
Method Article
Quantitativer Nachweis von DNA-Protein-Vernetzungen und deren posttranslationale Modifikationen
In diesem Artikel
Zusammenfassung
Das vorliegende Protokoll zeigt eine modifizierte Methode zum Nachweis und zur Quantifizierung von DNA-Protein-Crosslinks (DPCs) und deren posttranslationalen Modifikationen (PTMs), einschließlich Ubiquitylierung, SUMOylierung und ADP-Ribosylierung, die durch Topoisomerase-Inhibitoren und Formaldehyd induziert werden, wodurch die Bildung und Reparatur von DPCs und ihren PTMs untersucht werden kann.
Zusammenfassung
DNA-Protein-Crosslinks (DPCs) sind häufige, allgegenwärtige und schädliche DNA-Läsionen, die durch endogene DNA-Schäden, Fehlfunktionen von Enzymen (Topoisomerasen, Methyltransferasen usw.) oder exogene Wirkstoffe wie Chemotherapeutika und Vernetzungsmittel entstehen. Sobald DPCs induziert sind, werden mehrere Arten von posttranslationalen Modifikationen (PTMs) als frühe Reaktionsmechanismen sofort mit ihnen konjugiert. Es wurde gezeigt, dass DPCs durch Ubiquitin, kleine Ubiquitin-ähnliche Modifikatoren (SUMO) und Poly-ADP-Ribose modifiziert werden können, die die Substrate darauf vorbereiten, ihre jeweiligen Reparaturenzyme zu signalisieren und in einigen Fällen die Reparatur auf sequentielle Weise zu koordinieren. Da PTMs schnell transpirieren und hochgradig reversibel sind, war es schwierig, PTM-konjugierte DPCs zu isolieren und nachzuweisen, die normalerweise auf niedrigen Niveaus verbleiben. Hier wird ein Immunoassay zur Reinigung und quantitativen Detektion von ubiquitylierten, SUMOylierten und ADP-ribosylierten DPCs (arzneimittelinduzierte Topoisomerase-DPCs und Aldehyd-induzierte unspezifische DPCs) in vivo vorgestellt. Dieser Assay leitet sich vom RADAR-Assay (Rapid Approach to DNA Adduct Recovery) ab, der für die Isolierung von genomischer DNA, die DPCs enthält, durch Ethanolfällung verwendet wird. Nach Normalisierung und Nukleaseverdauung werden PTMs von DPCs, einschließlich Ubiquitylierung, SUMOylierung und ADP-Ribosylierung, durch Immunoblot mit den entsprechenden Antikörpern nachgewiesen. Dieser robuste Assay kann verwendet werden, um neue molekulare Mechanismen zu identifizieren und zu charakterisieren, die enzymatische und nicht-enzymatische DPCs reparieren, und hat das Potenzial, niedermolekulare Inhibitoren zu entdecken, die auf spezifische Faktoren abzielen, die PTMs zur Reparatur von DPCs regulieren.
Einleitung
Genomische DNA-Schäden entstehen durch spontanen Zerfall, innere Schäden und Umweltfaktoren1. Die daraus resultierenden DNA-Läsionen umfassen beschädigte Basen, Fehlanpassungen, Einzel- und Doppelstrangbrüche, Inter- und Intrastrang-Querverbindungen sowie DNA-Protein-Querverbindungen (DPCs). Ein DPC wird gebildet, wenn ein Chromatin-gebundenes Protein durch kovalente Bindung auf der DNA gefangen wird. DPCs werden durch endogene DNA-Läsionen und reaktive Metaboliten sowie durch exogene Wirkstoffe wie Chemotherapeutika und bifunktionelle Vernetzungsmittel induziert. Unter Umständen kann auch eine Enzymdysfunktion zur Bildung von DPCsführen 2. Der große Unterschied in den DPC-Induktoren führt zu einem Unterschied in der Identität des kovalent gebundenen Proteins, der Chromosomenregion, in der das DPC gebildet wird, dem Strukturtyp der DNA, die mit dem Protein vernetzt ist, und der chemischen Eigenschaft der kovalenten Bindung zwischen dem Protein und der DNA 2,3,4.
Basierend auf ihrer chemischen Natur werden DPCs im Allgemeinen in zwei Gruppen eingeteilt: enzymatische DPCs und nicht-enzymatische DPCs. Bestimmte Enzyme wie Topoisomerasen, Glykosylasen und Methyl-/Acyltransferasen wirken, indem sie während ihrer normalen katalytischen Reaktionen reversible kovalente Enzym-DNA-Zwischenprodukte bilden. Diese sind kurzlebige Enzym-DNA-Zwischenprodukte und können in langlebige enzymatische DPCs umgewandelt werden, wenn sie durch endogene oder exogene Wirkstoffe, insbesondere durch Chemotherapeutika, eingefangen werden3. Topoisomerase-DPCs gehören zu den häufigsten enzymatischen DPCs in eukaryotischen Zellen, die durch klinisch nützliche Topoisomerase-Inhibitoren (Topotecan und Irinotecan für Topoisomerase I [TOP1] und Etoposid und Doxorubicin für Topoisomerase II [TOP2]) gebildet werden können und die primären therapeutischen Mechanismen dieser Inhibitoren sind 5,6. Die DNA-Methyltransferasen (DNMT) 1, 3A und 3B sind das Ziel von 5-Aza-2'-Desoxycytidin (auch bekannt als Decitabin) und bilden DPCs bei Exposition gegenüber dem Medikament7. Reaktive Wirkstoffe sowie ultraviolettes Licht und ionisierende Strahlung induzieren nicht-enzymatische DPCs, indem sie Proteine unspezifisch mit der DNA vernetzen. Reaktive Aldehyde wie Acetaldehyd und Formaldehyd (FA) entstehen häufig als Nebenprodukte des Zellstoffwechsels, unter denen FA in mikromolaren Konzentrationen während des Methanolstoffwechsels, der Lipidperoxidation und der Histondemethylierung produziert wird. Außerdem ist FA eine weltweit hergestellte Produktionschemikalie in großen Mengen, der viele Menschen sowohl umwelt- als auch beruflich ausgesetzt sind 8,9.
Sowohl enzymatische als auch nicht-enzymatische DPCs sind hochtoxisch für Zellen, da ihre sperrigen Proteinbestandteile fast alle chromatinbasierten Prozesse, einschließlich Replikation und Transkription, effizient behindern und zu Zellzyklusarrest und Apoptose führen, wenn sie nicht repariert werden. In den letzten zwei Jahrzehnten wurde die Reparatur von DPCs intensiv untersucht, und es wurden mehrere Proteine/Signalwege als Schlüsselfaktoren identifiziert, die DPCs entweder direkt reparieren oder ihre Reparaturprozesse modulieren. Zum Beispiel ist bekannt, dass die Proteolyse der Proteinmasse eines DPC ein zentraler Schritt der DPC-Reparatur ist und dass die Proteolyse durch die Proteasen SPRTN 10,11,12,13,14, FAM111A15, GCNA 16,17 oder den 26S-Proteasom-Komplex 18,19,20,21,22 katalysiert werden kann,23,24,25,26,27 zelltyp- oder zellkontextabhängig. Die Identifizierung und Charakterisierung dieser Proteasen stützte sich weitgehend auf den In-vivo-Komplex des Enzyms (ICE) Assay28,29 und den Rapid Approach to DNA Adduct Recovery (RADAR) Assay30,31, die beide DNA-Moleküle und ihre kovalent gebundenen Proteine aus freien zellulären Proteinen isolieren, um den Nachweis von DPCs durch Slot-Blot unter Verwendung von Antikörpern zu ermöglichen, die auf die vernetzten Proteine abzielen. Außerdem wurde der TARDIS-Assay (Trapped-in Agarose DNA Immunostaining) als Mittel zum Nachweis und zur Quantifizierung von DPCs auf Einzelzellebene verwendet32. Derzeit entscheiden sich die Forscher für die Messung von DPCs für den RADAR-Assay gegenüber dem ICE-Assay, da der ICE-Assay auf der Reinigung von Nukleinsäuren mittels Cäsiumchlorid-Gradienten-Ultrazentrifugation beruht, die extrem zeitaufwändig ist, während der RADAR-Assay Nukleinsäuren mit Ethanol innerhalb eines viel kürzeren Zeitraums ausfällt.
In den letzten Jahren mehren sich die Hinweise, dass multiple posttranslationale Modifikationen (PTMs) an der Signalübertragung und Rekrutierung von DPC-gerichteten Proteasen beteiligt sind 3,33,34,35. So wurde beispielsweise festgestellt, dass sowohl TOP1- als auch TOP2-DPCs unabhängig von der DNA-Replikation und Transkription durch den kleinen Ubiquitin-ähnlichen Modifikator (SUMO)-2/3 und dann SUMO-1 durch die SUMO E3-Ligase PIAS4 konjugiert werden. Die sequentiellen SUMO-Modifikationen scheinen ein Ziel von Ubiquitin zu sein, das an die SUMOylierten TOP-DPCs abgelagert wird und durch seinen Lysin-48-Rest durch eine SUMO-gerichtete Ubiquitin-Ligase, die als RNF4 bezeichnet wird, polymere Ketten bildet. Anschließend löst das Ubiquitin-Polymer ein Signal an das 26S-Proteasom aus und rekrutiert es an TOP-DPCs23,36. Derselbe SUMO-Ubiquitin-Signalweg wurde kürzlich gezeigt, dass er sowohl auf DNMT1-DPCs als auch auf PARP-DNA-Komplexe für deren Reparatur wirkt37,38. Darüber hinaus wurde berichtet, dass die SUMO-unabhängige Ubiquitylierung durch die Ubiquitin-E3-Ligase TRAIP DPCs für den proteasomalen Abbau in einer replikationsgekoppelten Weise vorbereitet39. Ähnlich wie der proteasomale Abbau von TOP-DPCs erfordert auch die Proteolyse von enzymatischen und nicht-enzymatischen DPCs durch die replikationsgekoppelte Metalloprotease SPRTN eine Ubiquitylierung der DPC-Substrate als Mechanismus zur Aktivierung von SPRTN40,41. Die Abgrenzung der Rolle von SUMOylierung und Ubiquitylierung erfordert den Nachweis von DPCs, die mit diesen PTMs markiert sind. Da der ursprüngliche ICE-Assay und der RADAR-Assay auf Slot-Blot-/Dot-Blot-Apparaturen zur Messung unverdauter DNA-Proben angewiesen sind, ist keiner dieser beiden Assays in der Lage, PTM-konjugierte DPC-Spezies mit unterschiedlichen Molekulargewichten aufzulösen und zu visualisieren. Um dieses Problem zu lösen, verdauten wir die DNA-Proben nach ihrer Reinigung durch Ethanolfällung und Probennormalisierung mit Mikrokokken-Nuklease, einer DNA- und RNA-Endo-Exonuklease, um die vernetzten Proteine freizusetzen, was es uns ermöglichte, die Proteine sowie ihre kovalenten PTMs mit Natriumdodecylsulfat-Polyacrylamid-Gelelektrophorese (SDS-PAGE) aufzulösen. Die Elektrophorese ermöglichte es uns, PTM-konjugierte DPCs mit spezifischen Antikörpern, die auf die PTMs abzielen, zu detektieren und zu quantifizieren. Wir nannten diese verbesserte Methode zunächst DUST-Assay, um ihre Robustheit bei der Detektion von ubiquitylierten und SUMOylierten TOP-DPCshervorzuheben 23. Später erweiterten wir die Verwendung des Assays, um die ADP-Ribosylierung von TOP1-DPCs in vivo quantitativ zu bewerten, indem wir Antikörper gegen Poly-ADP-Ribose-Polymere verwendeten20.
Hier wird ein detailliertes Protokoll für den Assay vorgestellt, der ubiquitylierte, SUMOylierte und ADP-ribosylierte DPCs detektiert und misst, das für die modifizierten TOP-DPCs, die durch ihre Inhibitoren induziert werden, und die unspezifischen/nicht-enzymatischen DPCs, die durch FA induziert werden, optimiert wurde. Dieser Assay isoliert PTM-konjugierte DPCs, indem Zellen mit einem chaotropen Wirkstoff lysiert, DNA mit Ethanol ausgefällt und die ansonsten vernetzten Proteine und ihre Modifikatoren mit Mikrokokken-Nuklease freigesetzt werden. Die ansonsten DNA-gebundenen Proteine und ihre PTMs werden durch Immunoblotting mit spezifischen Antikörpern quantifiziert. Dieser Assay ebnet einen neuen Weg zur Aufklärung der molekularen Mechanismen, mit denen die Zelle sowohl enzymatische als auch nicht-enzymatische DPCs repariert. Insbesondere ermöglicht er detaillierte Untersuchungen der Induktion und Kinetik von PTMs, die für die Regulation des Abbaus und der Reparatur von TOP-DPC wichtig sind, und ermöglicht somit die Entdeckung neuer Faktoren wie E3-Ligasen, die die PTMs bestimmen. sowie Inhibitoren, die auf diese Faktoren abzielen. Da einige der PTMs, die für die TOP-DPC-Reparatur verantwortlich sind, wahrscheinlich an der Reparatur von DPCs beteiligt sind, die durch andere Chemotherapeutika, wie z. B. platinbasierte Medikamente22, induziert werden, hat dieser Assay auch das Potenzial für die Entdeckung neuer Medikamente und die rationale Optimierung kombinatorischer Therapien mit Topoisomerase-Inhibitoren oder platinbasierten Antineoplastiken in Patientenzellen, um Behandlungsschemata zu steuern.
Protokoll
1. Zellkultur und medikamentöse Behandlung in der humanen embryonalen Nierenzelllinie 293 (HEK293)
- Bereiten Sie das Nährmedium, das modifizierte Eagle-Medium (DMEM) von Dulbecco, vor, ergänzt mit 10 % fötalem Kälberserum, 1 % 2 mM L-Glutamin und 100 Einheiten/ml Penicillin-Streptomycin.
- Säen Sie 1 x 10 6 Zellen in einer 60-mm-Platte oder einer 6-Well-Platte pro Behandlungsbedingung plus Kontrolle.
- Behandeln Sie die Zellen am nächsten Tag mit DPC-Induktoren Ihrer Wahl.
- Um TOP1-DPCs und deren Ubiquitylierung und SUMOylierung zu induzieren, wird der TOP1-Inhibitor Camptothecin bei 20 μM zu den Zellen gegeben und die Zellen nach 20, 60 und 180 min gesammelt.
- Um TOP2α- und β-DPCs und deren Ubiquitylierung und SUMOylierung zu induzieren, werden die Zellen exponiert, um den TOP2-Inhibitor Etoposid bei 200 μM zu den Zellen hinzuzufügen und die Zellen nach 20, 60 und 180 Minuten zu sammeln.
- Um nicht-enzymatische DPCs und ihre Ubiquitylierung und SUMOylierung zu induzieren, fügen Sie FA in 1 mM hinzu und sammeln Sie die Zellen 2 Stunden nach der Exposition.
- Um die PARylierung von TOP1-DPCs zu induzieren, werden die Zellen 1 h lang vorbehandelt, um die DePARylierung mit Poly(ADP-Ribose)-Glykohydrolase (PARG)-Inhibitor PDD00017273 bei 10 μM zu blockieren, gefolgt von einer gleichzeitigen Behandlung mit 20 μM Camptothecin für 20, 60 und 180 min.
2. Isolierung und Normalisierung von DNA, die vernetzte Proteine enthält
- Saugen Sie das Medium nach der Behandlung schnell mit einer Saugpipette an und spülen Sie die Zellen mit eiskalter 1x phosphatgepufferter Kochsalzlösung (PBS) ab. Die Zellen werden sofort in 600 μl DNAzol-Reagenz lysiert, das 1x Protease-Cocktail-Inhibitor, 1 mM Dithiothreitol (DTT) und 20 mM N-Ethylmaleimid (Inhibitor von deSUMOylierenden und deubiquitylierenden Enzymen) enthält.
- Die Platte auf einer vibrierenden Plattform 10 min bei 4 °C langsam bewegen.
- Geben Sie 1/2 Volumen 100% kaltes Ethanol (0,3 ml) direkt auf die Platte und wiederholen Sie Schritt 2.2, bis ein undurchsichtiges Nukleinsäureaggregat sichtbar wird. Das Zelllysat wird in ein 1,5-ml-Mikrozentrifugenröhrchen überführt und das Röhrchen 15 Minuten lang bei 4 °C mit maximaler Geschwindigkeit (20.000 x g) zentrifugiert, um die Nukleinsäure und ihre vernetzten Proteine auszufällen.
- Der Überstand wird mit einer Saugpipette abgesaugt und das Nukleinsäurepellet in 1 ml 75%igem Ethanol gewaschen, gefolgt von einer 2-minütigen Zentrifugation mit 20.000 x g bei 4 °C.
- Saugen Sie den Überstand an, schleudern Sie ihn mit der gleichen Geschwindigkeit herunter und entfernen Sie die restliche Flüssigkeit mit einer P20-Pipette. Lassen Sie das Pellet 5 Minuten lang an der Luft trocknen.
- Lösen Sie das Nukleinsäure-Pellet schnell in 0,1 ml ddH2O auf. Resuspendieren Sie das Pellet durch wiederholtes Pipettieren und inkubieren Sie es dann in einem 37 °C warmen Wasserbad, bis das Pellet mindestens dreimal so groß ist (ca. 30 min).
- Beschallen Sie die Proben mit einer Ultraschallprozessorsonde mit einer Amplitude von 30 % für 10 Sekunden, um das Pellet vollständig aufzulösen.
- Optionaler Schritt: Behandeln Sie die Proben mit RNase A/T1 Mix (10 μg RNase A und 25 U RNase T1) und inkubieren Sie bei 37 °C für 15 min. Geben Sie 1/10 Volumen 3 M Natriumacetat und zwei Volumen eiskaltes 100%iges Ethanol in das Röhrchen, gefolgt von einer Zentrifugation bei 20.000 x g für 10 Minuten, um die DNA zu gewinnen. Entfernen Sie den Überstand und lösen Sie die präzipitierte DNA in 0,1 mlddH2Oauf.
- Optionaler Schritt: Zentrifugieren Sie die Probe für 5 min bei 20.000 x g und überführen Sie den Überstand in ein neues Röhrchen.
- Quantifizieren Sie die DNA-Konzentration mit einem UV-Vis-Spektrometer. Die typische DNA-Ausbeute liegt bei etwa 600-800 ng/μl. Das A260/A280-Verhältnis sollte nach RNA-Entfernung von 2,0-2,1 auf 1,8-1,9 reduziert werden.
- Stellen Sie die Konzentration der DNA auf 400-500 ng/μl in 0,12 ml ddH 2 O ein. 20 μl der Probe werden in ein neues Mikrozentrifugenröhrchen als unverdaute DNA-Beladungskontrolle überführt (siehe Schritt2.4).
- Um die in den verbleibenden 100 μlddH2Ogelöste DNA zu verdauen, fügen Sie der Probe 2.000 Geleinheiten Mikrokokkenuklease zusammen mit 1/10 Volumen (~11 μl) 10x Calcium-Mikrokokken-Nuklease-Reaktionspuffer hinzu. Bei 37 °C 30 min inkubieren.
3. Western Blot von verdauten DNA-Proben
- 4x Laemmli Probenpuffer zugeben und die Probe 5 min kochen.
- Laden Sie 5-6 μg der aufgeschlossenen Probe (~15 μl) auf 4%-20% Polyacrylamid-Gel, gefolgt von SDS-PAGE42 , um unmodifizierte und PTM-konjugierte DPCs aufzulösen.
- Um FA-induzierte nicht-enzymatische DPC-Spezies nachzuweisen, inkubieren Sie das Gel mit Coomassie-Blaufärbung über Nacht bei Raumtemperatur. Waschen Sie das Gel 2 h lang mit ddH2O und nehmen Sie ein Bild mit einem Bildgebungssystem auf.
- Das Gel wird überführt und die Membranen über Nacht bei 4 °C mit geeigneten Verdünnungen des Primärantikörpers in Blockierungspuffer inkubiert.
- Um eine Ubiquitylierung nachzuweisen, wird der Anti-Ubiquitin-Antikörper im Verhältnis 1:100 verdünnt.
- Um eine SUMO-1- oder SUMO-2/3-Modifikation nachzuweisen, wird der Anti-SUMO-1- oder Anti-SUMO-2/3-Antikörper im Verhältnis 1:250 verdünnt.
- Um eine ADP-Ribosylierung nachzuweisen, wird der Anti-PAR-Antikörper im Verhältnis 1:500 verdünnt.
- Um die Gesamtmenge an TOP1-, TOP2α- oder TOP2β-DPCs nachzuweisen, nehmen Sie eine 1:500-Verdünnung von Anti-TOP1-, Anti-TOP2α- oder Anti-TOP2β-Antikörpern vor.
HINWEIS: Einzelheiten zur Verdünnung von Antikörpern finden Sie in der Materialtabelle .
- Inkubieren Sie eine 1x PBS-T (0,1% Tween 20) gewaschene Membran mit einem 5.000-fach verdünnten Sekundärantikörper in Blocking-Puffer für 60 min bei Raumtemperatur.
- Entwickeln Sie die Membran mit einem verstärkten Chemilumineszenz-Reagenz (ECL) und nehmen Sie mit dem Bildgebungssystem ein Bild auf.
4. Slot-Blotting von unverdauten DNA-Proben
- Die unverdaute DNA-Probe von 20 μl wird in 180 μl Natriumphosphatpuffer (25 mM, pH 6,6) verdünnt.
- Die Nitrozellulosemembran durchtrennen (0,45 μm) und 5 min im Natriumphosphatpuffer ausgleichen.
- Montieren Sie das Schlitzgerät gemäß den Anweisungen des Herstellers und schließen Sie es an ein Vakuumsystem an.
- Waschen Sie die Vertiefungen mit Natriumphosphatpuffer, indem Sie das Vakuum anwenden. Stellen Sie sicher, dass die Brunnen nicht undicht sind.
- Stoppen Sie das Vakuum und laden Sie 200 μl DNA pro Probe (1 μg). Füllen Sie die leeren Vertiefungen mit 200 μl Natriumphosphatpuffer.
- Wenden Sie das Vakuum an.
- Wenn alle Vertiefungen vollständig leer sind, wird das Vakuum gestoppt, 200 μl Natriumphosphatpuffer in jede Vertiefung gegeben und Schritt 4.6 wiederholt.
- Holen Sie die Membran und blockieren Sie sie mit 5 % Blockierpuffer für 0,5 h bei Raumtemperatur.
- Sonde mit Anti-Doppelstrang-DNA (dsDNA)-Antikörper in einer Verdünnung von 1:5.000 über Nacht bei 4 °C.
- 3x mit 1x PBS-T waschen und mit 1:5.000 verdünntem Meerrettichperoxidase (HRP)-gebundenem Anti-Maus-Sekundärantikörper inkubieren.
- Entwickeln Sie die Membran mit einem verstärkten Chemilumineszenz-Reagenz (ECL) und nehmen Sie mit dem Bildgebungssystem ein Bild auf.
5. Densitometrische Analyse
- Berechnen Sie mit ImageJ das Verhältnis der Intensität jeder Bande/jedes Abstrichs relativ zur Intensität des Schlitzes unverdauter DNA und normalisieren Sie das Verhältnis zu dem von Zellen ohne/vor medikamentöser Behandlung.
Ergebnisse
Die repräsentativen Ergebnisse in Abbildung 1 zeigen die Bildung und Kinetik von arzneimittelinduzierten TOP1-DPCs und deren SUMOylierung und Ubiquitylierung. TOP1 spaltete einen Strang des DNA-Duplex und bildete ein kovalentes Enzym-DNA-Zwischenprodukt, das als TOP1-Spaltungskomplex (TOP1cc) bezeichnet wird. Die Behandlung mit Camptothecin (CPT), einem TOP1-Inhibitor, bindet und stabilisiert TOP1cc, was zur Bildung von langlebigen TOP1-DPCs führt. Es wurde beobachtet, dass TOP1-DPCs induziert wurden und 20 Minuten nach der CPT-Exposition ihren Höhepunkt erreichten. Gleichzeitig wurden die TOP1-DPCs durch SUMO-2/3 modifiziert, das ebenfalls 20 Minuten nach der CPT-Behandlung seinen Höhepunkt erreichte. Da SUMO-2 und SUMO-3 zu 95 % identisch sind, unterscheidet der Antikörper nicht voneinander. Nach 60 Minuten nahmen die TOP1-DPCs und ihre SUMO-2/3-Modifikation ab, begleitet von der Kulmination ihrer SUMO-1-Modifikation und Ubiquitylierung. Nach der 60-minütigen medikamentösen Behandlung begannen die Konzentrationen der TOP1-DPC SUMO-1-Modifikation und Ubiquitylierung zu sinken. In Säugetieren wirken TOP2-Isozyme α und β durch die Einführung eines DNA-Doppelstrangbruchs sowie durch die Bildung eines transienten und reversiblen kovalenten Enzym-DNA-Komplexes (TOP2cc). TOP2-Inhibitoren, wie z.B. Etoposid (ETOP), wandeln TOP2cc in TOP2-DPCs um und induzieren deren SUMOylierung und Ubiquitylierung. Ähnlich wie bei der Kinetik von TOP1-DPCs und ihren PTMs erreichten TOP2α- und β-DPCs und ihre SUMO-2/3-Modifikation nach 20 Minuten einen Höhepunkt und begannen dann zu sinken; In der Zwischenzeit erreichten ihre SUMO-1- und Ubiquitin-Modifikationen nach 60 Minuten ihren Höhepunkt (Abbildung 2). Es wurde gezeigt, dass die Clearance von TOP-DPCs aus dem proteasomalen Abbau resultiert, und die Clearance von TOP-DPC SUMOylierung und Ubiquitylierung ist wahrscheinlich auf das Recycling durch DeSUMOylierung bzw. Deubiquitylierung durch ihre umkehrenden Enzyme zurückzuführen. Die Experimente in Abbildung 3 untersuchten FA-induzierte nicht-enzymatische DPCs und ihre PTMs. Es wurde beobachtet, dass sich die DPCs und ihre SUMO-2/3, SUMO-1 und Ubiquitylierung dosisabhängig bildeten und akkumulierten. Abschließend wurde die PARylierung von TOP1-DPCs mit einem Anti-PAR-Antikörper mit der gleichen Methode quantitativ nachgewiesen (Abbildung 4). Die PARylierung von TOP1-DPC war nicht nachweisbar, es sei denn, der Zelle wurde ein PARP-Inhibitor zugesetzt, was darauf hindeutet, dass die PARylierung schnell transpiriert und hochdynamisch ist. In Übereinstimmung mit dem vorherigen Befund schien die Hemmung der dePARylierung durch PARGi TOP1-DPCs zu akkumulieren, wahrscheinlich durch die Blockierung des proteolytischen Abbaus.

Abbildung 1: Quantitative Analysen der Bildung und Kinetik von TOP1-DPCs und deren SUMOylierung und Ubiquitylierung nach CPT-Behandlung in HEK293-Zellen. (A) HEK293-Zellen wurden für bestimmte Zeiträume mit 20 μM CPT behandelt. Die Zelllysate wurden geerntet und dem modifizierten RADAR-Assay und dem Western Blot mit indizierten Antikörpern unterzogen. Unverdaute DNA-Proben wurden einem Slot-Blotting unterzogen, bei dem Anti-dsDNA-Antikörper als Beladungskontrolle verwendet wurden. (B) Die Bandintensitäten wurden mit der Software ImageJ quantifiziert und mit der Software Prism aufgetragen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 2: Quantitative Analysen der Bildung und Kinetik von TOP2-DPCs und deren SUMOylierung und Ubiquitylierung nach ETOP-Behandlung in HEK293-Zellen. (A) HEK293-Zellen wurden für bestimmte Zeiträume mit 200 μM ETOP behandelt. Die Zelllysate wurden geerntet und dem modifizierten RADAR-Assay und dem Western Blot mit indizierten Antikörpern unterzogen. Unverdaute DNA-Proben wurden einem Slot-Blotting unterzogen, bei dem Anti-dsDNA-Antikörper als Beladungskontrolle verwendet wurden. (B) Die Bandintensitäten wurden mit der Software ImageJ quantifiziert und mit der Software Prism aufgetragen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 3: Quantitative Analysen von nicht-enzymatischen DPCs und deren SUMOylierung und Ubiquitylierung nach FA-Behandlung in HEK293-Zellen. (A) HEK293-Zellen wurden 2 h lang mit FA der angegebenen Konzentrationen behandelt. Die Zelllysate wurden geerntet und dem modifizierten RADAR-Assay und dem Western Blot mit indizierten Antikörpern unterzogen. Unverdaute DNA-Proben wurden einem Slot-Blotting unterzogen, bei dem Anti-dsDNA-Antikörper als Beladungskontrolle verwendet wurden. (B) Die Bandintensitäten wurden mit der Software ImageJ quantifiziert und mit der Software Prism aufgetragen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
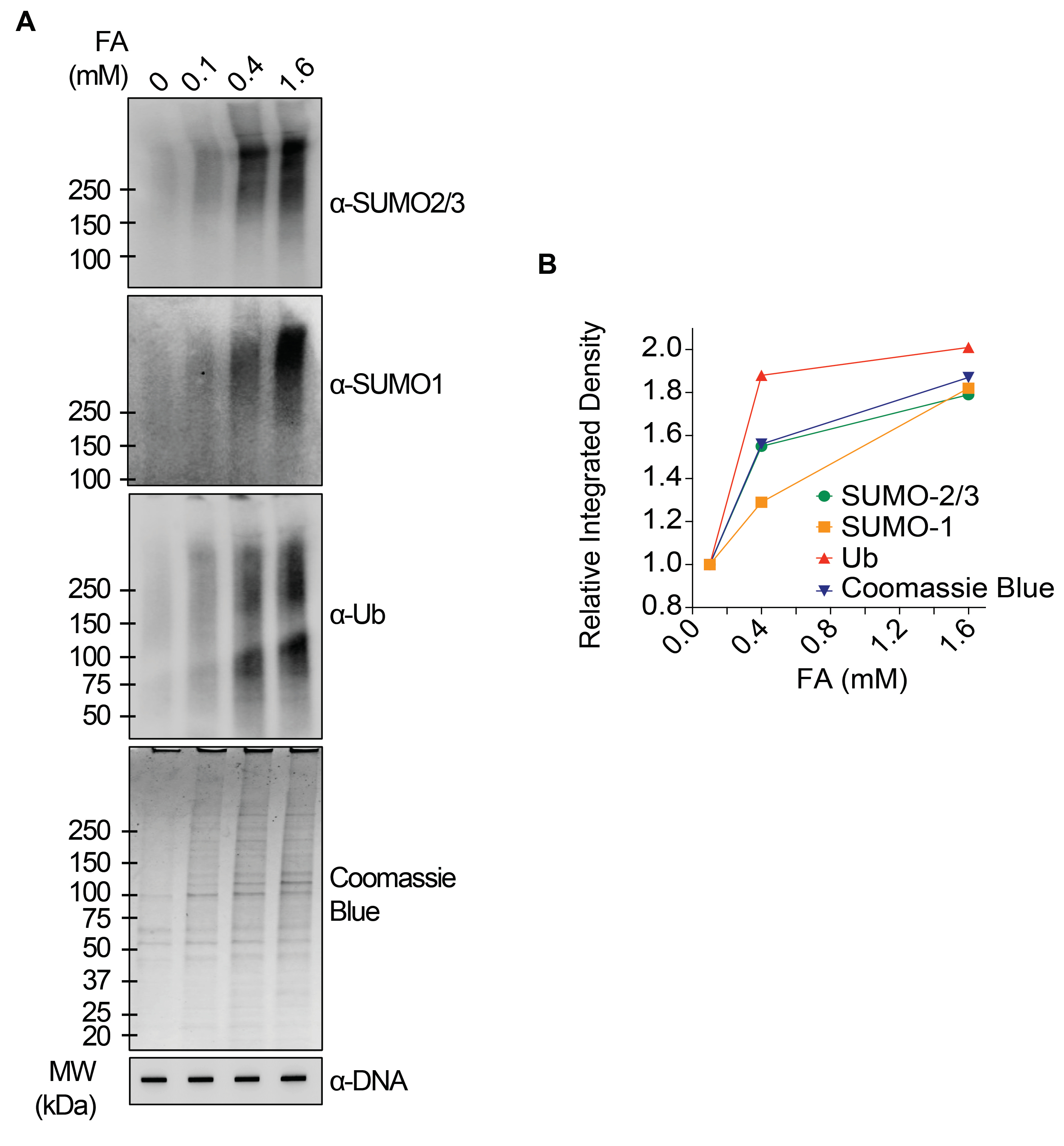{kind=link}

Abbildung 4: Quantitative Analysen von TOP1-DPCs und deren PARylierung nach CPT-Behandlung in HEK293-Zellen. (A) HEK293-Zellen wurden 1 h lang mit 10 μM PARGi vorbehandelt und dann für bestimmte Zeiträume mit CPT kobehandelt. Die Zelllysate wurden geerntet und dem modifizierten RADAR-Assay und dem Western Blot mit indizierten Antikörpern unterzogen. Unverdaute DNA-Proben wurden einem Slot-Blotting unterzogen, bei dem Anti-dsDNA-Antikörper als Beladungskontrolle verwendet wurden. (B) Die Bandintensitäten wurden mit der Software ImageJ quantifiziert und mit der Software Prism aufgetragen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Diskussion
Die beschriebene Methode ermöglicht die Messung von enzymatischen und nicht-enzymatischen DNA-Protein-Querverbindungen in Säugetierzellen und ist der einzige geeignete Ansatz, um deren Ubiquitylierung, SUMOylierung und ADP-Ribosylierung zu untersuchen. Das Slot-Blotting nach dem ICE- oder RADAR-Assay ermöglicht den schnellen Nachweis spezifischer enzymatischer DPCs wie z.B. TOP-DPCs anhand ihrer Antikörper. Ein Nachteil dieser Methode ist jedoch ihre Unfähigkeit, Proteine mit unterschiedlichem Molekulargewicht zu trennen, was es unmöglich macht, die Größe von PTM-konjugierten DPCs zu bestimmen. Die beschriebene Methode löst das Problem, indem vernetzte Proteine mit Mikrokokken-Nuklease freigesetzt werden, die DNA zu Oligonukleotiden mit terminalen 3'-Phosphaten abbauen, wodurch eine vollständige Trennung der Proteine (konjugiert mit Oligonukleotiden) durch SDS-PAGE ermöglicht wird. DPCs, die mit Ubiquitin-, SUMO- oder ADP-Ribose-Monomeren und Polymeren unterschiedlicher Größe modifiziert sind, können daher durch Antikörper gegen diese PTMs sichtbar gemacht und quantifiziert werden, was eine detaillierte Untersuchung ihrer Bildung und Kinetik ermöglicht. Um die Reproduzierbarkeit zu gewährleisten und die statistische Signifikanz zu berechnen, sind biologische Replikate der Experimente erforderlich.
Eines der häufigsten Probleme dieses Assays ist die geringe DNA-Ausbeute nach der Ethanolfällung. Einerseits kann die DNA-Ausbeute mit mehr Ausgangsmaterial (Zellen) erhöht werden. Andererseits kann die Inkubation von Zelllysaten mit Ethanol in einer flachen Platte anstelle eines Eppendorf-Röhrchens die Aggregation von DNA-Molekülen deutlich verbessern und damit deren Ausfällung erleichtern. Unspezifische Signale, die in Proben ohne medikamentöse Behandlung beobachtet werden, können auf eine nicht-kovalente Proteinkontamination hinweisen. Wenn dies der Fall ist, kann man in Betracht ziehen, DNA-Pellets mit salzreichem Puffer zu waschen, um die Verunreinigungen vor der Beschallung zu entfernen. Es wird auch empfohlen, DNA-Proben nach der Beschallung und dem Verdau von Mikrokokken-Nuklease abzuschleudern und alle unlöslichen Proben zu verwerfen. Im Falle eines schlechten oder fehlenden Signals können mehrere mögliche Lösungen ausprobiert werden. Erstens kann man die Beladungsmenge der DNA für SDS-PAGE und Immunoblotting erhöhen. Um SUMOylierte und ubiquitylierte DPC-Spezies nachweisbar zu machen, wird empfohlen, mindestens 4 μg DNA auf das Gel zu laden. Zweitens kann man die Wirkstoffkonzentrationen erhöhen, um höhere Konzentrationen von DPCs und den damit verbundenen PTMs zu induzieren. Drittens wird empfohlen, Blots mit primären Antikörpern für einen weiteren Tag zu inkubieren, wenn die Banden/Abstriche schwach zu sein scheinen. Eine 2-tägige Inkubation kann das Signal signifikant potenzieren und reduziert so die biologische Variabilität aus unabhängigen Experimenten23. Das Stripping von Membranen zur erneuten Färbung führt unweigerlich zum Verlust einer bestimmten Anzahl von PTM-konjugierten DPC-Spezies, die bereits in geringer Häufigkeit vorhanden sind. Daher wird dringend empfohlen, separate Gele für den Ubiquitin- und SUMO-Nachweis zu verwenden, anstatt einen Blot erneut zu untersuchen. Darüber hinaus müssen DNA-Pellets mit 75%igem Ethanol gewaschen werden, um das verbleibende DNAzol, das Guanidinsalz enthält, vor der Auflösung in H2O oder anderen Lösungsmitteln zu entfernen, was andernfalls zu einer Kristallisation der Probe nach der Zugabe von Laemmli-Ladepuffer führt.
Der Arbeitsablauf der beschriebenen Methode ist im Vergleich zum umständlichen ICE-Assay viel zeiteffizienter, da er auf eine schnelle Ethanolfällung anstelle der zeitaufwändigen Cäsiumchlorid-Ultrazentrifugation zur Isolierung genomischer DNA setzt. Die Reinigung auf Ethanolbasis führt zu einem geringen Preis zu einer geringen Menge an Proteinverunreinigungen, die normalerweise für den Immunnachweis vernachlässigbar sind. Wenn es jedoch um analytische Studien geht, wie z. B. die auf Massenspektrometrie basierende Proteomanalyse oder die Sequenzierung der nächsten Generation, die Genauigkeit und Präzision erfordern, ist die Cäsiumchlorid-Dichtegradientenzentrifugation immer noch ein zuverlässigerer Ansatz zur Isolierung reiner DNA mit hoher Häufigkeit. Diese Methode kann möglicherweise auch für die Profilierung von Modifikationsstellen auf vernetzten Proteinen und die Bestimmung von Kopplungstypen der Poly-Ubiquitylierung und Poly-SUMOylierung mit geeigneten massenspektrometriebasierten Methoden angewendet werden.
Bemerkenswert ist, dass dieser Assay die Identifizierung und Charakterisierung von Faktoren ermöglicht, die PTMs für die Reparatur von DPCs regulieren. Zum Beispiel sind unvoreingenommene Hochdurchsatz-Screening-Methoden (RNA-Interferenz und CRISPR) leistungsstarke Werkzeuge, um Ubiquitin-E3-Ligasen, SUMO E3-Ligasen und ihre assoziierten Cofaktoren zu entdecken, die die Zytotoxizität von DPC-Induktoren abschwächen. Die beschriebene Methode ermöglicht die molekulare Validierung dieser Proteine, indem festgestellt wird, ob sie Zellen helfen, DPC-Induktoren zu überleben, indem sie die DPCs reparieren. Neuartige niedermolekulare Inhibitoren, die auf diese Proteine abzielen, die beispielsweise durch virtuelles Screening identifiziert wurden, können ebenfalls mit diesem Protokoll validiert werden. Angesichts der Tatsache, dass Topoisomerase-Inhibitoren zu den am häufigsten verschriebenen Chemotherapeutika gehören, kann dieser robuste Assay als Werkzeug für die Entwicklung von Medikamenten entwickelt werden, die mit klinischen Topoisomerase-Inhibitoren synergistisch wirken.
Offenlegungen
Der Autor erklärt, dass keine konkurrierenden Interessen bestehen.
Danksagungen
Diese Arbeit wurde teilweise durch den National Cancer Institute Center for Cancer ResearchExcellence in Postdoctoral Research Transition Award unterstützt.
Materialien
| Name | Company | Catalog Number | Comments |
| 10x Phosphate buffered saline (PBS) | Thermo Fisher | 70011069 | |
| 4–20% precast polyacrylamide gel | Bio-Rad | 4561096 | |
| 4x Laemmli Sample Buffer | Bio-Rad | 1610747 | |
| AcquaStain (coomassie blue) | Bulldog Bio | AS001000 | |
| anti-dsDNA (mouse monoclonal) | Abcam | 27156 | 1: 5,000 dilution is recommended |
| anti-PAR (mouse monoclonal) | R&D systems | 4335-MC-100 | 1: 500 dilution is recommended |
| anti-SUMO-1(rabbit monoclonal) | Cell Signaling Technology | 4940 | 1: 250 dilution is recommended |
| anti-SUMO-2/3 (rabbit monoclonal) | Cell Signaling Technology | 4971 | 1: 250 dilution is recommended |
| anti-TOP1 (mouse monoclonal) | BD Biosciences | 556597 | 1: 500 dilution is recommended |
| anti-TOP2α (mouse monoclonal) | Santa Cruz Biotechnology | SC-365799 | 1: 250 dilution is recommended |
| anti-TOP2β (mouse monoclonal) | Santa Cruz Biotechnology | SC-25330 | 1: 250 dilution is recommended |
| anti-ubiquitin (mouse monoclonal) | Santa Cruz Biotechnology | SC-8017 | 1: 100 dilution is recommended |
| Calcium chloride | Sigma-Aldrich | 499609 | Used for micrococcal nuclease digestion |
| Camptothecin | Sigma-Aldrich | PHL89593 | |
| ChemiDo MP imaging system | Bio-Rad | 12003154 | |
| Disodium phosphate | Sigma-Aldrich | 5438380100 | Used to make sodium phosphate buffer |
| DNAzol | Thermo Fisher | 10503027 | |
| DTT (dithiothreitol) | Thermo Fisher | R0861 | |
| Dulbecco's modified eagle's medium | Sigma-Aldrich | 11965084 | |
| Ethyl alcohol, 200 proof | Sigma-Aldrich | E7023 | |
| Etoposide | Sigma-Aldrich | 1268808 | |
| Formaldehyde | Sigma-Aldrich | 47608 | |
| Graphpad Prism Software | GraphStats | Prism 9.0.0 | |
| HRP-linked Mouse IgG | Cytiva | NA931 | 1: 5,000 dilution is recommended |
| HRP-linked Rabbit IgG | Cytiva | NA934 | 1: 5,000 dilution is recommended |
| ImageJ Software | NIH, USA | ImageJ 1.53e | |
| L-Glutamine | Fisher Scientific | 25030081 | |
| Maximum sensitivity ECL substrate | Thermo Fisher | 34095 | |
| Micrococcal nuclease | New England BioLabs | M0247S | |
| Monosodium phosphate | Sigma-Aldrich | S3139 | Used to make sodium phosphate buffer |
| NanoDrop 2000 spectrophotometer | Thermo Scientific | ND-2000 | |
| N-ethylmaleimide | Thermo Fisher | 23030 | DeSUMOylation/deubiquitylation inhibitor |
| Nitrocellulose membrane, 0.45 µm | Bio-Rad | 1620115 | |
| Non-fat dry milk | Bio-Rad | 1706404XTU | |
| PDD00017273 | Selleckchem | S8862 | Poly(ADP-ribose) glycohydrolase inhibitor |
| Penicillin-Streptomycin | Thermo Fisher | 15140122 | |
| Protease inhibitor cocktail | Thermo Fisher | 78430 | |
| Q700 sonicator | Qsonica | Q700-110 | |
| Ready-to-assemble PVDF transfer kit | Bio-Rad | 1704274 | |
| Slot-blot apparatus | Bio-Rad | 1706542 | |
| Slot-blot filter paper | Bio-Rad | 1620161 | |
| Trans-Blot turbo transfer system | Bio-Rad | 1704150 | |
| Tris/Glycine/SDS electrophoresis buffer | Bio-Rad | 1610732 | |
| Tween-20 | Sigma-Aldrich | P3179 | |
| Vertical electrophoresis cell | Bio-Rad | 1658004 |
Referenzen
- Jackson, S. P., Bartek, J. The DNA-damage response in human biology and disease. Nature. 461 (7267), 1071-1078 (2009).
- Klages-Mundt, N. L., Li, L. Formation and repair of DNA-protein crosslink damage. Science China. Life Sciences. 60 (10), 1065-1076 (2017).
- Weickert, P., Stingele, J. DNA-protein crosslinks and their resolution. Annual Review of Biochemistry. 91, 157-181 (2022).
- Tretyakova, N. Y., Groehler, A., Ji, S. DNA-Protein cross-links: formation, structural identities, and biological outcomes. Accounts of Chemical Research. 48 (6), 1631-1644 (2015).
- Pommier, Y., Nussenzweig, A., Takeda, S., Austin, C. Human topoisomerases and their roles in genome stability and organization. Nature Reviews Molecular Cell Biology. 23 (6), 407-427 (2022).
- Pommier, Y., Sun, Y., Huang, S. N., Nitiss, J. L. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nature Reviews Molecular Cell Biology. 17 (11), 703-721 (2016).
- Ruggiano, A., Ramadan, K. DNA-protein crosslink proteases in genome stability. Communications Biology. 4 (1), 11 (2021).
- Hoffman, E. A., Frey, B. L., Smith, L. M., Auble, D. T. Formaldehyde crosslinking: a tool for the study of chromatin complexes. The Journal of Biological Chemistry. 290 (44), 26404-26411 (2015).
- Moretton, A., Loizou, J. I. Interplay between cellular metabolism and the DNA damage response in cancer. Cancers. 12 (8), 2051 (2020).
- Stingele, J., Schwarz, M. S., Bloemeke, N., Wolf, P. G., Jentsch, S. A DNA-dependent protease involved in DNA-protein crosslink repair. Cell. 158 (2), 327-338 (2014).
- Maskey, R. S., et al. Spartan deficiency causes accumulation of topoisomerase 1 cleavage complexes and tumorigenesis. Nucleic Acids Research. 45 (8), 4564-4576 (2017).
- Stingele, J., et al. Mechanism and regulation of DNA-protein crosslink repair by the DNA-dependent metalloprotease SPRTN. Molecular Cell. 64 (4), 688-703 (2016).
- Vaz, B., et al. Metalloprotease SPRTN/DVC1 orchestrates replication-coupled DNA-protein crosslink repair. Molecular Cell. 64 (4), 704-719 (2016).
- Lopez-Mosqueda, J., et al. SPRTN is a mammalian DNA-binding metalloprotease that resolves DNA-protein crosslinks. eLife. 5, e21491 (2016).
- Kojima, Y., et al. FAM111A protects replication forks from protein obstacles via its trypsin-like domain. Nature Communications. 11 (1), 1318 (2020).
- Dokshin, G. A., et al. GCNA interacts with spartan and topoisomerase II to regulate genome stability. Developmental Cell. 52 (1), 53-68 (2020).
- Borgermann, N., et al. SUMOylation promotes protective responses to DNA-protein crosslinks. The EMBO Journal. 38 (8), e101496 (2019).
- Sun, Y., Zhang, Y., Schultz, C. W., Pommier, Y., Thomas, A. CDK7 inhibition synergizes with topoisomerase I inhibition in small cell lung cancer cells by inducing ubiquitin-mediated proteolysis of RNA polymerase II. Molecular Cancer Therapeutics. 21 (9), 1430-1438 (2022).
- Sun, Y., et al. Requirements for MRN endonuclease processing of topoisomerase II-mediated DNA damage in mammalian cells. Frontiers in Molecular Biosciences. 9, 1007064 (2022).
- Sun, Y., et al. PARylation prevents the proteasomal degradation of topoisomerase I DNA-protein crosslinks and induces their deubiquitylation. Nature Communications. 12 (1), 5010 (2021).
- Sun, Y., et al. Excision repair of topoisomerase DNA-protein crosslinks (TOP-DPC). DNA Repair. 89, 102837 (2020).
- Sun, Y., Saha, L. K., Saha, S., Jo, U., Pommier, Y. Debulking of topoisomerase DNA-protein crosslinks (TOP-DPC) by the proteasome, non-proteasomal and non-proteolytic pathways. DNA Repair. 94, 102926 (2020).
- Sun, Y., et al. A conserved SUMO pathway repairs topoisomerase DNA-protein cross-links by engaging ubiquitin-mediated proteasomal degradation. Science Advances. 6 (46), (2020).
- Saha, S., et al. DNA and RNA cleavage complexes and repair pathway for TOP3B RNA- and DNA-protein crosslinks. Cell Reports. 33 (13), 108569 (2020).
- Swan, R. L., Cowell, I. G., Austin, C. A. Mechanisms to repair stalled topoisomerase II-DNA covalent complexes. Molecular Pharmacology. 101 (1), 24-32 (2022).
- Swan, R. L., Poh, L. L. K., Cowell, I. G., Austin, C. A. Small molecule inhibitors confirm ubiquitin-dependent removal of TOP2-DNA covalent complexes. Molecular Pharmacology. 98 (3), 222-233 (2020).
- Sciascia, N., et al. Suppressing proteasome mediated processing of topoisomerase II DNA-protein complexes preserves genome integrity. eLife. 9, e53447 (2020).
- Anand, J., Sun, Y., Zhao, Y., Nitiss, K. C., Nitiss, J. L. Detection of topoisomerase covalent complexes in eukaryotic cells. Methods in Molecular Biology. 1703, 283-299 (2018).
- Subramanian, D., Furbee, C. S., Muller, M. T. ICE bioassay. Isolating in vivo complexes of enzyme to DNA. Methods in Molecular Biology. 95, 137-147 (2001).
- Nitiss, J. L., Kiianitsa, K., Sun, Y., Nitiss, K. C., Maizels, N. Topoisomerase assays. Current Protocols. 1 (10), e250 (2021).
- Kiianitsa, K., Maizels, N. A rapid and sensitive assay for DNA-protein covalent complexes in living cells. Nucleic Acids Research. 41 (9), e104 (2013).
- Cowell, I. G., Tilby, M. J., Austin, C. A. An overview of the visualisation and quantitation of low and high MW DNA adducts using the trapped in agarose DNA immunostaining (TARDIS) assay. Mutagenesis. 26 (2), 253-260 (2011).
- Leng, X., Duxin, J. P. Targeting DNA-protein crosslinks via post-translational modifications. Frontiers in Molecular Biosciences. 9, 944775 (2022).
- Kuhbacher, U., Duxin, J. P. How to fix DNA-protein crosslinks. DNA Repair. 94, 102924 (2020).
- Stingele, J., Bellelli, R., Boulton, S. J. Mechanisms of DNA-protein crosslink repair. Nature Reviews Molecular Cell Biology. 18 (9), 563-573 (2017).
- Sun, Y., Nitiss, J. L., Pommier, Y. SUMO: A Swiss army knife for eukaryotic topoisomerases. Frontiers in Molecular Biosciences. 9, 871161 (2022).
- Krastev, D. B., et al. The ubiquitin-dependent ATPase p97 removes cytotoxic trapped PARP1 from chromatin. Nature Cell Biology. 24 (1), 62-73 (2022).
- Liu, J. C. Y., et al. Mechanism and function of DNA replication-independent DNA-protein crosslink repair via the SUMO-RNF4 pathway. The EMBO Journal. 40 (18), e107413 (2021).
- Larsen, N. B., et al. Replication-coupled DNA-protein crosslink repair by SPRTN and the proteasome in Xenopus egg extracts. Molecular Cell. 73 (3), 574-588 (2019).
- Zhao, S., et al. A ubiquitin switch controls autocatalytic inactivation of the DNA-protein crosslink repair protease SPRTN. Nucleic Acids Research. 49 (2), 902-915 (2021).
- Ruggiano, A., et al. The protease SPRTN and SUMOylation coordinate DNA-protein crosslink repair to prevent genome instability. Cell Reports. 37 (10), 110080 (2021).
- Mahmood, T., Yang, P. C. Western blot: technique, theory, and trouble shooting. North American Journal of Medical Sciences. 4 (9), 429-434 (2012).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten