
Method Article
Detección cuantitativa de enlaces cruzados ADN-proteína y sus modificaciones postraduccionales
En este artículo
Resumen
El presente protocolo destaca un método modificado para detectar y cuantificar los enlaces cruzados ADN-proteína (DPCs) y sus modificaciones postraduccionales (PTMs), incluyendo la ubiquitilación, la SUMOilación y la ADP-ribosilación inducida por inhibidores de la topoisomerasa y por formaldehído, permitiendo así el estudio de la formación y reparación de DPCs y sus PTMs.
Resumen
Los enlaces cruzados ADN-proteínas (CPD) son lesiones frecuentes, ubicuas y deletéreas del ADN, que surgen del daño endógeno del ADN, del mal funcionamiento de las enzimas (topoisomerasas, metiltransferasas, etc.) o de agentes exógenos como los quimioterápicos y los agentes reticulantes. Una vez que se inducen los CPD, se les conjugan rápidamente varios tipos de modificaciones postraduccionales (PTM) como mecanismos de respuesta temprana. Se ha demostrado que los DPC pueden ser modificados por ubiquitina, un pequeño modificador similar a la ubiquitina (SUMO) y poli-ADP-ribosa, que preparan los sustratos para señalar sus respectivas enzimas de reparación designadas y, en algunos casos, coordinan la reparación de manera secuencial. Dado que los PTM transpiran rápidamente y son altamente reversibles, ha sido un desafío aislar y detectar los CPD conjugados con PTM que generalmente permanecen en niveles bajos. Aquí se presenta un inmunoensayo para purificar y detectar cuantitativamente DPC ubiquitiladas, SUMOiladas y ADP-ribosiladas (DPC topoisomerasas inducidas por fármacos y DPC inespecíficas inducidas por aldehído) in vivo. Este ensayo se deriva del ensayo RADAR (enfoque rápido para la recuperación de aductos de ADN) que se utiliza para el aislamiento de ADN genómico que contiene DPC mediante precipitación de etanol. Después de la normalización y la digestión de las nucleasas, los PTM de las CPD, incluida la ubiquitilación, la SUMOilación y la ribosilación de ADP, se detectan mediante inmunotransferencia utilizando sus anticuerpos correspondientes. Este robusto ensayo se puede utilizar para identificar y caracterizar nuevos mecanismos moleculares que reparan los CPD enzimáticos y no enzimáticos y tiene el potencial de descubrir inhibidores de moléculas pequeñas dirigidos a factores específicos que regulan los PTM para reparar los CPD.
Introducción
El daño genómico del ADN ocurre debido a la descomposición espontánea, el daño interno y los factores ambientales1. Las lesiones de ADN resultantes comprenden bases dañadas, desajustes, roturas de cadena simple y doble, enlaces cruzados entre y dentro de la cadena y enlaces cruzados de ADN y proteínas (DPC). Un DPC se forma cuando una proteína unida a la cromatina queda atrapada en el ADN a través de un enlace covalente. Las CPD son inducidas por lesiones endógenas de ADN y metabolitos reactivos, así como por agentes exógenos como quimioterápicos y agentes reticulantes bifuncionales. En determinadas circunstancias, la disfunción enzimática también puede conducir a la formación de CPD2. La gran diferencia en los inductores de DPC da como resultado una diferencia en la identidad de la proteína unida a la covalente, la región cromosómica donde se forma la DPC, el tipo de estructura del ADN reticulado a la proteína y la propiedad química del enlace covalente entre la proteína y el ADN 2,3,4.
En función de su naturaleza química, los CPD generalmente se clasifican en dos grupos: CPD enzimáticos y CPD no enzimáticos. Ciertas enzimas como las topoisomerasas, las glicosilasas y las metil/aciltransferasas actúan formando intermediarios covalentes enzima-ADN reversibles durante sus reacciones catalíticas normales. Estos son intermediarios enzima-ADN de vida corta y pueden convertirse en CPD enzimáticos de larga vida al ser atrapados por agentes endógenos o exógenos, en particular por quimioterápicos3. Las CPD topoisomerasas se encuentran entre las CPD enzimáticas más frecuentes en las células eucariotas, que pueden ser generadas por inhibidores de la topoisomerasa clínicamente útiles (topotecán e irinotecán para la topoisomerasa I [TOP1] y etopósido y doxorrubicina para la topoisomerasa II [TOP2]) y son los principales mecanismos terapéuticos de estos inhibidores 5,6. Las ADN metiltransferasas (DNMT) 1, 3A y 3B son el objetivo de la 5-aza-2'-desoxicitidina (también conocida como decitabina) y forman DPC tras la exposición al fármaco7. Los agentes reactivos, así como la luz ultravioleta y la radiación ionizante, inducen CPD no enzimáticas mediante la reticulación no específica de proteínas al ADN. Los aldehídos reactivos como el acetaldehído y el formaldehído (FA) a menudo se generan como subproductos del metabolismo celular, entre los cuales el AG se produce en concentraciones micromolares durante el metabolismo del metanol, la peroxidación lipídica y la desmetilación de histonas. Además, el AG es un producto químico de producción de alto volumen fabricado en todo el mundo, al que muchas personas están expuestas tanto ambiental como ocupacionalmente 8,9.
Tanto los CPD enzimáticos como los no enzimáticos son altamente tóxicos para las células, ya que sus voluminosos componentes proteicos dificultan eficazmente casi todos los procesos basados en la cromatina, incluida la replicación y la transcripción, lo que conduce a la detención del ciclo celular y la apoptosis si no se reparan. A lo largo de las últimas dos décadas, se ha estudiado intensamente la reparación de las CPD, y se han identificado varias proteínas/vías como factores clave que reparan directamente las CPD o modulan sus procesos de reparación. Por ejemplo, se ha establecido que la proteólisis de la masa proteica de un DPC es un paso fundamental de la reparación de DPC, y que la proteólisis puede ser catalizada por las proteasas SPRTN 10,11,12,13,14, FAM111A15, GCNA 16,17 o el complejo proteasoma26S 18,19,20,21,22 ,23,24,25,26,27 de una manera dependiente del tipo de célula o del contexto celular. La identificación y caracterización de estas proteasas se ha basado en gran medida en el ensayo in vivo del complejo enzimático (ICE)28,29 y en el ensayo de enfoque rápido para la recuperación de aductos de ADN (RADAR)30,31, que aíslan las moléculas de ADN y sus proteínas unidas a covalentes de las proteínas celulares libres para permitir la detección de DPC por transferencia de ranura utilizando anticuerpos dirigidos a las proteínas reticuladas. Además, se utilizó el ensayo de inmunotinción de ADN de agarosa atrapada (TARDIS) como medio para detectar y cuantificar las CPD a nivel de una sola célula32. En la actualidad, los investigadores eligen el ensayo RADAR en lugar del ensayo ICE para medir los CPD, ya que el ensayo ICE se basa en la purificación de ácidos nucleicos mediante ultracentrifugación en gradiente de cloruro de cesio, que requiere mucho tiempo, mientras que el ensayo RADAR precipita los ácidos nucleicos utilizando etanol en un período mucho más corto.
En los últimos años, ha surgido una creciente evidencia de que múltiples modificaciones postraduccionales (PTM) están involucradas en la señalización y el reclutamiento de proteasas dirigidas a DPC 3,33,34,35. Por ejemplo, se encontró que tanto los DPC TOP1 como los TOP2 estaban conjugados por un pequeño modificador similar a la ubiquitina (SUMO)-2/3 y luego SUMO-1 por la ligasa SUMO E3 PIAS4, independientemente de la replicación y transcripción del ADN. Las modificaciones secuenciales de SUMO parecen ser un objetivo de la ubiquitina, que se deposita en los TOP-DPC SUMOilados y forma cadenas poliméricas a través de su residuo de lisina 48 por una ubiquitina ligasa dirigida a SUMO denominada RNF4. Posteriormente, el polímero de ubiquitina provoca una señal y recluta el proteasoma 26S a los TOP-DPC23,36. Recientemente se ha demostrado que la misma vía SUMO-ubiquitina actúa sobre DNMT1-DPCs, así como sobre complejos PARP-ADN para su reparación37,38. Además, se ha informado que la ubiquitilación independiente de SUMO por la ubiquitina E3 ligasa TRAIP prepara a los DPC para la degradación proteasómica de una manera acoplada a la replicación39. De manera similar a la degradación proteasómica de los TOP-DPC, la proteólisis de los DPC enzimáticos y no enzimáticos por la metaloproteasa acoplada a la replicación SPRTN también requiere la ubiquitilación de los sustratos DPC como mecanismo para activar SPRTN40,41. La delimitación del papel de la SUMOilación y la ubiquitilación requiere la detección de DPC que están marcados con estos PTM. Dado que el ensayo ICE y el ensayo RADAR originales se basan en el aparato de transferencia de ranuras/puntos para medir muestras de ADN no digeridas, ninguno de estos dos ensayos es capaz de resolver y visualizar especies de DPC conjugadas con PTM con diferentes pesos moleculares. Para superar este problema, digerimos las muestras de ADN tras su purificación mediante precipitación de etanol y normalización de la muestra con nucleasa microcócica, una endo-exonucleasa de ADN y ARN para liberar las proteínas reticuladas, lo que nos permitió resolver las proteínas así como sus PTM covalentes con electroforesis en gel de dodecil-sulfato de poliacrilamida de sodio (SDS-PAGE). La electroforesis nos permitió detectar y cuantificar las CPD conjugadas con PTM utilizando anticuerpos específicos dirigidos a las PTM. Inicialmente, denominamos a este método mejorado el ensayo DUST, para resaltar su robustez en la detección de TOP-DPCs ubiquitilados y SUMOilados23. Posteriormente, ampliamos el uso del ensayo para evaluar cuantitativamente la ribosilación de ADP-TOP1-DPCs in vivo, utilizando anticuerpos contra polímeros de poli-ADP-ribosa20.
Aquí se presenta un protocolo detallado para el ensayo que detecta y mide las CPD ubiquitiladas, SUMOiladas y ADP-ribosiladas, que se optimizó para las CPD TOP modificadas que son inducidas por sus inhibidores y las CPD no específicas/no enzimáticas que son inducidas por la AF. Este ensayo aísla las CPD conjugadas con PTM mediante la lisis de células con un agente caotrópico, precipitando el ADN con etanol y liberando las proteínas reticuladas y sus modificadores con nucleasa microcócica. Las proteínas que de otro modo se unirían al ADN y sus PTM se cuantifican mediante inmunotransferencia utilizando anticuerpos específicos. Este ensayo allana una nueva vía para dilucidar los mecanismos moleculares por los que la célula repara las CPD enzimáticas y no enzimáticas. En concreto, permite realizar estudios detallados de la inducción y la cinética de las PTM importantes para la regulación de la degradación y reparación de las TOP-DPC y, por tanto, permite descubrir nuevos factores como las ligasas E3 que dictan las PTM. así como inhibidores dirigidos a estos factores. Dado que es probable que algunos de los PTM responsables de la reparación de TOP-DPC estén implicados en la reparación de DPC inducidos por otros quimioterapéuticos, como los fármacos basados en platino22, este ensayo también tiene el potencial de aplicarse al descubrimiento de nuevos fármacos y a la optimización racional de terapias combinatorias con inhibidores de la topoisomerasa o antineoplásicos basados en platino en células de pacientes para guiar los regímenes de tratamiento.
Protocolo
1. Cultivo celular y tratamiento farmacológico en la línea celular de riñón embrionario humano 293 (HEK293)
- Prepare el medio de cultivo, el medio de Eagle modificado de Dulbecco (DMEM), suplementado con suero bovino fetal al 10%, L-glutamina al 1% de 2 mM y 100 unidades/ml de penicilina-estreptomicina.
- Siembre 1 x 10 6 celdas en una placa de 60 mm o una placa de6 pocillos por condición de tratamiento más control.
- Al día siguiente, trate las células con los inductores de DPC de su elección.
- Para inducir TOP1-DPCs y su ubiquitilación y SUMOilación, añadir el inhibidor de TOP1 camptotecina a 20 μM a las células y recoger las células a los 20, 60 y 180 min.
- Para inducir TOP2α y β-DPC y su ubiquitilación y SUMOilación, exponga las células para agregar el inhibidor de TOP2 etopósido a 200 μM a las células y recoja las células a los 20, 60 y 180 min.
- Para inducir DPC no enzimáticas y su ubiquitilación y SUMOilación, añadir AG a 1 mM y recoger las células 2 h después de la exposición.
- Para inducir la parilación de TOP1-DPC, pretratar las células durante 1 h para bloquear la desparilación con un inhibidor de la poli(ADP-ribosa) glicohidrolasa (PARG) PDD00017273 a 10 μM, seguido de un cotratamiento con 20 μM de camptotecina durante 20, 60 y 180 min.
2. Aislamiento y normalización de ADN que contiene proteínas reticuladas
- Aspire rápidamente el medio con una pipeta de succión después del tratamiento y enjuague las células con solución salina tamponada con fosfato (PBS) 1x helada. Lisar inmediatamente las células en 600 μL de reactivo de DNAzol que contiene 1x inhibidor del cóctel de proteasa, 1 mM de ditiotreitol (DTT) y 20 mM de N-etilmaleimida (inhibidor de las enzimas deSUMOilantes y desubbiquitilantes).
- Agite lentamente la placa sobre una plataforma vibratoria durante 10 minutos a 4 °C.
- Agregue 1/2 volumen de etanol 100% frío (0,3 ml) directamente a la placa y repita el paso 2.2 hasta que el agregado de ácido nucleico opaco se haga visible. Transfiera el lisado celular a un tubo de microcentrífuga de 1,5 ml y someta el tubo a una centrifugación de velocidad máxima (20.000 x g) durante 15 min a 4 °C para precipitar el ácido nucleico y sus proteínas reticuladas.
- Aspirar el sobrenadante con una pipeta de aspiración y lavar el gránulo de ácido nucleico en 1 ml de etanol al 75%, seguido de 2 min de centrifugación de 20.000 x g a 4 °C.
- Aspire el sobrenadante, gire hacia abajo a la misma velocidad y elimine el líquido restante con una pipeta P20. Seque el pellet al aire durante 5 min.
- Disuelva rápidamente el gránulo de ácido nucleico en 0,1 ml de ddH2O. Vuelva a suspender el gránulo mediante pipeteo repetido y luego incube en un baño de agua a 37 °C hasta que el gránulo se hinche al menos tres veces más grande (aproximadamente 30 minutos).
- Sonicar las muestras con una sonda de procesador ultrasónico al 30% de amplitud durante 10 s para disolver completamente el gránulo.
- Paso opcional: Tratar las muestras con una mezcla de ARNasa A/T1 (10 μg de ARNasa A y 25 U de ARNasa T1) e incubar a 37 °C durante 15 min. Añadir 1/10 de volumen de acetato de sodio 3 M y dos volúmenes de etanol 100% helado al tubo, seguido de centrifugación a 20.000 x g durante 10 minutos para recuperar el ADN. Eliminar el sobrenadante y disolver el ADN precipitado en 0,1 mL de ddH2O.
- Paso opcional: Centrifugar la muestra durante 5 minutos a 20.000 x g y transferir el sobrenadante a un nuevo tubo.
- Cuantifique la concentración de ADN utilizando un espectrómetro ultravioleta-visible (UV-Vis). El rendimiento típico de ADN es de alrededor de 600-800 ng/μL. La relación A260/A280 debe reducirse de 2,0-2,1 a 1,8-1,9 después de la eliminación del ARN.
- Ajustar la concentración del ADN a 400-500 ng/μL en 0,12 mL deddH2O. Transferir 20 μL de la muestra a un nuevo tubo de microcentrífuga como control de la carga de ADN no digerido (consultar el paso 2.4).
- Para digerir el ADN disuelto en los 100 μL restantes deddH2O, agregue 2,000 unidades de gel de nucleasa microcócica junto con 1/10 de volumen (~11 μL) de tampón de reacción de nucleasa microcócica de calcio 10x a la muestra. Incubar a 37 °C durante 30 min.
3. Western blot de muestras de ADN digeridas
- Agregue 4 tampones de muestra Laemmli, luego hierva la muestra durante 5 minutos.
- Cargue 5-6 μg de muestra digerida (~15 μL) en gel de poliacrilamida al 4%-20%, seguido de SDS-PAGE42 para resolver los CPD no modificados y conjugados con PTM.
- Para detectar especies de DPC no enzimáticas inducidas por FA, incubar el gel con tinción azul de Coomassie durante la noche a temperatura ambiente. Lavar el gel con ddH 2 O durante2h y adquirir una imagen utilizando un sistema de imagen.
- Transferir el gel e incubar las membranas durante la noche a 4 °C con diluciones apropiadas del anticuerpo primario en tampón de bloqueo.
- Para detectar la ubiquitilación, haga una dilución 1:100 del anticuerpo anti-ubiquitina.
- Para detectar la modificación de SUMO-1 o SUMO-2/3, realice una dilución 1:250 del anticuerpo anti-SUMO-1 o anti-SUMO-2/3.
- Para detectar la ribosilación de ADP, realice una dilución 1:500 del anticuerpo anti-PAR.
- Para detectar los DPC TOP1, TOP2α o TOP2β totales, realice una dilución 1:500 de anticuerpos anti-TOP1, anti-TOP2α o anti-TOP2β.
NOTA: Consulte la Tabla de materiales para obtener detalles sobre la dilución de anticuerpos.
- Incubar una membrana lavada con 1x PBS-T (0,1% tween 20) con un anticuerpo secundario diluido 5.000 veces en tampón de bloqueo durante 60 min a temperatura ambiente.
- Desarrolle la membrana con el reactivo de quimioluminiscencia mejorada (ECL) y adquiera una imagen utilizando el sistema de imágenes.
4. Transferencia de ranuras de muestras de ADN no digeridas
- Diluir la muestra de ADN no digerido de 20 μL en 180 μL de tampón fosfato sódico (25 mM, pH 6,6).
- Cortar la membrana de nitrocelulosa (0,45 μm) y equilibrar durante 5 min en el tampón de fosfato de sodio.
- Ensamble el aparato de ranura de acuerdo con las instrucciones del fabricante y conéctelo a un sistema de vacío.
- Lave los pocillos con tampón de fosfato de sodio aplicando la aspiradora. Asegúrese de que no haya fugas en los pozos.
- Detenga el vacío y cargue 200 μL de ADN por muestra (1 μg). Llene los pocillos vacíos con 200 μL de tampón fosfato de sodio.
- Aplica la aspiradora.
- Cuando todos los pocillos estén completamente vacíos, detenga el vacío, cargue 200 μL de tampón de fosfato de sodio en cada pocillo y repita el paso 4.6.
- Recupere la membrana y bloquee con un tampón de bloqueo al 5% durante 0,5 h a temperatura ambiente.
- Sonda con anticuerpo anti-ADN de doble cadena (dsDNA) a una dilución de 1:5.000 durante la noche a 4 °C.
- Lavar 3 veces con 1x PBS-T e incubar con 1:5.000 anticuerpo secundario anti-ratón diluido unido a peroxidasa de rábano picante (HRP).
- Desarrolle la membrana con el reactivo de quimioluminiscencia mejorada (ECL) y adquiera una imagen utilizando el sistema de imágenes.
5. Análisis densitométrico
- Con ImageJ, calcule la relación entre la intensidad de cada banda/frotis en relación con la intensidad de la ranura de ADN no digerido y normalice la proporción con la de las células sin/antes del tratamiento farmacológico.
Resultados
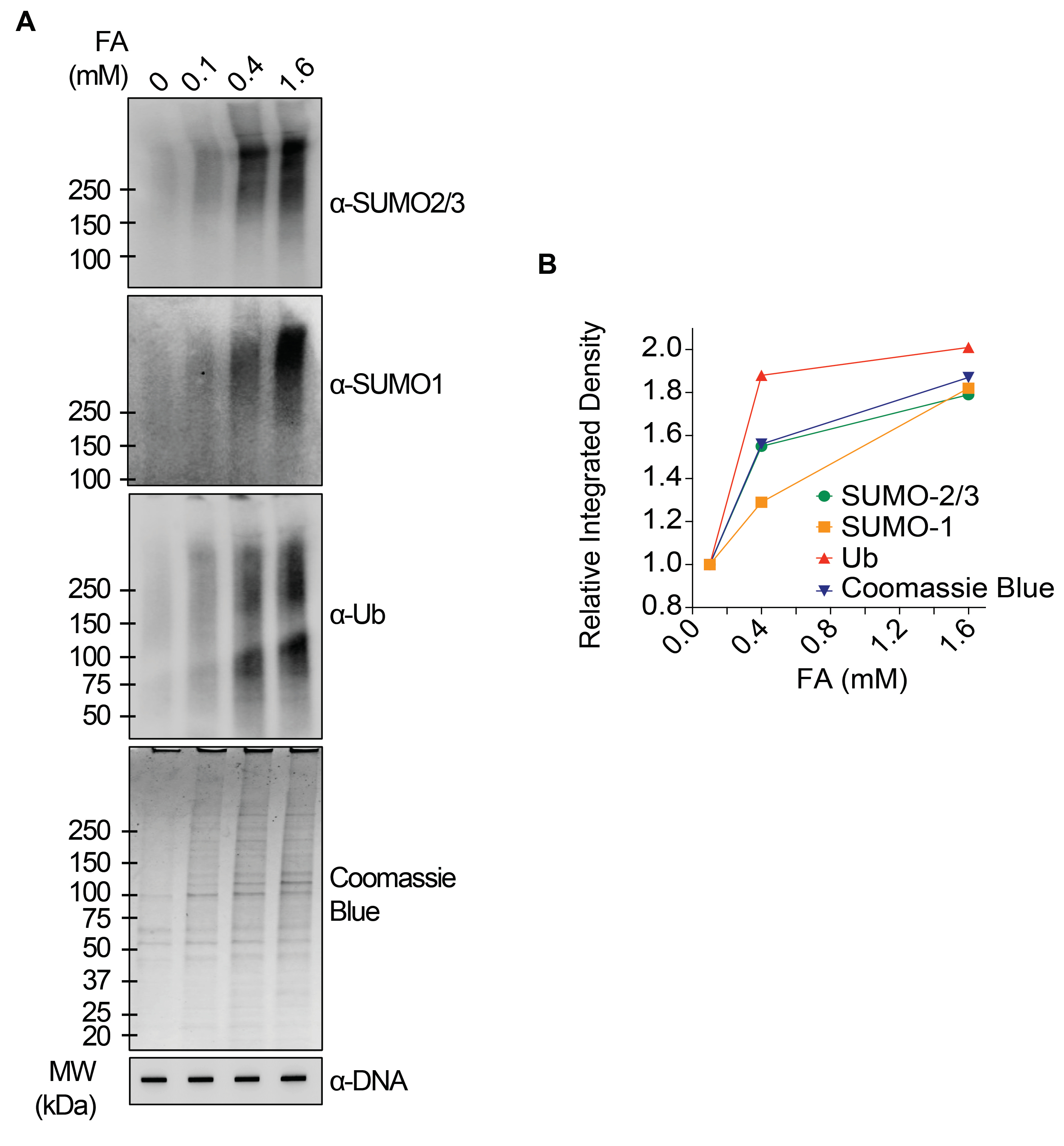
Los resultados representativos presentados en la Figura 1 muestran la formación y cinética de las TOP1-DPCs inducidas por fármacos y su SUMOilación y ubiquitilación. TOP1 escindió una hebra del dúplex de ADN y formó un intermedio covalente enzima-ADN, denominado complejo de escisión TOP1 (TOP1cc). El tratamiento con camptotecina (CPT), un inhibidor de TOP1, se unió a TOP1cc y lo estabilizó, lo que condujo a la formación de TOP1-DPC de larga duración. Se observó que los TOP1-DPC eran inducidos y alcanzaban su punto máximo 20 min después de la exposición a CPT. Al mismo tiempo, los TOP1-DPC fueron modificados por SUMO-2/3, que también alcanzó su punto máximo 20 min después del tratamiento con CPT. Como SUMO-2 y SUMO-3 comparten un 95% de identidad de secuencia, el anticuerpo no distingue uno del otro. A los 60 min, los TOP1-DPC y su modificación SUMO-2/3 disminuyeron, acompañados de la culminación de su modificación y ubiquitilación SUMO-1. Después del tratamiento farmacológico de 60 minutos, los niveles de modificación y ubiquitilación de TOP1-DPC SUMO-1 comenzaron a disminuir. En los mamíferos, las isoenzimas TOP2 α y β actúan mediante la introducción de una ruptura de doble cadena de ADN, así como a través de la formación de un complejo covalente enzima-ADN transitorio y reversible (TOP2cc). Los inhibidores de TOP2, como el etopósido (ETOP), convierten TOP2cc en TOP2-DPC e inducen su SUMOilación y ubiquitilación. Al igual que la cinética de los TOP1-DPC y sus PTM, los TOP2α- y β-DPC y su modificación SUMO-2/3 alcanzaron un pico a los 20 minutos, luego comenzaron a disminuir; mientras tanto, sus modificaciones de SUMO-1 y ubiquitina alcanzaron su punto máximo a los 60 min (Figura 2). Se ha demostrado que el aclaramiento de los TOP-DPC es el resultado de la degradación proteasómica, y el aclaramiento de la SUMOilación y la ubiquitilación de TOP-DPC se debe probablemente al reciclaje por desSUMOilación y desubiquitilación, respectivamente, por sus enzimas de reversión. Los experimentos de la Figura 3 examinaron los CPD no enzimáticos inducidos por AF y sus PTM. Se observó que los CPD y sus SUMO-2/3, SUMO-1 y ubiquitilación se formaron y acumularon de manera dependiente de la dosis de AF. Finalmente, la PARilación de TOP1-DPCs se detectó cuantitativamente con un anticuerpo anti-PAR utilizando el mismo método (Figura 4). La parilación de TOP1-DPC no fue detectable a menos que se añadiera un inhibidor de PARG a la célula, lo que sugiere que la parilación se produce rápidamente y es muy dinámica. De acuerdo con el hallazgo anterior, la inhibición de la dePARilación por PARGi pareció acumular TOP1-DPC, probablemente bloqueando la degradación proteolítica.

Figura 1: Análisis cuantitativo de la formación y cinética de TOP1-DPCs y su SUMOilación y ubiquitilación tras el tratamiento con CPT en células HEK293. (A) Las células HEK293 se trataron con 20 μM de CPT durante los períodos de tiempo indicados. Los lisados celulares se recolectaron y se sometieron al ensayo RADAR modificado y Western blot con anticuerpos indicados. Las muestras de ADN no digeridas se sometieron a transferencia de ranuras utilizando anticuerpos anti-dsDNA como control de carga. (B) Las intensidades de banda se cuantificaron con el software ImageJ y se trazaron con el software Prism. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2: Análisis cuantitativo de la formación y cinética de TOP2-DPCs y su SUMOilación y ubiquitilación tras el tratamiento con ETOP en células HEK293. (A) Las células HEK293 se trataron con 200 μM de ETOP durante los períodos de tiempo indicados. Los lisados celulares se recolectaron y se sometieron al ensayo RADAR modificado y Western blot con anticuerpos indicados. Las muestras de ADN no digeridas se sometieron a transferencia de ranuras utilizando anticuerpos anti-dsDNA como control de carga. (B) Las intensidades de banda se cuantificaron con el software ImageJ y se trazaron con el software Prism. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3: Análisis cuantitativo de DPCs no enzimáticas y su SUMOilación y ubiquitilación tras el tratamiento con AF en células HEK293. (A) Las células HEK293 fueron tratadas con AG de las concentraciones indicadas durante 2 h. Los lisados celulares se recolectaron y se sometieron al ensayo RADAR modificado y Western blot con anticuerpos indicados. Las muestras de ADN no digeridas se sometieron a transferencia de ranuras utilizando anticuerpos anti-dsDNA como control de carga. (B) Las intensidades de banda se cuantificaron con el software ImageJ y se trazaron con el software Prism. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 4: Análisis cuantitativo de TOP1-DPCs y su PARilación tras el tratamiento con CPT en células HEK293. (A) Las células HEK293 se trataron previamente con 10 μM de PARGi durante 1 h y luego se trataron conjuntamente con CPT durante períodos de tiempo indicados. Los lisados celulares se recolectaron y se sometieron al ensayo RADAR modificado y Western blot con anticuerpos indicados. Las muestras de ADN no digeridas se sometieron a transferencia de ranuras utilizando anticuerpos anti-dsDNA como control de carga. (B) Las intensidades de banda se cuantificaron con el software ImageJ y se trazaron con el software Prism. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Discusión
El método descrito permite la medición de enlaces cruzados enzimáticos y no enzimáticos ADN-proteína en células de mamíferos y es el único enfoque adecuado para estudiar su ubiquitilación, SUMOilación y ADP-ribosilación. La transferencia de ranuras después del ensayo ICE o RADAR permite la detección rápida de DPC enzimáticos específicos, como los TOP-DPC, utilizando sus anticuerpos. Sin embargo, una advertencia a este método es su incapacidad para separar proteínas de diferentes pesos moleculares, lo que hace imposible determinar los tamaños de los CPD conjugados con PTM. El método descrito resuelve el problema mediante la liberación de proteínas reticuladas con nucleasa microcócica, que degrada el ADN a oligonucleótidos con 3'-fosfatos terminales, lo que permite la separación completa de las proteínas (conjugadas con oligonucleótidos) por SDS-PAGE. Por lo tanto, los DPC modificados con monómeros y polímeros de ubiquitina, SUMO o ADP-ribosa de diferentes tamaños pueden visualizarse y cuantificarse mediante anticuerpos dirigidos a estos PTM, lo que permite una investigación detallada de su formación y cinética. Para garantizar la reproducibilidad y calcular la significación estadística, se requieren réplicas biológicas de los experimentos.
Uno de los problemas más comunes de este ensayo es el bajo rendimiento de ADN después de la precipitación de etanol. Por un lado, el rendimiento de ADN se puede aumentar con más material de partida (células). Por otro lado, la incubación de lisados celulares con etanol en una placa plana en lugar de un tubo Eppendorf puede mejorar notablemente la agregación de moléculas de ADN y, por lo tanto, facilitar su precipitación. Las señales inespecíficas observadas en muestras sin tratamientos farmacológicos pueden indicar contaminación por proteínas no covalentes. Si este es el caso, se puede considerar lavar los gránulos de ADN con un tampón con alto contenido de sal para eliminar los contaminantes antes de la sonicación. También se recomienda centrifugar las muestras de ADN después de la sonicación y la digestión de la nucleasa microcócica y descartar las insolubles. En el caso de que la señal sea deficiente o nula, se pueden intentar varias soluciones posibles. En primer lugar, se puede aumentar la cantidad de carga de ADN para SDS-PAGE y la inmunotransferencia. Para que las especies de DPC SUMOiladas y ubiquitiladas sean detectables, se recomienda cargar al menos 4 μg de ADN en el gel. En segundo lugar, se pueden aumentar las concentraciones del fármaco para inducir niveles más altos de CPD y sus PTM asociados. En tercer lugar, se sugiere incubar las transferencias con anticuerpos primarios durante otro día si las bandas/frotis parecen ser débiles. Una incubación de 2 días puede potenciar significativamente la señal y, por lo tanto, reducir la variabilidad biológica de los experimentos independientes23. El desprendimiento de la membrana para volver a teñir inevitablemente resulta en la pérdida de una cierta cantidad de especies de DPC conjugadas con PTM que ya son en baja abundancia. Por lo tanto, se recomienda encarecidamente utilizar geles separados para la detección de ubiquitina y SUMO en lugar de volver a sondear una sola mancha. Además, los gránulos de ADN deben lavarse con etanol al 75% para eliminar el ADN restante que contiene sal de guanidina antes de disolverlo en H2O o cualquier otro solvente, lo que de otro modo causa la cristalización de la muestra después de la adición del tampón de carga Laemmli.
El flujo de trabajo del método descrito es mucho más eficiente en términos de tiempo en comparación con el engorroso ensayo ICE, ya que se basa en la precipitación rápida de etanol en lugar de la ultracentrifugación de cloruro de cesio, que consume mucho tiempo, para aislar el ADN genómico. A un precio, la purificación a base de etanol produce una baja cantidad de contaminantes proteicos que normalmente son insignificantes para la inmunodetección. Sin embargo, cuando se trata de estudios analíticos, como el análisis proteómico basado en espectrometría de masas o la secuenciación de próxima generación que requieren exactitud y precisión, la centrifugación en gradiente de densidad de cloruro de cesio sigue siendo un enfoque más confiable para aislar ADN puro y de alta abundancia. Este método también puede aplicarse potencialmente a la elaboración de perfiles de sitios de modificación en proteínas reticuladas y a la determinación de tipos de enlace de poliubiquitilación y polisumosilación utilizando métodos adecuados basados en espectrometría de masas.
Cabe destacar que este ensayo permite la identificación y caracterización de los factores que regulan los PTM para la reparación de CPD. Por ejemplo, los métodos de cribado imparciales de alto rendimiento (ARN de interferencia y CRISPR) son herramientas poderosas para descubrir ligasas de ubiquitina E3, ligasas SUMO E3 y sus cofactores asociados que mitigan la citotoxicidad de los inductores de DPC. El método descrito permite la validación molecular de estas proteínas mediante la determinación de si ayudan a las células a sobrevivir a los inductores de DPC mediante la reparación de las DPC. Los nuevos inhibidores de moléculas pequeñas dirigidos a estas proteínas identificados, por ejemplo, mediante cribado virtual, también pueden validarse utilizando este protocolo. Dado que los inhibidores de la topoisomerasa se encuentran entre los quimioterapéuticos más prescritos, este ensayo robusto puede desarrollarse como una herramienta para el desarrollo de fármacos que hagan sinergia con los inhibidores clínicos de la topoisomerasa.
Divulgaciones
El autor declara no tener intereses contrapuestos.
Agradecimientos
Este trabajo fue financiado en parte por el Premio a la Excelencia en la Transición de la Investigación Postdoctoral del Centro para la Investigación del Cáncer del Instituto Nacional del Cáncer.
Materiales
| Name | Company | Catalog Number | Comments |
| 10x Phosphate buffered saline (PBS) | Thermo Fisher | 70011069 | |
| 4–20% precast polyacrylamide gel | Bio-Rad | 4561096 | |
| 4x Laemmli Sample Buffer | Bio-Rad | 1610747 | |
| AcquaStain (coomassie blue) | Bulldog Bio | AS001000 | |
| anti-dsDNA (mouse monoclonal) | Abcam | 27156 | 1: 5,000 dilution is recommended |
| anti-PAR (mouse monoclonal) | R&D systems | 4335-MC-100 | 1: 500 dilution is recommended |
| anti-SUMO-1(rabbit monoclonal) | Cell Signaling Technology | 4940 | 1: 250 dilution is recommended |
| anti-SUMO-2/3 (rabbit monoclonal) | Cell Signaling Technology | 4971 | 1: 250 dilution is recommended |
| anti-TOP1 (mouse monoclonal) | BD Biosciences | 556597 | 1: 500 dilution is recommended |
| anti-TOP2α (mouse monoclonal) | Santa Cruz Biotechnology | SC-365799 | 1: 250 dilution is recommended |
| anti-TOP2β (mouse monoclonal) | Santa Cruz Biotechnology | SC-25330 | 1: 250 dilution is recommended |
| anti-ubiquitin (mouse monoclonal) | Santa Cruz Biotechnology | SC-8017 | 1: 100 dilution is recommended |
| Calcium chloride | Sigma-Aldrich | 499609 | Used for micrococcal nuclease digestion |
| Camptothecin | Sigma-Aldrich | PHL89593 | |
| ChemiDo MP imaging system | Bio-Rad | 12003154 | |
| Disodium phosphate | Sigma-Aldrich | 5438380100 | Used to make sodium phosphate buffer |
| DNAzol | Thermo Fisher | 10503027 | |
| DTT (dithiothreitol) | Thermo Fisher | R0861 | |
| Dulbecco's modified eagle's medium | Sigma-Aldrich | 11965084 | |
| Ethyl alcohol, 200 proof | Sigma-Aldrich | E7023 | |
| Etoposide | Sigma-Aldrich | 1268808 | |
| Formaldehyde | Sigma-Aldrich | 47608 | |
| Graphpad Prism Software | GraphStats | Prism 9.0.0 | |
| HRP-linked Mouse IgG | Cytiva | NA931 | 1: 5,000 dilution is recommended |
| HRP-linked Rabbit IgG | Cytiva | NA934 | 1: 5,000 dilution is recommended |
| ImageJ Software | NIH, USA | ImageJ 1.53e | |
| L-Glutamine | Fisher Scientific | 25030081 | |
| Maximum sensitivity ECL substrate | Thermo Fisher | 34095 | |
| Micrococcal nuclease | New England BioLabs | M0247S | |
| Monosodium phosphate | Sigma-Aldrich | S3139 | Used to make sodium phosphate buffer |
| NanoDrop 2000 spectrophotometer | Thermo Scientific | ND-2000 | |
| N-ethylmaleimide | Thermo Fisher | 23030 | DeSUMOylation/deubiquitylation inhibitor |
| Nitrocellulose membrane, 0.45 µm | Bio-Rad | 1620115 | |
| Non-fat dry milk | Bio-Rad | 1706404XTU | |
| PDD00017273 | Selleckchem | S8862 | Poly(ADP-ribose) glycohydrolase inhibitor |
| Penicillin-Streptomycin | Thermo Fisher | 15140122 | |
| Protease inhibitor cocktail | Thermo Fisher | 78430 | |
| Q700 sonicator | Qsonica | Q700-110 | |
| Ready-to-assemble PVDF transfer kit | Bio-Rad | 1704274 | |
| Slot-blot apparatus | Bio-Rad | 1706542 | |
| Slot-blot filter paper | Bio-Rad | 1620161 | |
| Trans-Blot turbo transfer system | Bio-Rad | 1704150 | |
| Tris/Glycine/SDS electrophoresis buffer | Bio-Rad | 1610732 | |
| Tween-20 | Sigma-Aldrich | P3179 | |
| Vertical electrophoresis cell | Bio-Rad | 1658004 |
Referencias
- Jackson, S. P., Bartek, J. The DNA-damage response in human biology and disease. Nature. 461 (7267), 1071-1078 (2009).
- Klages-Mundt, N. L., Li, L. Formation and repair of DNA-protein crosslink damage. Science China. Life Sciences. 60 (10), 1065-1076 (2017).
- Weickert, P., Stingele, J. DNA-protein crosslinks and their resolution. Annual Review of Biochemistry. 91, 157-181 (2022).
- Tretyakova, N. Y., Groehler, A., Ji, S. DNA-Protein cross-links: formation, structural identities, and biological outcomes. Accounts of Chemical Research. 48 (6), 1631-1644 (2015).
- Pommier, Y., Nussenzweig, A., Takeda, S., Austin, C. Human topoisomerases and their roles in genome stability and organization. Nature Reviews Molecular Cell Biology. 23 (6), 407-427 (2022).
- Pommier, Y., Sun, Y., Huang, S. N., Nitiss, J. L. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nature Reviews Molecular Cell Biology. 17 (11), 703-721 (2016).
- Ruggiano, A., Ramadan, K. DNA-protein crosslink proteases in genome stability. Communications Biology. 4 (1), 11 (2021).
- Hoffman, E. A., Frey, B. L., Smith, L. M., Auble, D. T. Formaldehyde crosslinking: a tool for the study of chromatin complexes. The Journal of Biological Chemistry. 290 (44), 26404-26411 (2015).
- Moretton, A., Loizou, J. I. Interplay between cellular metabolism and the DNA damage response in cancer. Cancers. 12 (8), 2051 (2020).
- Stingele, J., Schwarz, M. S., Bloemeke, N., Wolf, P. G., Jentsch, S. A DNA-dependent protease involved in DNA-protein crosslink repair. Cell. 158 (2), 327-338 (2014).
- Maskey, R. S., et al. Spartan deficiency causes accumulation of topoisomerase 1 cleavage complexes and tumorigenesis. Nucleic Acids Research. 45 (8), 4564-4576 (2017).
- Stingele, J., et al. Mechanism and regulation of DNA-protein crosslink repair by the DNA-dependent metalloprotease SPRTN. Molecular Cell. 64 (4), 688-703 (2016).
- Vaz, B., et al. Metalloprotease SPRTN/DVC1 orchestrates replication-coupled DNA-protein crosslink repair. Molecular Cell. 64 (4), 704-719 (2016).
- Lopez-Mosqueda, J., et al. SPRTN is a mammalian DNA-binding metalloprotease that resolves DNA-protein crosslinks. eLife. 5, e21491 (2016).
- Kojima, Y., et al. FAM111A protects replication forks from protein obstacles via its trypsin-like domain. Nature Communications. 11 (1), 1318 (2020).
- Dokshin, G. A., et al. GCNA interacts with spartan and topoisomerase II to regulate genome stability. Developmental Cell. 52 (1), 53-68 (2020).
- Borgermann, N., et al. SUMOylation promotes protective responses to DNA-protein crosslinks. The EMBO Journal. 38 (8), e101496 (2019).
- Sun, Y., Zhang, Y., Schultz, C. W., Pommier, Y., Thomas, A. CDK7 inhibition synergizes with topoisomerase I inhibition in small cell lung cancer cells by inducing ubiquitin-mediated proteolysis of RNA polymerase II. Molecular Cancer Therapeutics. 21 (9), 1430-1438 (2022).
- Sun, Y., et al. Requirements for MRN endonuclease processing of topoisomerase II-mediated DNA damage in mammalian cells. Frontiers in Molecular Biosciences. 9, 1007064 (2022).
- Sun, Y., et al. PARylation prevents the proteasomal degradation of topoisomerase I DNA-protein crosslinks and induces their deubiquitylation. Nature Communications. 12 (1), 5010 (2021).
- Sun, Y., et al. Excision repair of topoisomerase DNA-protein crosslinks (TOP-DPC). DNA Repair. 89, 102837 (2020).
- Sun, Y., Saha, L. K., Saha, S., Jo, U., Pommier, Y. Debulking of topoisomerase DNA-protein crosslinks (TOP-DPC) by the proteasome, non-proteasomal and non-proteolytic pathways. DNA Repair. 94, 102926 (2020).
- Sun, Y., et al. A conserved SUMO pathway repairs topoisomerase DNA-protein cross-links by engaging ubiquitin-mediated proteasomal degradation. Science Advances. 6 (46), (2020).
- Saha, S., et al. DNA and RNA cleavage complexes and repair pathway for TOP3B RNA- and DNA-protein crosslinks. Cell Reports. 33 (13), 108569 (2020).
- Swan, R. L., Cowell, I. G., Austin, C. A. Mechanisms to repair stalled topoisomerase II-DNA covalent complexes. Molecular Pharmacology. 101 (1), 24-32 (2022).
- Swan, R. L., Poh, L. L. K., Cowell, I. G., Austin, C. A. Small molecule inhibitors confirm ubiquitin-dependent removal of TOP2-DNA covalent complexes. Molecular Pharmacology. 98 (3), 222-233 (2020).
- Sciascia, N., et al. Suppressing proteasome mediated processing of topoisomerase II DNA-protein complexes preserves genome integrity. eLife. 9, e53447 (2020).
- Anand, J., Sun, Y., Zhao, Y., Nitiss, K. C., Nitiss, J. L. Detection of topoisomerase covalent complexes in eukaryotic cells. Methods in Molecular Biology. 1703, 283-299 (2018).
- Subramanian, D., Furbee, C. S., Muller, M. T. ICE bioassay. Isolating in vivo complexes of enzyme to DNA. Methods in Molecular Biology. 95, 137-147 (2001).
- Nitiss, J. L., Kiianitsa, K., Sun, Y., Nitiss, K. C., Maizels, N. Topoisomerase assays. Current Protocols. 1 (10), e250 (2021).
- Kiianitsa, K., Maizels, N. A rapid and sensitive assay for DNA-protein covalent complexes in living cells. Nucleic Acids Research. 41 (9), e104 (2013).
- Cowell, I. G., Tilby, M. J., Austin, C. A. An overview of the visualisation and quantitation of low and high MW DNA adducts using the trapped in agarose DNA immunostaining (TARDIS) assay. Mutagenesis. 26 (2), 253-260 (2011).
- Leng, X., Duxin, J. P. Targeting DNA-protein crosslinks via post-translational modifications. Frontiers in Molecular Biosciences. 9, 944775 (2022).
- Kuhbacher, U., Duxin, J. P. How to fix DNA-protein crosslinks. DNA Repair. 94, 102924 (2020).
- Stingele, J., Bellelli, R., Boulton, S. J. Mechanisms of DNA-protein crosslink repair. Nature Reviews Molecular Cell Biology. 18 (9), 563-573 (2017).
- Sun, Y., Nitiss, J. L., Pommier, Y. SUMO: A Swiss army knife for eukaryotic topoisomerases. Frontiers in Molecular Biosciences. 9, 871161 (2022).
- Krastev, D. B., et al. The ubiquitin-dependent ATPase p97 removes cytotoxic trapped PARP1 from chromatin. Nature Cell Biology. 24 (1), 62-73 (2022).
- Liu, J. C. Y., et al. Mechanism and function of DNA replication-independent DNA-protein crosslink repair via the SUMO-RNF4 pathway. The EMBO Journal. 40 (18), e107413 (2021).
- Larsen, N. B., et al. Replication-coupled DNA-protein crosslink repair by SPRTN and the proteasome in Xenopus egg extracts. Molecular Cell. 73 (3), 574-588 (2019).
- Zhao, S., et al. A ubiquitin switch controls autocatalytic inactivation of the DNA-protein crosslink repair protease SPRTN. Nucleic Acids Research. 49 (2), 902-915 (2021).
- Ruggiano, A., et al. The protease SPRTN and SUMOylation coordinate DNA-protein crosslink repair to prevent genome instability. Cell Reports. 37 (10), 110080 (2021).
- Mahmood, T., Yang, P. C. Western blot: technique, theory, and trouble shooting. North American Journal of Medical Sciences. 4 (9), 429-434 (2012).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoExplorar más artículos
This article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados