einen Antikörpertest verwendet wird, ist es nicht erforderlich, OCT-Medium auszuschließen. Die Verwendung von beschichteten Glasobjektträgern wird nicht empfohlen. Beschichtete Objektträger üben zu viel Haftkraft auf das Gewebe aus und behindern so in den folgenden Schritten die Translokation der Gewebeprobe von den Objektträgern in das Mikrozentrifugenröhrchen.
3. Freihanddissektion von Hirnschnitten (~ 30 min pro Maus)
- Legen Sie die Glasobjektträger mit den Hirnschnitten unter einem Dissektionsmikroskop auf Trockeneis (Abbildung 1C - 1).
- Bereiten Sie die Mikrozentrifugenröhrchen auf Trockeneis vor und stellen Sie sicher, dass die Röhrchen mindestens 1 min auf Trockeneis bleiben, um vor dem Probentransfer ausreichend kalt zu sein.
HINWEIS: Verwenden Sie Mikrozentrifugenröhrchen von hoher Qualität, da einige minderwertige Röhrchen in den nachfolgenden Gewebeaufschlussschritten, die mit MS-Messungen verbunden sind, Kunststoff abwerfen können. - Heben Sie die Scheiben für 15-30 s aus dem Trockeneis, um ein kurzes, unvollständiges Auftauen zu erreichen, um das kompakte Myelin des Striatums als dichte weiße Punkte beobachtbar zu machen.
HINWEIS: Die Lokalisierung der Grenze zwischen der SWZ und dem Striatum wird möglich (Abbildung 1C - 2, siehe Abbildung 2A zum Ausschluss von Myelin und einem Vergleich mit der Wholemount-Methode). Wenn das Auftauen zu lange dauert, kann der Prozess beschleunigt werden, indem ein mit Handschuhen bedeckter Finger auf die gegenüberliegende Seite des Glasobjektträgers gedrückt wird. Dieses Manöver sollte jedoch geübt werden, da übermäßiges Auftauen leicht auftritt. - Trennen Sie die SwZ mit einem vorgekühlten Skalpell vom benachbarten Striatum (Abbildung 1C,D).
- Übertragen Sie die SEZ entweder als ganzes Stück oder in 2-4 Teile geschnitten in ein Mikrozentrifugenrohr, indem Sie die stumpfe Kante des gekühlten Skalpells verwenden. Wenn das Gewebe für eine andere Art von Analyse als MS verwendet werden soll, übertragen Sie die Gewebeprobe stattdessen in den entsprechenden Behälter (z. B. eine 96-Well-Platte).
HINWEIS: Das Schneiden des vollständig gefrorenen Gewebes kann dazu führen, dass Gewebe schnell abbricht und vom Objektträger fällt. Das Schneiden von vollständig aufgetautem Gewebe führt zum Zerfall des Gewebes. Stellen Sie sicher, dass das Gewebe weder vollständig eingefroren noch vollständig aufgetaut ist.
Ergebnisse
Wenn Sie die oben genannten Schritte ausführen, sind die Gewebeproben in den Mikrozentrifugenröhrchen bereit und kompatibel mit der MS-Probenvorbereitung. Nach der Probenvorbereitung erhielten wir ~ 5-7 μg Peptide pro Probe von entweder SEZ oder MEZ pro Maus. Die endgültigen Mengen der Peptide können jedoch von der MS-Zubereitungsmethode abhängen. In den folgenden Proteomvergleichen wurde die Proteinidentifikations- und Quantifizierungstiefe (500-1.000 Proteine pro Probe) erhöht, indem die Peptidspektren rechnerisch mit den für jede Geweberegion erstellten Peptidspektrenbibliotheken abgeglichen wurden25,27. Bemerkenswert ist, dass die hier verwendete verlustfreie Nanofraktionierungsmethode zur Erstellung der Peptidspektrenbibliotheken derzeit nicht kommerziell verfügbar ist. Die MS-Rohdaten wurden mit der MaxQuant-Software28 analysiert, wodurch Massengenauigkeiten im Bereich von Teilen pro Milliarde29 erreicht wurden. Die Max Quant-Umgebung ermöglicht den Abgleich zwischen MS-Läufen. Die Proteinhäufigkeit wurde mit einem markierungsfreien Quantifizierungsalgorithmus quantifiziert30. Die immunhistochemische Färbung wurde an frischem gefrorenem Gewebe durchgeführt und wie zuvor berichtet durchgeführt25 (siehe Materialtabelle).
Kryo-Sektions-Dissektion
Die vollständige SEZ und MEZ von erwachsenen Mäusen (n = 4) wurde mit CSD erhalten (siehe Abbildung 1 und Protokoll). Der somatosensorische Kortex (Cx) wurde mit einer chirurgischen Schere seziert. Weitere 4 Mäuse wurden auf die gleiche Weise seziert; Das sezierte Gewebe wurde jedoch in einer Probe pro Region zusammengefasst, um die Proteombibliothek (10.923 identifizierte Proteine) für eine erhöhte Proteinidentifizierung und Quantifizierung in den einzelnen Proben zu erstellen25. In den vier Einzelproben (Mittelwert ± SD) wurden 6.673 ± 317,4 Proteine in der SWZ und 6.747 ± 37,7 in der MEZ quantifiziert. Alle MS-Proteomik-Daten wurden über das PRIDE31-Partner-Repository im ProteomeXchange-Konsortium hinterlegt, und die Hier gemeldete Zugangsnummer für die Proteome ist ProteomeXchange: PXD016632 (http://proteomecentral.proteomexchange.org).
Vergleich zur Wholemount-Dissektion
Die Wholemount-Dissektion wurde nach einem Standardprotokoll durchgeführt26. Die Wholemount-Dissektion ergab eine ähnliche Anzahl von Proteinen (etwa 6.000 für SEZ und 6.000 für Cx, n = 4 pro Gruppe) im Vergleich zu CSD25. Eine der beabsichtigten Verbesserungen bei der Verwendung von CSD für die SEZ anstelle eines Wholemount-Dissektionsprotokolls ist die Reduzierung einer potenziellen striatalen Kontamination. In SEZ-Proben, die mit Gewebe aus einer anderen Region kontaminiert sind, können nachgewiesene Kandidatenproteine keiner Region zugeordnet werden, da sich eine signifikante Anreicherung aus der interessierenden Region und dem Kontaminator ergeben kann. Immunhistochemisch wurden die Myelin-assoziierten Glykoprotein (MAG)-positiven myelinreichen inneren Kapseln des Striatums in den Wholemount-Proben, aber selten in den CSD-Proben identifiziert (Abbildung 2A). Die striatale Kontamination in den Wholemount-Proben konnte durch die Identifizierung der Anreicherung von Myelinproteinen in der SWZ im Vergleich zu den Proben der somatosensorischen Kortex (Cx) grauen Substanz (GM) bestätigt werden (Abbildung 2B). Beachten Sie, dass große Teile des Cx GM, insbesondere die oberen Cx-Schichten, unmyelinisiert sind32.
Da große Faserbündel das Striatum passieren, führte die Kontamination durch diese Region zur Anreicherung von Myelinproteinen im Vergleich zum Cx. Die Myelinproteine, die als Marker für die striatale Kontamination in den SEZ-Proben verwendet wurden, waren das Myelin-Basisprotein (MBP), das Myelin-assoziierte Glykoprotein (MAG), das Proteolipid-Protein 1 (Plp1) und die 2',3'-cyclische Nukleotid-3'-Phosphodiesterase (Cnp). Alle Myelin-Marker-Proteine waren in der SEZ im Vergleich zur Cx signifikant angereichert. Umgekehrt ergaben Vergleiche für die vier Myelin-Markerproteine im CSD-Datensatz keine signifikanten Unterschiede beim Vergleich von SEZ mit Cx (Abbildung 2B). Proteomische Daten des Striatums33 stützen die Hypothese, dass die Anreicherung von Myelinproteinen in den SEZ-Proben der Wholemount-Dissektion durch die Kontamination mit striatalem Gewebe verursacht wurde. Daher verhinderte der CSD weitgehend eine Kontamination durch striatales Gewebe (reich an kompaktem Myelin) im Vergleich zu einer Wholemount-Dissektion.
Eine unvoreingenommene Proteomanalyse von nicht dissoziiertem Gewebe kann interessante extrazelluläre Proteine aufdecken. Bei verbesserter Dissektion unter Verwendung des CSD wurden extrazellulär assoziierte Proteine in den Proben im Vergleich zu den Wholemount-Proben signifikant angereichert (Abbildung 2C, Annotationsanreicherungstest). Die CSD- und Wholemount-Dissektion zeigen eine vergleichbare Anreicherung der Genontologie (GO) Begriffe "extrazelluläres vesikuläres Exosom" und "extrazellulärer Regionsteil". Allerdings ist der GO-Begriff "Matrisomen-assoziiert" im CSD etwas stärker angereichert als in der Wholemount-Dissektion. Dementsprechend wurden das ECM-Kreuzbindungsenzym und der kürzlich entdeckte Neurogeneseregulator Transglutaminase-2 (Tgm2) in der SEZ im Vergleich zu Cx unter Verwendung des CSD25 angereichert befunden. Im Gegensatz dazu wurde kein Unterschied zwischen SEZ- und Cx-Proben gefunden, die durch die Wholemount-Dissektion erhalten wurden (Abbildung 2D). Proteomische Daten des Striatums33 stützen die Hypothese, dass der Nachweis des Neurogeneseregulators Tgm2 durch Wholemount-Dissektion durch die Kontamination mit striatalem Gewebe behindert wurde. Insgesamt ist die Kryoschnitt-Dissektion somit eine gelungene, aber auch notwendige Verbesserung gegenüber der Standarddissektion für die nischenspezifische Proteomanalyse.
Vergleich zur Laser-Capture-Mikroskopie
Die vordere Hälfte der SEZ und die MEZ von 3 erwachsenen Mäusen wurden für LCM erhalten (Abbildung 3A). Insgesamt weist die LCM-Methode einige Nachteile auf, insbesondere in Bezug auf Gewebestörung und Effizienz. Um die Region von Interesse unter dem Dissektionsmikroskop zu visualisieren, ist eine Hintergrundfärbung notwendig, die möglicherweise kleine oder lösliche Proteine von Interesse wegspült, z. B. Wachstumsfaktoren, Zytokine oder ECM-Regulatoren wie Enzyme. Darüber hinaus verbringen Objektträger während der Laserentfernung unterschiedliche Zeiten bei Raumtemperatur. Darüber hinaus könnte der Laser selbst interessante Proteine denaturieren.
CSD hat einen erheblichen Vorteil gegenüber LCM in Bezug auf den Zeit- und Arbeitsaufwand, der für die Durchführung der Dissektion erforderlich ist: Schritt 1 des Protokolls muss sowohl für CSD als auch für LCM ähnlich durchgeführt werden; Ohne diesen Schritt bleiben die ventrikulären Wände haften, was die Trennung von MEZ- und SEZ-Proben erschwert. Da die CSD-Abschnitte (100 μm) 6-7-mal dicker sind als die maximale Dicke34 der LCM-Abschnitte (15 μm), dauert Schritt 2 (Schneiden des Gehirns) und Schritt 3 (Entfernen von MEZ und SEZ aus jedem koronalen Abschnitt) mindestens 6-7-mal länger für LCM. Die notwendige Hintergrundfärbung und das Einrichten des Lasermikroskops nehmen zusätzliche Zeit in Anspruch. Hier dauerte es dreimal länger, 50% der SWZ und MEZ von 3 Tieren von LCM zu ernten, verglichen mit 100% der SWZ und MEZ von 4 Tieren von CSD, was einen achtfachen Geschwindigkeitsvorteil von CSD darstellt. Zusammenfassend lässt sich sagen, dass LCM nicht nur einen erheblichen zusätzlichen Aufwand erfordert, sondern das Gewebe auch einer wesentlich längeren Manipulationsdauer und Temperaturänderungen ausgesetzt ist, die die Dynamik und Zuverlässigkeit der durch nachfolgende Analysen generierten Daten beeinträchtigen kann.
Die MS-Ergebnisse von CSD wurden mit den Ergebnissen der Laser-Capture-Mikrodissektion (LCM) verglichen. Beide Datensätze wurden mit der proteomischen Bibliothek abgeglichen, die durch das Pooling von CSD-Proben generiert wurde. Im Durchschnitt ergab LCM 3.441 ± 270,0 und 3.613 ± 238,7 einzelne Proteine in der SEZ bzw. der medialen ventrikulären Zone (Abbildung 3B). Angesichts des bemerkenswerten Unterschieds in der Proteinidentifizierung zeigte die Hauptkomponentenanalyse (PCA) eine deutliche Trennung nach der Dissektionsmethode (Komponente 1: 62,7%, nicht gezeigt). Komponente 2 zeigte den größten Abstand für SEZ und MEZ unter den LCM-Proben (8,5%, Abbildung 3C). Komponente 3 scheint auch LCM und CSD zu trennen; Dieser Unterschied könnte jedoch eher auf methodische Unterschiede als auf die Anzahl der identifizierten Proteine (6,4%) zurückzuführen sein. Dennoch blieb die regionale Trennung insgesamt für die Kryo-Dissektionsdaten auffallend deutlich und weitaus besser als für LCM. Diese Diskrepanz in der Datendynamik kann sich aus unterschiedlichen Zeiten ergeben, die die Proben während der Laserdissektion bei Raumtemperatur verbringen, oder eine höhere Anfälligkeit für kleine Gewebe führt zu einer Variabilität in den nachfolgenden Proteomikprotokollen und Massenspektrometriemessungen.
Um nach Unterschieden im Proteomprofil des ECM zu suchen, wurde für SEZ und MEZ ein 2D-Annotationsanreicherungstest zwischen CSD und LCM durchgeführt (Abbildung 3D). Die Berechnung der relativen Anreicherung von GO-Termen zwischen LCM- und CSD-Proben ermöglicht den Vergleich der relativen Proteomdynamik der ECM-Proteincluster zwischen den beiden Methoden trotz der ungleichen Gewebemenge und der Unterschiede im Dissektionsprotokoll. Die Plots zeigen eine gute Korrelation zwischen LCM und CSD. Die Annotationen "extrazellulärer Regionsteil" und "extrazelluläre membrangebundene Organelle" sind sowohl in Methoden als auch in Regionen ähnlich angereichert. Daher scheint der erhöhte Zeitbedarf von LCM nicht durch eine relativ höhere Sensitivität für ECM-assoziierte Proteine kompensiert zu werden. Stattdessen bietet CSD eine robustere Identifizierung/Quantifizierung beim Vergleich der Probendaten für die Neurogenese und die SEZ-assoziierten ECM-Proteine Tgm2, Thrombospondin-4 (Thbs4), S100a6 und Tenacin-C (Tnc) (Abbildung 3E). Im Fall von TnC zeigte zwar in allen Proben quantifiziert, aber nur CSD zeigte eine Anreicherung für SEZ im Vergleich zu MEZ. Dennoch zeigten die SEZ-assoziierten Basalmembranproteine Nidogen-1 (Nid1), Laminin-Untereinheit beta-2 (Lamb2) und Basalmembran-spezifisches Heparansulfat-Proteoglykan-Kernprotein (Hspg2)35 in den LCM-Proben eine noch robustere Anreicherung (im Vergleich zu MEZ) als in den CSD-Proben (nicht gezeigt). Daher kann CSD Gewebeproben liefern, die ein genaues und tiefes quantitatives Proteom für die SEZ-Charakterisierung in einem angemessenen Zeitrahmen liefern, ohne sich Gedanken über eine beeinträchtigte Gewebeintegrität oder einen Proteinverlust machen zu müssen.
Statistik
Statistische Tests, 2D-Annotationsanreicherungstests und PCA wurden in der Perseus-Umgebung durchgeführt. Proteine wurden in die Analyse einbezogen, wenn für jede Methode in mindestens einer Probe ein gültiger Wert nachgewiesen wurde. Proteinhäufigkeits- und Zahlenvergleiche wurden mittels Datenanalyse-Software visualisiert (siehe Materialtabelle). Für Proteinvergleiche wurde eine permutationsbasierte Kontrolle der False Discovery Rate (FDR) (FDR wurde auf 0,05, 250 Randomisierungen gesetzt) verwendet. Für die 2D-Annotationsanreicherungstests36 werden die angezeigten GO-Terme deutlich angereichert (FDR wurde mit der Benjamini-Hochberg FDR-Kontrollmethode auf 0,02 gesetzt).

Abbildung 1: Die Kryoschnitt-Dissektionsmethode. (A) Überblick über die interessierende Region: den lateralen Ventrikel mit der neurogenen SEZ und der nicht-neurogenen MEZ. Neuroblasten, die mit Dcx immungefärbt sind. (B) Schrittweise Entfernung des OB, des Vorderpols, des Kortex und des Corpus callosum oberhalb der Ventrikel und des Aderhautplexus: 1. Platzierung im Dissektionsmedium, 2. Entfernung von OB, 3. Entfernung des vorderen Pols des Kortex, 4. sagittale Schnitte der ventrikulären Oberseite, 5. Entfernung der ventrikulären Oberseite, 6. Ausbreitung der Ventrikelwände. (C) 100 μm koronale Scheiben des frisch gefrorenen Mausgehirns, (1.) vor und (2.) nach der Entfernung der Ventrikelwände mit einem eiskalten Skalpell. Skalenbalken = 4 mm (D) Färbung eines koronalen Schnitts eines lateralen Ventrikels (GFAP: grün; DAPI: blau), zeigt die SEZ und MEZ, die mit dem CSD seziert wurden. Maßstabsbalken = 300 μm (A), 200 μm (D). Abkürzungen: CSD = Kryo-Sektions-Dissektion; SWZ = subependymale Zone; MEZ = mediale ependymale Zone; Dcx = Doublecortin; OB = Riechkolben; GFAP = Gliafibrillen-saures Protein; DAPI = 4′,6-Diamidino-2-phenylindol. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 2: Überlegene Dissektionsgenauigkeit bei der Kryo-Schnitt-Dissektion im Vergleich zur Wholemount-Dissektion. (A) Immunhistochemisches Bild einer SEZ-Probe, erhalten durch Wholemount-Dissektion (links). Der Einschluss von myelinreichem Striatalgewebe wird durch Färbung gegen MAG (grün) sichtbar gemacht. Färbung einer mit dem CSD sezierten SEZ (rechts). Bei CSD ist fast das gesamte striatale Myelin (Färbung gegen MAG, grün) aus dem Probenband ausgeschlossen. Kerne wurden mit DAPI (blau) visualisiert. (B) Vergleich der Myelinmarkeranreicherung in SWZ vs. Cx aus der Gesamtenmenge (MBP: p = 0,0074; MAG: p = 0,0016; Plp1: p = 0,0011; CNP: p = 0,0029) und CSD (MBP: p = 0,0667; MAG: p = 0,0236; Plp1: p = 0,3420; CNP: p = 0,1842). (C) 2D-Annotationsanreicherungstest, bei dem die gesamte SEZ mit den CSD-SEZ-Proben verglichen wird. Die GO-Begriffe extrazellulärer Raum und Matrisomen-assoziiert sind in den CSD-Daten stärker angereichert als in den Wholemount-Daten. (D) Die Proteinhäufigkeit des NSC-Regulators Tgm225, dargestellt für die Wholemount-Dissektion und den CSD. Tgm2 ist in der SWZ im Vergleich zum Cx im CSD signifikant angereichert (CSD: p = 0,0029; Wholemount: p = 0,1775). Für B und D: Als Referenz Proteomdaten von Sharma et al.33 mit Messungen von Striatum und Cortex, die für die entsprechenden Proteine in den Wholemount- und CSD-Proben dargestellt wurden. Maßstabsbalken = 200 μm (A). Abkürzungen: CSD = Kryo-Sektions-Dissektion; SWZ = subependymale Zone; MAG = Myelin-assoziiertes Glykoprotein; Cx = somatosensorischer Kortex; MBP = Myelin-Basisprotein; Plp1 = Proteolipid-Protein 1; CNP = 2',3'-cyclische Nukleotid-3'-phosphodiesterase; GO = Genontologie; NSC = neurale Stammzelle; Tgm2 = Tranglutaminase 2; DAPI = 4′,6-Diamidino-2-phenylindol; LFQ = markierungsfreie Quantifizierung. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 3: Verbesserte extrazelluläre Proteinquantifizierung mit Kryoschnittdissektion im Vergleich zu LCM. (A) Kresylviolettfärbung eines lateralen Ventrikels vor und nach der Lasererfassung der SEZ und MEZ (links). Zum Vergleich der CSD-Schnitt der SWZ und MEZ (rechts). Skalenbalken = 150 μm. (B) Vergleich der Anzahl der nachgewiesenen Proteine in den SEZ- und MEZ-Proben aus CSD und LCM. Die Daten werden als Mittelwert ± SD dargestellt. (C) Hauptkomponentenanalyse der SEZ- und MEZ-Proben im Vergleich von CSD und LCM (Komponente 2: 8,5% der Varianz; Komponente 3: 6,4%). (D) 2D-Annotationsanreicherung der kryokonierten und laserdisparierten MEZ (oben) und SEZ (unten). Die GO-Begriffe extrazelluläre Organelle und extrazellulärer Regionsteil sind signifikant angereichert (rote Punkte). (E) Häufigkeiten extrazellulärer SEZ-assoziierter Markerproteine in SEZ und MEZ für LCM (Tnc: p = 0,3789) und den CSD-Proben (Tgm2: p = 0,2940; S100a6: p = 0,0218; THBS4: p = 0,3941; Tnc: p = 0,0004). Abkürzungen: CSD = Kryo-Sektions-Dissektion; LCM = Laser-Capture-Mikrodissektion; SWZ = subependymale Zone; MEZ = mediale ependymale Zone; GO = Genontologie; Tnc = Tenacin-C; Tgm2 = Transglutaminase 2; S100a6 = S100 Calcium-bindendes Protein A6; THBS4 = Thrombospondin-4; LFQ = markierungsfreie Quantifizierung. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Diskussion
Die CSD-Methode ermöglichte es, SEZ-Gewebe präzise zu extrahieren und mit MS ein zuverlässiges Proteom mit signifikanter Tiefe zu erzeugen. CSD zeigt einen klaren Vorteil gegenüber der Wholemount-Dissektion in Bezug auf eine stark reduzierte striatale Kontamination von SEZ-Proben und extrazelluläre Proteinanreicherung. Da es auch möglich ist, mit CSD und Wholemount-Dissektion eine ähnliche Anzahl von Proteinen in Einzelproben (~6.500 Proteine pro Probe) nachzuweisen, lohnt sich die zusätzliche Zeit für CSD. LCM bietet eine präzisere SEZ-Dissektion, erreichte aber eine niedrigere Proteomtiefe mit nur 3.500 Proteinen pro Probe, obwohl das gleiche MS-Protokoll wie CSD (Library Matching und labelfreie Quantifizierung) verwendet wurde. Wichtig ist, dass die Variabilität viel größer war, wahrscheinlich aufgrund der achtmal längeren Vorbereitungszeit pro Probe. PcA der von LCM und CSD gewonnenen Proben zeigt eine klare Trennung beider Methoden mit engen regionsspezifischen Clustern, die robust voneinander getrennt sind. Im Gegensatz dazu zeigten die LCM-Proben eine verstreutere Verteilung, was wahrscheinlich zum Teil auf die Länge der Vorbereitung zurückzuführen ist. Es ist unklar, ob das Sammeln von weit mehr Proben über einen längeren Zeitraum ein Proteom von gleicher Robustheit und Tiefe wie LCM ergeben hätte. Bei der Berechnung einer Schätzung würde das Sammeln eines ähnlichen Probenvolumens wie bei CSD mit LCM 5-8-mal länger dauern, sogar bis zu 15-mal länger, wenn Proben für die Peptidspektrenbibliotheken einbezogen würden, und ein Großteil davon unter aufgetauten Bedingungen. Darüber hinaus lieferte LCM in Anbetracht der zusätzlichen Störungen des Gewebes, die für LCM notwendig sind (Hintergrundfärbung, Laserdissektion), wenig, wenn überhaupt, gewinnbringend gegenüber CSD. Daher kann CSD als besser geeignet für die extrazelluläre Proteomforschung, insbesondere für die SEZ, angesehen werden.
Insbesondere, wenn die Region von Interesse kleiner als die SEZ ist (z. B. nur die ependymale Zellschicht untersuchen), fällt ein Freihandansatz hinter die Genauigkeit des LCM zurück. Zum Beispiel ist die Verwendung von CSD zur Trennung der ependymalen von der subependymalen Schicht schwierig, da die ependymale Schicht nur einen Zelldurchmesser breit ist und die Abgrenzung zur subependymalen Schicht für das bloße Auge in frischem gefrorenem Gewebe nicht sichtbar ist. Daher ist LCM eine bessere Wahl als CSD, wenn eine präzise Dissektion auf einer Skala unter 50 μm wichtiger ist als ungestörtes Gewebe oder die Dissektionszeit kurz gehalten wird. Für Regionen mit einer Breite von 50 μm und mehr ist die Präzision von CSD jedoch vergleichbar mit der von LCM für die ECM-Proteinanalyse.
CSD hat sich bereits als nützlich erwiesen, indem er zur Untersuchung der funktionellen Rolle des ECM in der neurogenen Nische beiträgt25. Daher könnte die fortgesetzte Anwendung von CSD in der SEZ für verschiedene Protein- und Proteomuntersuchungen (oder sogar die Einzelkern-RNA-Sequenzierung) zum Nachweis weiterer Neurogenese-Regulatoren, Stammzellaktivierungsmarker und einem tieferen Verständnis der SEZ-Stammzell-Nischenphysiologie führen. In Anbetracht des Rückgangs der Neurogenese in der alternden SEZ37 könnte eine prägnante Analyse der ECM-Veränderungen der SWZ von älteren vs. jungen Mäusen das Verständnis der genauen Nischenmechanismen fördern, die die Entwicklung und Aufrechterhaltung von NSC fördern38,39. Darüber hinaus ist der Einfluss von Entzündungen und Verletzungen auf die Neurogenese der SWZ gut belegt40,41,42,43. Die Anreicherung von blutabgeleitetem Fibrinogen in der SWZ nach einer kortikalen Hirnverletzung und ihr Einfluss auf die Astrogliogenese und Narbenbildung der SWZ44 unterstreicht den potenziellen Einfluss traumainduzierter Mikroumgebungsveränderungen auf die Physiologie der SEZ-Stammzellen. Daher könnte die Untersuchung des SEZ-ECM-Proteoms in Verbindung mit Hirnverletzungen unter Verwendung von CSD dazu beitragen, die Mechanismen aufzuklären, durch die Verletzungen und Entzündungen die Neurogenese beeinflussen. Wichtig ist, dass die Methode auch auf neurogene Nischen des menschlichen Gehirns in Gesundheit und Krankheit anwendbar sein könnte, da frisches gefrorenes Gewebe oft aus Operationen gewonnen werden kann. Darüber hinaus wäre es angesichts der Speziesunterschiede in der adulten Neurogenese auch faszinierend, die CSD-Methode auf andere Spezies anzuwenden, z.B. in Verbindung mit der striatalen Neurogenese. Darüber hinaus können mit anderen Proteinnachweismethoden Unterschiede in lokal produzierten Wachstumsfaktoren mit CSD für die SWZ und MEZ (z. B. ELISA) genau und effizient untersucht werden.
Schließlich könnte das Dissektionsverfahren möglicherweise für die genaue Extraktion anderer Hirnregionen modifiziert werden, auch für Forschungsfragen, die nicht mit der Neurogenese zusammenhängen. Zum Beispiel beinhaltet CSD einen kurzen Halbauftauschritt, bei dem kompaktes Myelin als weiße Bereiche sichtbar ist, die sich vom durchscheinenderen Restgehirngewebe unterscheiden. Mit einer einfachen Modifikation der Methode würde diese Eigenschaft die präzise Dissektion von nur Corpus callosum kompaktem Myelingewebe ermöglichen, das einer proteomischen Analyse verletzungsbedingter Veränderungen unterzogen werden könnte. Ein Vorschlag für eine Protokolländerung, die die korpuskalöse Dissektion ermöglichen würde, besteht darin, die Schritte 1.5-1.9 des Protokolls wegzulassen und direkt mit der Vorbereitung der koronalen Abschnitte fortzufahren, anstatt die Ventrikel zu öffnen, um die SEZ und MEZ zugänglich zu machen. Legen Sie dann die Abschnitte auf Trockeneis, heben Sie die Scheiben kurz an und tauen Sie sie halb auf und entfernen Sie einfach den Corpus callosum mit einem Skalpell. Dieses Präparat sollte nun für jede Analyse bereit sein, die eine effiziente Dissektion von nativem Corpus callosum-Gewebe erfordert.
Zusammenfassend stellt diese Studie eine Mikrodissektionsmethode vor, die für eine zuverlässige ventrikuläre neurogene Nischenproteomanalyse verwendet werden könnte. Die Daten unterstreichen die Kompatibilität und den Nutzen der CSD-Methode zusammen mit der MS-basierten proteomischen Analyse der SEZ-Mikroumgebung. Die Kombination aus Präzision, Effizienz und minimaler Gewebestörung machen den CSD zu einer wertvollen Erweiterung bestehender Methoden.
Offenlegungen
Die Autoren erklären keine konkurrierenden Interessen
Danksagungen
Wir danken Mathias Mann für die Durchführung großer Teile der Experimente in seinem Labor, Fabian Coscia für die Hilfe bei LCM und Proteomanalyse, Tatiana Simon-Ebert für die Ganzmount-Dissektion und Korbinian Mayr und Igor Paron für ihre technische Hilfe. Wir danken der Deutschen Forschungsgemeinschaft für die Förderung von MG (SFB870, TFR274), der EU (Eranet S-700982-5008-001), des ERC (aERC "NeuroCentro" an MG), der Swedish Society for Medical Research (SSMF, an JK) Postdoc-Stipendium und KI-Stiftungen und -Fonds (2020-01351, an JK).
Materialien
| Name | Company | Catalog Number | Comments |
| Cryostat CM3050S | Leica | ||
| Dissecting microscope | Leica | ||
| Dumont no. 5SF forceps, Inox super fine tip | Fine Science Tools | cat. no. 11252-00 | |
| Hank’s Balanced Salt Solution with CaCl2 and MgCl2 | Invitrogen | cat. no. 24020 | |
| HEPES buffer solution (1 M) | Invitrogen | cat. no. 15630 | |
| Microscope slides | RS France | cat. no. BPB018 | |
| Safe-lock tubes, PCR clean 2.0 mL | Eppendorf | cat. no. 0030123344 | |
| Spring scissors, Vannas-Tubingen 5 mm | Fine Science Tools | cat. no. 15003-08 | |
| Surgical disposable scalpels | B. Braun | cat. no. 5518083 | |
| Tissue culture dishes 60 mm | Greiner Bio-One | cat. no. 633180 | |
| Antibodies | |||
| Alexa Fluor secondary antibodies (488, 555) (1/1,000) | ThermoFisher Scientific | cat. no. A-11001 | |
| DAPI | Sigma | cat. no. D9542 | |
| guinea pig polyclonal anti-DCX 1:500 | Millipore | cat. no. AB2253, | |
| mouse monoclonal anti-GFAP 1:500 | Sigma | cat. no. G3893 | |
| mouse monoclonal anti-MAG 1:400 | Millipore | cat. no. MAB1567 | |
| Software | |||
| GraphPad Prism version 9 | GraphPad Software, San Diego California USA | www.graphpad.com | |
| Perseus Version 1.6.10.50 | Max-Planck Institute for Biochemistry, Munich Bavaria Germany | https://maxquant.net/perseus/ | |
| ZEN imaging software | Carl Zeiss |
Referenzen
- Goodman, T., Hajihosseini, M. K. Hypothalamic tanycytes-masters and servants of metabolic, neuroendocrine, and neurogenic functions. Frontiers in Neuroscience. 9, 387 (2015).
- Chaker, Z., et al. Hypothalamic neurogenesis persists in the aging brain and is controlled by energy-sensing IGF-I pathway. Neurobiology of Aging. 41, 64-72 (2016).
- Kuhn, H. G., Toda, T., Gage, F. H. Adult hippocampal neurogenesis: a coming-of-age story. Journal of Neuroscience. 38 (49), 10401-10410 (2018).
- Obernier, K., Alvarez-Buylla, A. Neural stem cells: origin, heterogeneity and regulation in the adult mammalian brain. Development. 146 (4), (2019).
- Obernier, K., et al. Adult neurogenesis is sustained by symmetric self-renewal and differentiation. Cell Stem Cell. 22 (2), 221-234 (2018).
- Bordiuk, O. L., Smith, K., Morin, P. J., Semënov, M. V. Cell proliferation and neurogenesis in adult mouse brain. PloS One. 9 (11), 111453 (2014).
- Doetsch, F., García-Verdugo, J. M., Alvarez-Buylla, A. Cellular composition and three-dimensional organization of the subventricular germinal zone in the adult mammalian brain. Journal of Neuroscience. 17 (13), 5046-5061 (1997).
- Lim, D. A., Alvarez-Buylla, A. The adult ventricular-subventricular zone (V-SVZ) and olfactory bulb (OB) neurogenesis. Cold Spring Harbor Perspectives in Biology. 8 (5), 018820 (2016).
- Sato, K. Effects of microglia on neurogenesis. Glia. 63 (8), 1394-1405 (2015).
- Tavazoie, M., et al. A specialized vascular niche for adult neural stem cells. Cell Stem Cell. 3 (3), 279-288 (2008).
- Sirko, S., et al. Chondroitin sulfates are required for fibroblast growth factor-2-dependent proliferation and maintenance in neural stem cells and for epidermal growth factor-dependent migration of their progeny. Stem Cells. 28 (4), 775-787 (2010).
- Mercier, F. Fractones: extracellular matrix niche controlling stem cell fate and growth factor activity in the brain in health and disease. Cellular and Molecular Life Sciences: CMLS. 73 (24), 4661-4674 (2016).
- Mercier, F., Kitasako, J. T., Hatton, G. I. Anatomy of the brain neurogenic zones revisited: fractones and the fibroblast/macrophage network. Journal of Comparative Neurology. 451 (2), 170-188 (2002).
- Nascimento, M. A., Sorokin, L., Coelho-Sampaio, T. Fractone bulbs derive from ependymal cells and their laminin composition influence the stem cell niche in the subventricular zone. Journal of Neuroscience. 38 (16), 3880-3889 (2018).
- Chen, G., et al. In vivo reprogramming for brain and spinal cord repair. eNeuro. 2 (5), 0106-0115 (2015).
- Zhang, Q., Chen, W., Tan, S., Lin, T. Stem cells for modeling and therapy of Parkinson's disease. Human Gene Therapy. 28 (1), 85-98 (2017).
- Wei, C., Xiong, S., Cheng, L. Reprogramming of fibroblasts to neural stem cells by a chemical cocktail. Methods in Molecular Biology. 2117, 265-270 (2020).
- Tian, Z., Zhao, Q., Biswas, S., Deng, W. Methods of reactivation and reprogramming of neural stem cells for neural repair. Methods. 133, 3-20 (2018).
- Heinrich, C., et al. Sox2-mediated conversion of NG2 glia into induced neurons in the injured adult cerebral cortex. Stem Cell Reports. 3 (6), 1000-1014 (2014).
- Masserdotti, G., et al. Transcriptional mechanisms of proneural factors and REST in regulating neuronal reprogramming of astrocytes. Cell Stem Cell. 17 (1), 74-88 (2015).
- Seidenfaden, R., Desoeuvre, A., Bosio, A., Virard, I., Cremer, H. Glial conversion of SVZ-derived committed neuronal precursors after ectopic grafting into the adult brain. Molecular and Cellular Neurosciences. 32 (1-2), 187-198 (2006).
- Lepko, T., et al. Choroid plexus-derived miR-204 regulates the number of quiescent neural stem cells in the adult brain. EMBO Journal. 38 (17), 100481 (2019).
- Silva-Vargas, V., Maldonado-Soto, A. R., Mizrak, D., Codega, P., Doetsch, F. Age-dependent niche signals from the choroid plexus regulate adult neural stem cells. Cell Stem Cell. 19 (5), 643-652 (2016).
- Angelidis, I., et al. An atlas of the aging lung mapped by single cell transcriptomics and deep tissue proteomics. Nature Communications. 10 (1), 963 (2019).
- Kjell, J., et al. Defining the adult neural stem cell niche proteome identifies key regulators of adult neurogenesis. Cell Stem Cell. 26 (2), 277-293 (2020).
- Mirzadeh, Z., Doetsch, F., Sawamoto, K., Wichterle, H., Alvarez-Buylla, A. The subventricular zone en-face: wholemount staining and ependymal flow. Journal of Visualized Experiments: JoVE. (39), e1938 (2010).
- Kulak, N. A., Geyer, P. E., Mann, M. Loss-less nano-fractionator for high sensitivity, high coverage proteomics. Molecular & Cellular Proteomics: MCP. 16 (4), 694-705 (2017).
- Tyanova, S., Temu, T., Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nature Protocols. 11 (12), 2301-2319 (2016).
- Cox, J., Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nature Biotechnology. 26 (12), 1367-1372 (2008).
- Cox, J., et al. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Molecular & Cellular Proteomics: MCP. 13 (9), 2513-2526 (2014).
- Perez-Riverol, Y., et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Research. 47 (1), 442-450 (2019).
- Tomassy, G. S., et al. Distinct profiles of myelin distribution along single axons of pyramidal neurons in the neocortex. Science. 344 (6181), 319-324 (2014).
- Sharma, K., et al. Cell type- and brain region-resolved mouse brain proteome. Nature Neuroscience. 18 (12), 1819-1831 (2015).
- Datta, S., et al. Laser capture microdissection: Big data from small samples. Histology and Histopathology. 30 (11), 1255-1269 (2015).
- Kerever, A., et al. Novel extracellular matrix structures in the neural stem cell niche capture the neurogenic factor fibroblast growth factor 2 from the extracellular milieu. Stem Cells. 25 (9), 2146-2157 (2007).
- Cox, J., Mann, M. 1D and 2D annotation enrichment: a statistical method integrating quantitative proteomics with complementary high-throughput data. BMC Bioinformatics. 13, 12 (2012).
- Daynac, M., Morizur, L., Chicheportiche, A., Mouthon, M. -. A., Boussin, F. D. Age-related neurogenesis decline in the subventricular zone is associated with specific cell cycle regulation changes in activated neural stem cells. Scientific Reports. 6, 21505 (2016).
- Navarro Negredo, P., Yeo, R. W., Brunet, A. Aging and rejuvenation of neural stem cells and their niches. Cell Stem Cell. 27 (2), 202-223 (2020).
- Smith, L. K., White, C. W., Villeda, S. A. The systemic environment: at the interface of aging and adult neurogenesis. Cell and Tissue Research. 371 (1), 105-113 (2018).
- Neuberger, E. J., Swietek, B., Corrubia, L., Prasanna, A., Santhakumar, V. Enhanced dentate neurogenesis after brain injury undermines long-term neurogenic potential and promotes seizure susceptibility. Stem Cell Reports. 9 (3), 972-984 (2017).
- Fisch, U., Brégère, C., Geier, F., Chicha, L., Guzman, R. Neonatal hypoxia-ischemia in rat elicits a region-specific neurotrophic response in SVZ microglia. Journal of Neuroinflammation. 17 (1), 26 (2020).
- Götz, M., Sirko, S., Beckers, J., Irmler, M. Reactive astrocytes as neural stem or progenitor cells: In vivo lineage, In vitro potential, and Genome-wide expression analysis. Glia. 63 (8), 1452-1468 (2015).
- Kernie, S. G., Parent, J. M. Forebrain neurogenesis after focal Ischemic and traumatic brain injury. Neurobiology of Disease. 37 (2), 267-274 (2010).
- Pous, L., et al. Fibrinogen induces neural stem cell differentiation into astrocytes in the subventricular zone via BMP signaling. Nature Communications. 11 (1), 630 (2020).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten