
Method Article
Determinante 3'-Termini y secuencias de las moléculas de ADN Viral naciente en HIV-1 Reverse transcripción en células infectadas
En este artículo
Resumen
Aquí presentamos un enfoque de secuenciación profunda que proporciona una determinación imparcial de naciente 3'-termini, así como perfiles mutacionales de moléculas de ADN monocatenario. La principal aplicación es la caracterización de los nacientes DNAs complementarios retrovirales (cADN), los intermedios generados durante el proceso de transcripción inversa retroviral.
Resumen
Monitoreo de productos intermedios ácidos nucleicos durante la replicación de virus proporciona penetraciones en los efectos y mecanismos de acción de compuestos antivirales y proteínas de la célula huésped en la síntesis de ADN viral. Aquí nos dirigimos a la falta de un análisis basado en la célula, alta cobertura y alta resolución que es capaz de definir productos intermedios de la transcripción inversa retroviral en el contexto fisiológico de infección por el virus. El método descrito captura 3'-termini de nacientes moléculas complementarias de ADN (cDNA) dentro de las células de infectados por VIH-1 en la resolución de un solo nucleótido. El protocolo consiste en extracción de ADN, enriquecimiento específica de ADN viral vía captura de híbrido, ligadura de adaptador, fraccionamiento tamaño gel purificación, amplificación de PCR, secuenciación profunda y análisis de datos de la célula entera. Un paso clave es la ligadura de las moléculas del adaptador para abrir termini de 3'-ADN eficiente e imparcial. Aplicación del método descrito determina la abundancia de transcripción inversa de cada longitud particular en una muestra determinada. También proporciona información sobre la variación de la secuencia (interno) en la transcripción inversa y de tal modo ninguna mutaciones posibles. En general, el ensayo es adecuado para cualquier pregunta relativa al ADN 3'-extensión, siempre y cuando se conoce la secuencia de la plantilla.
Introducción
Con el fin de diseccionar y comprender la replicación viral completamente, cada vez más refinadas técnicas que capturan la replicación intermedios son necesarios. En particular, la definición precisa de las especies de ácido nucleico viral en el contexto de las células infectadas puede proporcionar nuevas perspectivas, ya que muchos mecanismos de replicación viral a la fecha han sido examinados en reacciones aisladas en vitro . Un ejemplo es el proceso de transcripción inversa en retrovirus como el virus de inmunodeficiencia humana 1 (VIH-1). Las diferentes etapas de la transcripción reversa del VIH-1, durante el cual la enzima viral de la transcriptasa reversa (RT) copia el genoma de ARN monocatenario a ADN de doble hebra, han sido estudiados principalmente en ensayos de extensión de la cartilla con las proteínas purificadas y nucleicos ácidos1,2,3,4,5. Mientras que se establecieron los principios fundamentales, tales análisis no incorporan todos los componentes virales y celulares y pueden no reflejar los stoichiometries biológicamente relevantes de factores implicados. Por lo tanto, hemos diseñado una técnica poderosa para determinar el espectro de productos intermedios de la reversa de la transcripción precisa del cDNA 3'-termini (es decir, determinar su longitud exacta) y secuencias de nucleótidos en el contexto de las infecciones de la vida las células6. Recolección de datos de tiempo experimentos de curso pueden ser utilizados para comparar el perfil de las transcripciones en diversas condiciones, tales como la presencia de moléculas antivirales o proteínas, que pueden influir en la eficiencia y la procesividad de la síntesis de ADN y acumulación. Esto permite una comprensión más detallada del patógeno natural del ciclo de vida, que a menudo es la base para el diseño de fármacos específicos y la intervención terapéutica exitosa.
Transcripción reversa del VIH-1 compone de una serie de sucesivos eventos iniciados por el recocido de un primer tRNA a la plantilla de RNA genómica, que luego se extendió por RT para producir una transcripción de ADNc monocatenario corto llamada una cadena menos fuerte-parada (-sss) (véase Figura 1). Posteriormente, el cDNA - sss se transfiere desde el terminal 5' de largo repetición (LTR) hasta el litro 3' del ARN genómico, donde recuece y sirve como la cartilla para RT continuo mediada por alargamiento del menos filamento de la DNA (véase comentarios en transcripción reversa1 , 2 , 3 , 4). esta primera transferencia de cadena es una de las medidas de limitación de velocidad de reversa de la transcripción; por lo tanto, el cDNA - sss se sabe que se acumulan. El diseño básico del flujo de trabajo y biblioteca para capturar los productos de reversa de la transcripción en células infectadas se describe en la Figura 2a. Los iniciadores específicos y análisis de parámetros que se utilizan en el protocolo y listados en la tabla 1 de destino todos temprano revés transcripción cDNA intermedios dentro de la gama de longitud de 23 a ~ 650 nt, que incluye el ADN de 180-182 nt - sss. Sin embargo, las adaptaciones menores adecuadas a la estrategia permitirá aplicación no solo productos finales de reversa de la transcripción, pero también otros virus y sistemas, donde el objetivo es detectar que contiene extremos de ADN 3'-OH. Limitaciones importantes a considerar incluyen la gama de la longitud del producto final de PCR en la biblioteca; en particular, plantillas en las que la distancia entre el adaptador en el open 3'-terminal y el primer río arriba excede ~ 1000 nt serán probablemente ser menos eficientemente secuenciadas, introducir potencialmente engañosa prejuicios técnicos durante la preparación () Biblioteca Vea la discusión para más detalles y sugerencias de adaptación).
Previamente divulgadas técnicas para la determinación sistemática de 3'-termini de ácido nucleico se han centrado en RNA, no ADN, moléculas. Un ejemplo es 3' RACE (amplificación rápida del cDNA termina)7, que depende de la poliadenilación del mRNA. Además, se desarrollaron estrategias de ligadura base adaptador empleando ligasas de RNA, que han incluido RLM-raza (raza de RNA ligasa-mediada)8 o encaje (amplificación basada en la ligadura de los extremos del cDNA de)9. Es importante destacar que amplificaciones de ligadura son sensibles a cualquier sesgo introducido por la reacción de ligadura sí mismo. Por ejemplo, la ligadura puede ser más o menos eficiente dependiendo de un determinado nucleótido en la posición 3', la secuencia, longitud total de la molécula o estructura local. Tales preferencias ligasa conducen a captura incompleta de moléculas y falsedad en la lectura, que nosotros y otros hemos observado9,10. Para minimizar el sesgo de ligadura durante los pasos además de adaptador en el protocolo descrito en este documento, probado una serie de estrategias de la ligadura y el uso de T4 ADN ligasa con un adaptador de DNA monocatenario de horquilla (como se describe por Kwok et al. 11) ser el único procedimiento con cerca ligadura cuantitativa que no produjo diferencias significativas en la eficacia de la ligadura cuando se evaluó con un conjunto de control oligonucleótidos6específicamente seleccionado. La elección de esta estrategia de ligadura es, por lo tanto, una característica clave en el éxito de este protocolo.
Hasta la fecha, seguimiento de la progresión de la RT del VIH-1 en células infectadas principalmente se ha logrado mediante la medición de productos de la transcripción inversa de diferentes longitud con PCR cuantitativa (qPCR) haciendo uso de la cartilla-probe que únicamente miden la mayor o menor (principios y tarde, respectivamente) cDNA productos12,13,14. Mientras que este enfoque de qPCR es apropiado para determinar eficiencias intrínsecas del proceso de transcripción inversa en sistemas celulares, la salida es de resolución relativamente baja, sin información de la secuencia se deriva. Nuestro nuevo enfoque, basado en adaptador optimizado ligadura, generación de biblioteca mediada por PCR y secuenciación profunda direcciones la brecha tecnología y ofrece una oportunidad para monitorear la reversa de la transcripción durante la infección por VIH-1 cuantitativamente y en un solo nucleótido resolución.
Hemos ilustrado la utilidad de este método en un estudio que distingue entre dos modelos propuestos para la capacidad del VIH-1 factor de la restricción APOBEC3G (Apolipoproteína B mRNA edición enzima catalítico polipéptido-como 3G) para interferir con la producción de transcripción reversa viral6.
Protocolo
Nota: Consulte la Tabla de materiales reactivos específicos y equipos utilizados en el presente Protocolo.
1. virus de la producción y la infección de células
PRECAUCIÓN: Infecciosas VIH-1 debe manipularse sólo en laboratorios de contención de bioseguridad aprobados.
Nota: La producción de partículas VIH-1 por transfección transitoria de riñón embrionario humano (HEK) 293T células, como se describe en paso 1.1, es un procedimiento estándar y ha sido descrito previamente15,16. Cultura general de la célula son procedimientos descrito anteriormente17.
- Producción del virus VIH-1.
- Mantener las células 293T de Dulbecco modificación medio de Eagle (DMEM) suplementado con 10% suero bovino fetal (FBS) y 1% penicilina/estreptomicina (DMEM completo) en una incubadora de cultura de célula estándar a 37 ° C y 5% CO2 como se describe anteriormente17.
- En una campana de flujo laminar estándar tejido cultura Retire el medio de crecimiento y añadir 3 mL de tripsina precalentado (37 ° C) a una placa de cultivo de célula 10 cm cerca del confluente (~1.2 x 107 células) de las células 293T. Volver a poner el plato en la incubadora durante 2-3 minutos.
- Llevar el plato de la incubadora en la campana de cultivo de tejidos y Añadir 7 mL de medio completo. Pipetee hacia arriba y hacia abajo en el plato varias veces para resuspender las células. Dividir las células 1:4 añadiendo 2.5 mL de la suspensión de células a un nuevo plato de 10 cm y rellenarlo con 7,5 mL de medio completo.
- Al día siguiente, 10 μg de plásmido de HIV-1 proviral DNA (como pNL4.3) de la mezcla con 1 mL de medio esencial mínimo libre de suero y agregar solución de polietilenimina (PEI) (25.000 mW, 1 mg/mL pH 7) en 4.5 μL 1 μg de ADN. Incubar por 10 min a temperatura ambiente y añadir gota a gota a las células 293T.
- 24 h después de la transfección, quitarlo del medio y reemplazarlo con 6 mL de DMEM completo que contengan libre de Rnasa DNasa en medio 20 de U/mL. Después de 6 h, sustituir el medio con 10 mL de DMEM completo.
- 48 h después de la transfección, coseche el sobrenadante y filtrar con un filtro de 0,22 μm, usando una jeringa de 10 mL en un tubo de polipropileno de 15 mL.
- Añadir 2 mL de sacarosa al 20% estéril de 1 x de tampón fosfato salino (PBS) a un tubo abierto de paredes delgadas ultracentrífuga. Lentamente el recubrimiento la sacarosa con el sobrenadante filtrado de la célula.
- Centrifugar durante 1 h 15 min a 134.000 x g a 4 ° C utilizando una ultracentrífuga.
- Retirar cuidadosamente los tubos de la ultracentrífuga. Poco a poco sacar el sobrenadante y sacarosa con una aspiración o una pipeta. Utilice una pipeta más pequeño y el tubo de la inclinación al sacar la última solución de sacarosa. Deje el virus sedimentado en el fondo del tubo.
Nota: La pastilla no será visible. - Añadir 200 μL de PBS 1 x, dejar en la nevera de 4 a 12 h, suspender y congelar en alícuotas de μl 20 a-80 ° C.
- Determinar p24Gag contenido utilizando la un antígeno p24 de VIH-1 ELISA kit (siguientes instrucciones).
- Infección de la línea del T-cell.
- Una línea de células T inmortalizada de la cultura (por ej., células CEM-SS) en medio del Roswell Park Memorial Institute (RPMI) 1640 suplementado con 10% FBS y 1% penicilina/estreptomicina (RPMI completo). Contar las células usando un hemocitómetro18 y semilla 1 bien por la muestra con 1 mL de RPMI completo con 2 x 106 células/mL en una placa de cultivo celular de formato 12-bien.
- Añadir partículas de VIH-1 equivalente a 150 ng p24Gag y coloque el plato en un balanceo centrífuga cubo con tapas de biocontención para infectar a la vuelta por centrifugación en una centrífuga de sobremesa para 2 h a 2.000 x g a 30 ° C.
- Extraiga la placa de la centrifugadora y se deja reposar durante 1 hora en una incubadora de cultivo de tejidos estándar a 37 ° C y 5% CO2.
- Para limpiar virus de entrada, recogen las células por las suspensiones de células de transferencia a tubos de microcentrífuga y centrifugación en una microcentrífuga en RT (RT) a 500 x g durante 2 min. Sacar el sobrenadante sin perturbar el sedimento celulares.
- Resuspender el pellet celular en 1 mL de precalentado (37 ° C), estéril de 1 x PBS. Repetir la centrifugación, extracción sobrenadante y resuspensión pasos dos veces más.
- Centrifugar nuevamente, quite el sobrenadante y resuspender el pellet celular en 1 mL de RPMI completo. Añadir cada suspensión a un pozo en una nueva placa bien 12.
- En el post inicial además de 6 h del virus (centrifugación después de 4 h), cosechar las células por centrifugación como hecho en el paso 1.2.4. Retire y descarte el sobrenadante. Los pellets de células pueden ser congelados a-80 ° C o procesados directamente para la extracción de ADN.
2. ADN extracción, cuantificación de ADN de VIH-1 y enriquecimiento por captura de híbridos
- Extraer ADN de células enteras con un tejido y sangre total DNA kit de extracción, siguiendo el manual de kit de cultivo de tejidos las células. El único cambio es la elución en 200 μL de H libre de nucleasa2O en lugar del tampón de elución proporcionada.
Nota: Después de la adición del tampón de lisis caotrópico (del kit "AL buffer") y proteinasa, muestras pueden ser extraídas del laboratorio de contención de bioseguridad y manejadas en un laboratorio de nivel de seguridad estándar para el resto del protocolo. - Determinar el número de copia del cDNA de VIH-1 por qPCR.
- 17 μl de eluido en el paso 2.1 y añadir 2 μl de buffer de enzima de la restricción de x 10 con 1 μl de enzima de la restricción de DpnI. Incubar durante 1 h a 37 ° C para eliminar cualquier potencial residual entrada ADN plásmido de transfección.
- Realización de qPCR para filamento menos fuerte parada cDNA usando el siguiente conjunto de sonda de cartilla: oHC64 (5-taactagggaacccactgc-3 ') y oHC65 (5-gctagagattttccacactg-3 ') y sonda oHC66 (5′-FAM-acacaacagacgggcacacacta-TAMRA-3 '). configuración de qPCR y condiciones exactas pueden encontrarse en las referencias6,13. Llevar a lo largo de las muestras con una dilución seriada del plásmido proviral pNL4.3 como una curva estándar para determinar números de copia de moléculas de cDNA.
Nota: Vea la discusión para las cantidades esperadas.
- Enriquecimiento por captura de híbridos de ADN VIH-1.
Nota: De este paso, es preferible utilizar tubos de microcentrífuga con propiedades de enlace de baja ácidos nucleicos así como puntas de pipeta de filtro aerosol para todas las muestras de ADN. Si es posible, trabajar en una estación de trabajo PCR. Todos los pasos y los reactivos están en RT (RT) a menos que se indique lo contrario.- Para preparar una mezcla maestra de granos magnéticos estreptavidina, Pipetear 100 granos μl por muestra en un tubo de microcentrífuga sola. Coloque el tubo en un imán adecuado para tubos de microcentrífuga.
- Después de que los granos han colocado hacia el lado del imán del tubo (~ 1 min), sacar el buffer de almacenamiento, retire el tubo del imán y suspender granos en 500 μl de bind y lavado tampón (buffer BW, 5 mM Tris-HCL pH 7,5, EDTA 0,5 mM 1 M NaCl) para lavar.
- Coloque el tubo en imán, quite el sobrenadante y agregar 500 μl solución de caseína. Tomar del imán, resuspender e incubar por 10 min a temperatura ambiente, luego lavar con tampón de BW.
Nota: Un lavado se refiere a colocar el tubo en el imán, sacar el sobrenadante, teniendo el tubo el imán, agregando el almacenador intermediario y resuspender. - Coloque el tubo en el imán, sacar el sobrenadante y resuspender los granos en 500 μl de tampón de BW. Añadir 50 pmol de cada captura oligonucleótidos biotinilados (ver tabla 1, tres de oligos en este caso) por muestra. (Por ejemplo, si 5 muestras de ADN a procesar, utilizar 500 μl de granos magnéticos de paso 2.3.1 y 250 pmol de cada oligonucleótido).
- Incubar durante 30 min a temperatura ambiente mientras que la oscilación en un mezclador de extremo a extremo.
- Lavar los granos con los oligonucleótidos inmovilizados dos veces con 500 μl de tampón diez de 1 x (10 mM Tris HCl pH 8.0, 1 mM EDTA, 100 mM NaCl).
- Resuspender los granos en 10 μl de 1 Tampón 10 x por muestra.
- Para cada muestra, etiquetar un tubo de microcentrífuga y añadir 10 μl de suspensión de perlas, 170 μl de ADN (del paso 2.1) y 90 μl de 3 x 10 buffer. Incubar en un bloque de calor seco a 92 ° C por 2 min desnaturalizar el ADN.
- Hacia los tubos de un bloque de calor seco diferentes, que se encuentra a 52 ° C, e incubar durante 1 h. invertir para mezclar regularmente (~ cada 10 minutos) durante la incubación.
- Una vez lavado con 500 μl de 1 x buffer diez y resuspender en 35 μl libre de nucleasa H2O.
- Para eluir, incubar los tubos a 92 ° C en un bloque de calor seco por 2 min. A continuación, mover rápidamente los tubos sobre el imán (un tubo a la vez). Una vez que los granos están limitados al lado del tubo, transferir el sobrenadante que contiene el ADN del VIH-1 a un tubo nuevo.
- Opcional: Repita qPCR (como hace en el paso 2.2.2) para determinar el cDNA del VIH-1 recuperado.
Nota: Vea la discusión para las cantidades esperadas.
3. adaptador ligadura
- Preparando el adaptador
- Resuspender adaptador liofilizado (ver tabla 1 "completo Kwok + MiSeq") a 100 μm en libre de nucleasa H2O.
- Por ejemplo más muestra un control, combinar 0.45 μl de 10 x buffer ligasa de T4 ADN, 4 μL de adaptador y 0.05 μl de H libre de nucleasas cada2O. calor a 92 ° C por 2 min y dejar enfriarán lentamente.
Nota: Si la opción está disponible, use una máquina PCR con velocidad de enfriamiento ajustable (tasa de 2% de uso). Esto toma unos 30 min de 92 ° C a 16 ° C. Alternativamente, use un bloque de calor seco a 92 ° C y apagar. Saque el adaptador mastermix cuando el bloque de calor es volver a TA. Esto es dejar el adaptador forma una estructura de horquilla (ver figura 2b).
- Preparar una reacción de control con una serie de oligonucleótidos sintetizados (ver tabla 2) en lugar de ADN extraído de células.
- Realizar acciones de 100 μm de cada oligonucleótido. Mezclar 1 μl de cada uno de los 17 oligonucelotides y 8 μl de H2O para una relación equimolar en un volumen final de 25 μl.
- Diluir la mezcla 1:2,500 en libre de nucleasa H2O en una dilución seriada. 1 μl de la mezcla se combinan con 17.3 μl de H libre de nucleasa2O usar en la ligadura de la muestra de control en el paso 3.3.1 para que cada oligonucleótido está presente en el 1,6 fmol (equivalente a 0.026 nM en la reacción de 60 μL).
- Configuración de trompas
- Para reacciones de volumen final de 60 μL en tubos de PCR 6 μl de 10 x T4 ADN ligasa del almacenador intermediario, 24 μl de 40% PEG, 6 μl de 5 M betaína, μl 4.5 (400 pmol) de adaptador (pre-annelead como en el paso 3.1.2), 1,2 μl de T4 ADN ligasa (2.000.000 unidades/mL) y 18.3 μl de ADN (a partir de Paso 2.3.11)
Nota: Tenga especial cuidado con soluciones viscosas como 40% PEG para mantener volúmenes exactos. No hacen un mastermix. - Establecer la misma reacción ya que se realiza en el paso 3.3.1 pero con la mezcla de oligonucleótidos de control preparado en el paso 3.2.2.
- Mezclar bien las reacciones e incubar en una máquina PCR en 16 ° C durante la noche.
- Para reacciones de volumen final de 60 μL en tubos de PCR 6 μl de 10 x T4 ADN ligasa del almacenador intermediario, 24 μl de 40% PEG, 6 μl de 5 M betaína, μl 4.5 (400 pmol) de adaptador (pre-annelead como en el paso 3.1.2), 1,2 μl de T4 ADN ligasa (2.000.000 unidades/mL) y 18.3 μl de ADN (a partir de Paso 2.3.11)
4. adaptador extracción y separación de tamaño
- Desnaturalizando electroforesis del gel
- Añadir 30 μl de formamida que contiene gel de ADN cargando buffer a cada reacción de ligadura. Mezclar por pipeteo.
- Calentar por 2 min a 94 ° C en la máquina de la polimerización en cadena y colocar inmediatamente en hielo.
- Coloque un prefabricado 6% borato/Tris/EDTA (TBE) desnaturalización urea gel de poliacrilamida (peine de 10 pozos) en un depósito de gel apropiado. Añadir 1 buffer de corriente x TBE (89 mM Tris base, el ácido bórico 89 mM, EDTA 2 mM) y funcionamiento previo el gel por 20 min en constante V/max 250.
- Lavar las bolsas de gel con el funcionamiento de búfer mediante una jeringa y una aguja de 21G.
- Carga cada 90 μl de la muestra en tres pocillos (30 μL por pocillo) y correr durante 20 min (250 V/max) hasta el frente del tinte azul oscuro es sobre a medio camino a través del gel.
- Tinción y cortar los ácidos nucleicos del gel
- Preparar 3 tubos de microcentrífuga pequeña (0.5 mL) por ejemplo empujando los agujeros en la parte inferior utilizando una aguja de jeringa de 21 G (tenga cuidado al trabajar con objetos punzantes). Introduzca cada uno de los tubos preparados en un tubo de microcentrífuga de 2.0 mL y etiquetarlos con el nombre de la muestra más "bajo", "mediados" o "high".
- Sacar y abrir el cartucho de gel. Corte el gel verticalmente con una cuchilla de afeitar para generosamente la tira con 3 pozos de muestras cargados. Añadir la tira de gel a un recipiente con 1 x TBE (aproximadamente 30 mL) y 5 μl de tintura de Cianina ácidos nucleicos. Incubar durante 3-5 minutos.
Nota: El paso de extracción de gel es particularmente sensible a la contaminación cruzada. Es recomendable ejecutar sólo 1 ejemplo por gel y utilizando un recipiente separado, limpio para cada tinción del gel. Guantes deben cambiarse si las partículas de gel en contacto con los dedos enguantados. - Limpie la superficie de un transiluminador de luz azul con ddH2O. Tomar la pieza de gel en el recipiente de tinción y añadir a la caja de luz.
- Encender la caja de luz e inspeccione los ácidos nucleicos manchados a través del filtro naranja.
Nota: El adaptador normalmente aparece sobrecargado y funciona como gran "blob" ligados ADN VIH-1 que como un rayo. - Usando una hoja de afeitar nueva, corte los lados del gel si hay áreas con ninguna muestra cargado todavía presente. A continuación, corte apenas sobre el adaptador para quitar el adaptador y gel inferior partes. Finalmente, corte la parte superior del gel, incluyendo alrededor de 1 mm del gel de bolsillos, que a menudo tienen una señal intensa aguda de mayor peso molecular DNA.
- Dividir el pedazo restante de la gel que contiene la muestra, que es típicamente ~ 2 x 3 cm de tamaño, horizontalmente en tres trozos incluso: "baja", "mid" y "alto" peso molecular.
Nota: Cada pieza ahora se manejarán por separado [es decir., habrá tres tubos (bajo, medio y alto)] por la muestra original. - Cada uno de los fragmentos de tres gel cortadas en trozos más pequeños (~ 2 x 2 partículas mm) y transferirlas a los tubos de microcentrífuga de preparado 0,5 mL (paso 4.2.1).
- Vuelta a la máxima velocidad con las tapas abiertas para 1 minuto exprimir las piezas de gel a través del orificio en el tubo de 2 mL para crear un gel de aguanieve. Si las partículas de gel permanecen en la parte inferior del tubo de 0.5 mL, transferir al tubo 2 mL manualmente usando una punta de aguja o pipeta.
- Extracción de ADN
- Añadir 1 mL de tampón de extracción de gel de urea (0,5 M NH4CH3CO2, 1 mM EDTA, 0.2% SDS) a los granizados de gel. Gire los tubos para un mínimo de 3 h (durante la noche es aceptable) a temperatura ambiente con una batidora de extremo a extremo.
- Utilizar un sistema limpio de pinzas para agregar un filtro de fibra de vidrio redondo a columnas de centrífuga con filtros de membrana de acetato de celulosa (0.2 μm), que evita la obstrucción de membrana. Coloque el filtro en su lugar con una punta de pipeta invertida.
- Brevemente girar los tubos de 2 mL con tampón de aguanieve y extracción de gel en una microcentrífuga y transferir 700 μl del sobrenadante a las columnas de filtro preparado. Mantenga el gel aguanieve y el sobrenadante restante.
- Centrifugue las columnas de filtro en una microcentrífuga a la máxima velocidad durante 1 minuto transferencia el flujo en un tubo nuevo de microcentrífuga de 2.0 mL.
- Volver a cargar las columnas con el sobrenadante restante. Trate de obtener tanto líquido como sea posible de la extracción del aguanieve. Transferencia de piezas de gel no es un motivo de preocupación. Vuelta otra vez y flowthroughs de las mismas muestras de extracción se combinan.
- Precipitación del ADN
- Agregar 3 μl de polyA RNA (1 μg/μl; como un portador), 1 μl de glucógeno y 0,7 mL de isopropanol para el flujo de paso 4.3.5. Mezclar brevemente y congelar a-80 ° C durante la noche.
- Tomar muestras del congelador de-80 ° C y dejarlos descongelar brevemente. Ponerlas en una microcentrífuga refrigerada (4 ° C) y vuelta por 30 min a velocidad máxima.
- Retire y descarte el sobrenadante. Ser muy cuidado de no remover el sedimento. Dejar 30 a 50 μl de líquido si es incierto ese diábolo se eliminaría en caso contrario.
Nota: Por lo general todas las muestras de "altas" muestran un pellet más visible que las muestras de "mediadas" y "bajas". - Añadir 800 μl de etanol al 80%. Invierta los tubos y vuelta otra vez durante 1 min a velocidad máxima. Eliminar la mayoría del etanol con una pipeta, girar los tubos otra vez brevemente y quitar más etanol con una pipeta de volumen más pequeño.
- Que cualquier resto etanol se evaporan mediante la colocación de los tubos con una tapa abierta en un bloque de 55 ° C calor seco. Cuando las muestras están secas (2-4 minutos) Añadir 20 μl de libre de nucleasa H2O y la extensión alrededor de la parte inferior del tubo para asegurar el pellet de DNA se disuelve. La muestra de ADN puede ser almacenada a-20 ° C.
5. PCR amplificación y preparación de la biblioteca
- Una reacción de PCR 40 μl con 20 μl de ADN polimerasa pre-mezcla, 18 μL del precipitado y se redisuelve ADN de paso 4.4.5, 1 μl del primer avance "MP1.0 + 22HIV" (10) (ver tabla 1) y 1 μl de primers multiplex oligo (cartillas de índice 1 a 24) (véase mesa de Ma materiales).
Nota: Ejecute que las tres reacciones (bajo, medio, alto) de cada muestra en reacciones de PCR independientes, pero con el mismo indexadas cartilla. Utilizar un índice diferente para cada una de las muestras de la infección original.- Ejecutar las reacciones de PCR bajo las siguientes condiciones: desnaturalización de 94 ° C 2 min, luego 18 ciclos de PCR de 3 pasos; 15 s en desnaturalización de 94 ° C, 15 s de recocido a 55 ° C y extensión de s 30 a 68 ° C.
- Como una opción de control de calidad, analizar las reacciones de PCR con el sistema de electroforesis de gel de alta sensibilidad automatizada. Tomar 2 μl de una muestra de baja, media y alta para ejecutar según instrucciones del fabricante.
Nota: Los dos cebadores deben ser visible y a menudo funcionamiento en una longitud calculada de sobre 45 y 95 nt (diferencia de longitud real). Además, se debe detectar ADN entre 150 a 500 nt. Si no hay señal, es aconsejable añadir ciclos adicionales de PCR, entre 2 y 10 ciclos adicionales. No añadir ciclos adicionales para las muestras de control de oligonucleótidos creados en el punto 3.3.2. - Para quitar los iniciadores utilizan un sistema de limpieza basado en grano paramagnético PCR.
- Tomar 20 μl de cada reacción de PCR y piscina junto las muestras (mezclar todas las muestras en este punto). Congelar las reacciones de μl 20 restantes como copias de seguridad a-20 ° C.
- Deja las bolas paramagnéticas llegue a RT y las reacciones de PCR combinadas con 1,8 x el volumen de la solución de grano de la mezcla. Mezclar mediante pipeteo e incubar durante 5 minutos.
Nota: Como ejemplo, si 4 muestras se prepararon y cada uno tiene reacciones bajas, media y altas, el volumen sería de 4 x 3 x 20 μl = reacciones de PCR de 240 μl con una solución de grano 432 μl. - Poner los tubos en un imán de tubo de microcentrífuga, dejar que los granos se unen para ~ 1 min y sacar el sobrenadante para descartar. Dejar los tubos en el imán y agregar 500 μl de etanol al 80%.
- Deja el etanol por 30 s, luego despegar cuidadosamente y dejar que los granos seca al aire para ~ 5 min agregar 40 μl de H libre de nucleasa2O, los tubos fuera del imán y pipetear arriba y abajo varias veces.
- Dejar la suspensión durante 5 minutos poner el tubo en el imán, dejó las cuentas colocan al lado y transferir el sobrenadante a un tubo nuevo. Esta es la biblioteca. Tomar 10 μl alícuota para controles de calidad y congelar el resto a-20 ° C.
6. evaluación de la biblioteca
- Determinar la calidad de la biblioteca, la concentración y la molaridad.
- Use un método de cuantificación fluorométrica. Medir 1 μl y 3 μl de la biblioteca con un kit de ensayo de dsDNA de alta sensibilidad según las instrucciones del fabricante.
Nota: Concentraciones típicas están entre 1 y 10 ng/μl. - Medida el espectro de peso molecular de ADN de biblioteca por alta sensibilidad automatizado de electroforesis en gel como se describió anteriormente (paso 5.2).
- Uso el gel análisis de la electroforesis para determinar el peso molecular promedio de la biblioteca y calcular para diluir la biblioteca libre de nucleasa H2O a 4 nM. Varias bibliotecas se pueden combinar como todos los índices son únicos.
- Use un método de cuantificación fluorométrica. Medir 1 μl y 3 μl de la biblioteca con un kit de ensayo de dsDNA de alta sensibilidad según las instrucciones del fabricante.
- Control de calidad de bajo rendimiento opcional
- Perjuicio de la biblioteca de ADN TA19 de clonación para insertar las moléculas biblioteca en vectores para la amplificación. Siga las instrucciones del kit, crecen hacia fuera de ~ 10-20 colonias y extracto de ADN mediante miniprep protocolos, tal como se describe aquí20.
- Vector de secuencia usando los servicios locales de la secuencia y comprobar que los insertos contienen el VIH-1 deseado deriva secuencias y adaptadores específicos de biblioteca.
7. gestión de secuenciación de alto rendimiento de
- Crear una hoja de muestra de la secuencia con el software comercial con la plataforma de secuenciación.
- Indicar el juego de la secuencia seleccionada. Por lo general, elija un kit de 150 ciclos, pero otros son adecuados dependiendo de la longitud que desee leer.
- Seleccione "Sólo Fastq" como el flujo de trabajo de aplicación. Elija una de las plantillas que contiene los 24 índices presentes en el kit de multiplex de oligonucleótidos (indicado en el manual del kit).
- Seleccione "25 nt" para Read1 y "125 nt" de Read2. Mantener 6 nt índice solo leer.
Nota: En el análisis interno Read2 sólo se utiliza en el análisis. Mantener Read1 a un mínimo de 25 nt para propósitos de plataforma algoritmo de secuenciación.
- Siga las instrucciones del fabricante, precisamente para la preparación de la biblioteca pre-funcionamiento y configuración. Optar por el máximo 20 pM concentración y uso una 15% PhiX espiga, como la biblioteca es de muy baja complejidad.
8. Análisis de datos
- Compruebe si el pass filtro porcentaje y puntaje de calidad de Q30 es aceptables según las instrucciones del fabricante de la plataforma de secuenciación.
Nota: Filtro de paso suele ser > 90% y Q30 puntuaciones suelen ser > 80%. - Descargar el. fastq.gz los archivos del centro de secuenciación del fabricante.
- Configurar la secuencia de comandos de la secuencia
- Crear un nuevo directorio (carpeta) llamado "AnalysisXYZ" y vaya a https://github.com/malimlab/seqparse para descargar los archivos de código fuente (parse_sam.pl, rc_extract.pl, parse.sh) en este directorio.
- Descargar el alineador de lectura corto Bowtie, versión 1.1.2, de http://bowtie-bio.sourceforge.net/index.shtml en el mismo directorio.
- La descarga crea un subdirectorio dentro de "AnalysisXYZ" llamado "Bowtie-1.1.2". Dentro de este directorio subdirectorio "índices" de abrir y descargar las secuencias de plantilla provista de 6 archivos con extensiones .ebwt.
- Descargar las lecturas cortas FASTQ/A preproceso toolkit fastx-0.0.13 de http://hannonlab.cshl.edu/fastx_toolkit/download.html en el directorio "AnalysisXYZ".
- Descargar Samtools (https://sourceforge.net/projects/samtools/files/) y bam-readcount (https://github.com/genome/bam-readcount) en el directorio "Documentos".
- Mover el. fastq.gz archivos, descargados en el paso 8.2, de todo leen 2s (terminando en... _R2_001.fastq.gz) en el directorio "AnalysisXYZ".
- Abra la consola/terminal de comandos. Hacia el "AnalysisXYZ" como el directorio actual mediante los comandos cd. Tipo "./parse.sh." para ejecutar las secuencias de comandos.
- Encontrar los archivos .csv con resúmenes de todas las muestras en total leerlas cuentas, longitud ajustada Lee cuentas y normaliza cuentas leerlas, así como archivos con la variación de la base, para cada muestra en un directorio llamado parse_results dentro del directorio "Análisis XYZ".
Nota: Vea la discusión para más información sobre el proceso de análisis. La secuencia de comandos devuelve archivos csv con Lee total para cada nucleótido a lo largo de las secuencias de HIV-1NL4.3 fuerte parada y primer filamento transferencia hasta el nucleótido 635. Como una guía, Lee única de 50.000 a 100.000 típicamente se observa en muestras de infecciones con el número de celular indicado y el inóculo viral y sin proteínas antivirales o compuestos. La muestra de control de oligonucleótidos generalmente produce lecturas de 100.000 a 200.000.
Resultados
La técnica descrita en este artículo se aplica a un estudio más amplio para abordar los mecanismos subyacentes a la inhibición de la transcripción inversa de VIH-1 por la proteína humana de antirretrovirales APOBEC3G (A3G)6. La figura 3 muestra resultados representativos obtenidos después de emplear el protocolo en las muestras de CEM-SS-linfocitos T infectados con vif-deficiente VIH-1 en la ausencia o presencia de A3G. El número total de Lee único obtenidos de cada muestra después de filtrar cualquier duplicados PCR que tienen el mismo 6 nt código de barras y la misma longitud (realizada por el software de análisis proporcionado) se grafican en la figura 3un. Niveles crecientes de A3G reducen el número total de leer lo que refleja el efecto inhibitorio de A3G en la síntesis de cDNA RT mediado anteriormente descrito y medido por qPCR6,13,21,22. En la figura 3b, se muestran la fracción de moléculas en cada longitud posible dentro de los primeros 182 nt. Para infección por VIH-1 en la ausencia de A3G, la especie más abundante es el principal 180 nt fuerte parada molécula sí mismo, con alguna acumulación de lecturas en el rango más corto (23 a 40 nt) (top histogramas gráfico, azul). La adición de cambios A3G este perfil como un fuerte aumento del corto, truncado moléculas de cDNA en unos puestos muy específicos y reproducibles es detectado (gráficos de medios y bajos). A3G es una desaminasa citidina, citosina a uridina (identificado como C-a-T) mutaciones en el cDNA ocurren cuando A3G está presente en el infeccioso virions21,23,24. Con la información obtenida de la secuencia, el porcentaje de mutaciones de C-a-T fue trazado en el mismo gráfico (línea roja de puntos). Cabe señalar que el perfil mutacional se deriva de Lee todo único combinado y cobertura de cada nucleótido variará. Sin embargo, si se requiere información de secuencia puede ser relacionada a cada molécula y correlacionada con una específica 3'-terminal. Los datos fueron tomados de Pollpeter et al. 6 y la correlación entre mutacional y perfiles de longitud de cDNA fue demostrado para ser debido a la detección y escote de cDNA deaminated por celular maquinaria de reparación del ADN.
Control positivo para el enfoque de la asignación de 3' se puede producir fácilmente mediante el procesamiento de un grupo de oligonucleótidos sintéticos de secuencia conocida, la longitud y la concentración. Este control se añade en la ligadura del adaptador en el paso 3.3.2 y aconsejado para ser incluidos en todas las bibliotecas de multiplexado. Datos obtenidos de una muestra de control deben tener todos los oligonucleótidos en los cocientes de entrada previstos, con lecturas de fondo muy pequeñas. La figura 4 muestra los resultados de un sistema de control positivo de 17 oligonucleótidos sintetizados químicamente (secuencias, véase tabla 2), que se mezclaron en proporciones equimolares. Como era de esperarse, todas las moléculas aparecen en cerca de abundancia igual con sólo pequeñas variaciones (gráfico superior). Mientras que la mayoría de los puestos dentro de la secuencia de ADN - sss que no estaban representada por un oligonucleótido volver cero cuentas leerlas, observamos especies menores que son 1 o 2 nt más cortos que los oligonucleótidos de control real. No investigaron más estas especies menores, pero asumir que representan productos degradados o incompletos potencialmente presentes en las poblaciones de oligonucleótidos proporcionado en la compra (oligonucleótidos fueron ordenados como HPLC purificados, para que el fabricante indica > 80% de pureza). El gráfico abajo muestra la muestra de control de una biblioteca diferente de correr, donde la variación es ligeramente superior entre los 17 oligonucleótidos y se correlaciona con la longitud total en que más moléculas de control se detectan más eficientemente y más cortos. Esto puede deberse a un sesgo menor en reacciones de PCR o clustering durante la secuencia de MiSeq, que tiene un grado óptimo de tamaño de inserto y puede ocurrir con las bibliotecas llevan inserto particularmente amplias gamas. Una forma básica para abordar este sesgo es la aplicación de un factor de normalización basado en la pendiente que indica el sesgo de la correlación a la longitud de la molécula (línea rosa). Los cálculos necesarios se incluyen en el programa de análisis (ver paso 8.3 en el protocolo).

Figura 1: diagrama que muestra los primeros pasos del VIH-1 de transcripción reversa. El proceso comienza con la de recocido de tRNA(Lys,3) (naranja) para el sitio de unión del cebador (PBS) en RNA genómico viral (paso 1), que permite la iniciación y elongación del cDNA viral (azul, paso 2). Concomitante, el plantilla de ARN genómico se degrada por actividad RNaseH de RT (paso 3). El primer intermedio completo en el proceso de transcripción inversa es el filamento negativo fuerte parada (-) sss cDNA, que es completa cuando la polimerización catalizada RT alcanza 5'-terminal de la región (R) repetición de gRNA (paso 3). El sss (-) intermedias es transferido a la terminal 3' de la plantilla de RNA genómica por recocido a la región de repetición de largo 3' terminal complementaria (LTR) R. Desde aquí, la polimerización continúa (paso 4). En el método descrito la progresión de transcripción inversa se determina mediante la asignación de la longitud exacta del cDNA viral naciente (azul). PPT, tracto polypurine; U5, 5'-secuencia única; U3, 3'-secuencia única. Esta cifra se vuelve a publicar de una anterior publicación6. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2 : Esquema de flujo de trabajo y esquemas de la estrategia de amplificación de PCR y ligadura adaptador. (a) flujo de trabajo exponiendo los principales pasos de la técnica descrita para determinar 3'-termini de HIV-1 reverse transcritos en células infectadas. La figura es una adaptación de una anterior publicación6. (b) esquema de la estrategia de amplificación PCR y ligadura del adaptador. Moléculas de cDNA naciente de longitud variable que han sido purificadas en pasos anteriores se unen a un adaptador de ADN monocatenario con T4 ADN ligasa. El adaptador de la horquilla (llamado "full Kwok + MiSeq", ver tabla 1) el diseño fue inspirado por Kwok et al. 11. el adaptador lleva al azar 6 secuencia de código de barras de nt que permite la sincronización de base para facilitar la ligadura y al mismo tiempo sirve como un identificador para lecturas únicas. 3'-termini del adaptador lleva un espaciador (SpC3) para prevenir la uno mismo-ligadura. Ligados productos se separan del adaptador exceso desnaturalizando electroforesis del gel de poliacrilamida (PAGE). Los ácidos nucleicos en gel teñidos y cortados en tres piezas separadas, igual tamaño gel en la zona desde el adaptador al pozo como hecho en25. Después de elución, precipitación y resuspensión, los productos son PCR amplificado con iniciadores de recocido a la conocida secuencia del adaptador (cartilla 1, kit multiplex de oligonucleótidos, véase Tabla de materiales) y una cartilla llevando el primer 22 nt del VIH-1 Secuencia 5'-LTR inmediatamente después el tRNA (cartilla 2, MP1.0 + 22HIV). El termini 5' de los cebadores solicitados lleva adaptadores para la plataforma de secuenciación solicitadas (P5 y P7) así como una secuencia de índice para distinguir muestras individuales en la misma biblioteca. Puntos de partida de la secuencia leer cartillas están indicadas. El cuadro azul indica la región de interés para determinar el original 3'-termini de la molécula de capturados. Esta figura es una adaptación de una anterior publicación6. Haga clic aquí para ver una versión más grande de esta figura.
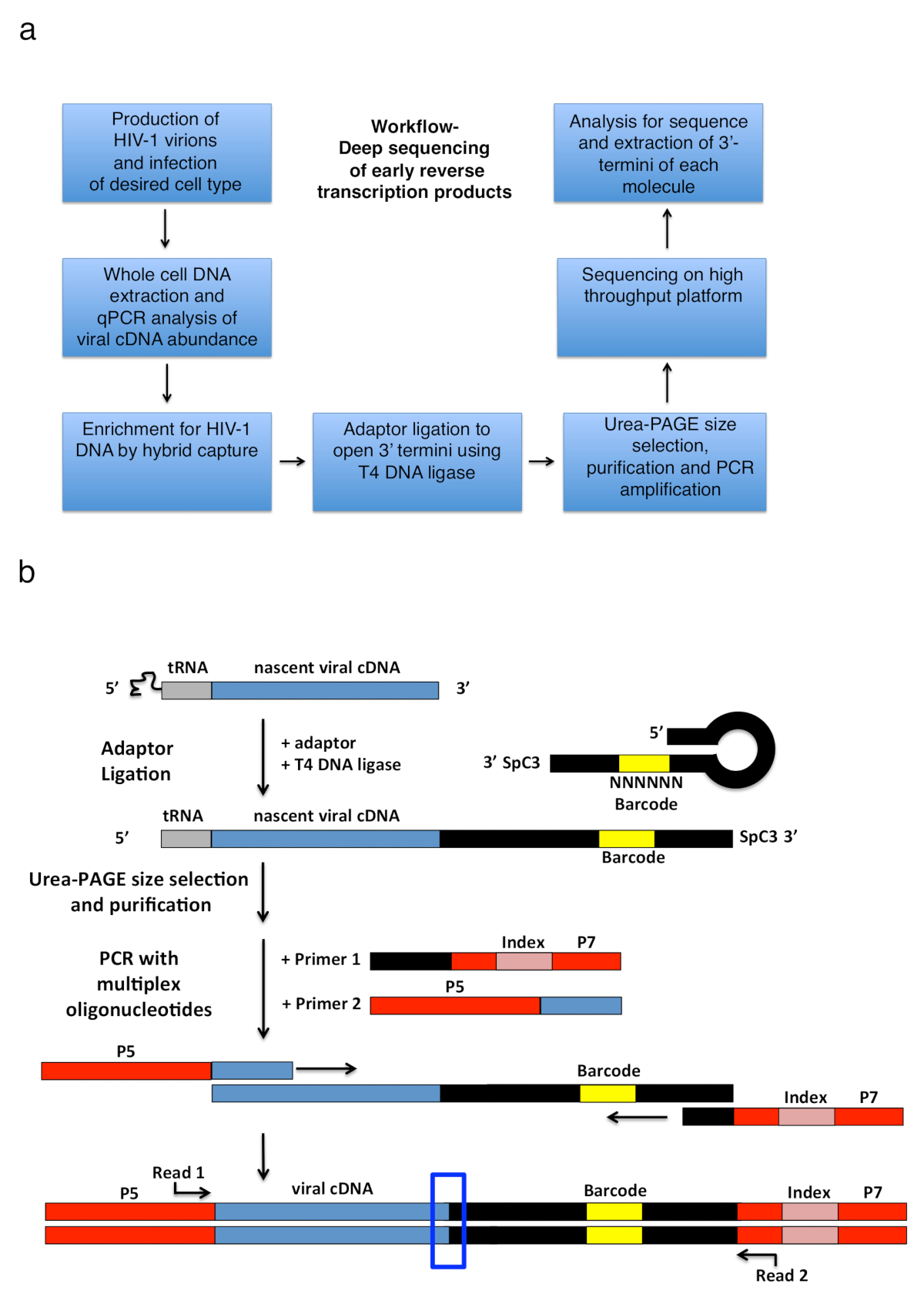{kind=link}

Figura 3 : Resultados representativos. (a) leer recuento total de muestras representativas con el protocolo descrito. Esto incluye todas las secuencias que fueron identificadas como Lee única de moléculas de VIH-1 con sus 3'-termini dentro de la primera 635 nt de menos strand cDNA (hasta el PPT, ver figura 1). Infección con el VIH-1 no llevar A3G rinde el mayor número de lecturas, mientras que A3G inhibe la síntesis de cDNA y así reduce el total cuenta de leer. Las células no infectadas sirven como un control negativo, mientras que una serie de oligonucleótidos sintéticos proporciona un control positivo. b) la abundancia relativa de cADN para cada longitud entre nt 23 y 182 (cDNA de larga duración - sss es 180 a 182 nt) de los VIH-1NL4.3 secuencia (eje x) se muestra en azul histogramas (escala en el eje izquierdo). Se calculó la abundancia relativa de cDNA del número absoluto de secuencias de terminación en un nucleótido determinado dentro de la secuencia de cDNA - sss dividido por la suma de todas las lecturas de medición 182nt o menos. En líneas rojas discontinuas muestran los porcentajes de lecturas con las mutaciones de C-a-T/U en la posición correspondiente (escala en los ejes de derecho y–). Figura 3 b se vuelve a publicar de una anterior publicación6. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 4 : Resultados representativos de las muestras control. Se muestran dos perfiles para piscinas que contienen cantidades equimolares de oligonucleótidos sintéticos de diversa longitud 17. Estos oligonucleótidos tienen secuencias de HIV-1NL4.3 y fueron seleccionados para cubrir varias longitudes y 4 todas las bases se presentan como un 3'-nucleótido (ver tabla 2). El gráfico superior muestra el control positivo de la figura 3un. No se detecta ningún sesgo significativo hacia la longitud de la molécula o la abierta 3'-termini. El gráfico inferior muestra una otra biblioteca, que produce un sesgo de menor longitud de la secuencia. En este caso, es aconsejable aplicar un factor de normalización, que se deriva de la cuesta (se muestra en color rosa) que representa el sesgo de tamaño. Esta cifra se vuelve a publicar de una anterior publicación6. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
| Nombre de OLIGO | Longitud en nt | Secuencia de | Propósito | Fabricante (purificación) | ||||||||||||||
| completo Kwok + MiSeq | 61 | 5'-PHO-tgaagagcctagtcgctgttcannnnnnctgcccatagagagatcggaagagcacacgtct-SpC3-3' | Adaptador | Tecnologías del ADN de la IDT (HPLC) | ||||||||||||||
| 2xBiotin SS cebo | 40 | 5'-biotina-cagtgtggaaaatctctagcagtggcgcccgaacagggac-biotina-3' | Captura de híbridos | Eurofins MWG (HPLC) | ||||||||||||||
| Ss de biotina 1-16 | 22 | 5'-cagtgtggaaaatctctagcag-BiTEG-3' | Captura de híbridos | Eurofins MWG (HPLC | ||||||||||||||
| TRNA de biotina + CTG | 16 | 5'-cagtggcgcccgaaca-BITEG-3' | Captura de híbridos | Eurofins MWG (HPLC) | ||||||||||||||
| MP1.0 + 22HIV | 82 | 5'-aatgatacggcgaccaccgagatctacactctttccctacacgacgctcttccgatctcactgctagagattttccacactg-3' | Amplificación por PCR | Eurofins MWG (HPLC | ||||||||||||||
Tabla 1: tabla de oligonucleótidos incluyendo longitud, secuencias y las modificaciones que se utilizan en el protocolo descrito. La tabla está adaptada de una anterior publicación6. Haga clic aquí para descargar esta tabla como un archivo de excel.
| Nombre de OLIGO | Longitud en nt | Secuencia de | Fabricante (purificación) | |||||||||||||
| HTP con mucho C | 120 | 5'-ctgctagagattttccacactgactaaaagggtctgagggatctctagttaccagagtcacacaacagacgggcacacactactttgagcactcaaggcaagctttattgaggcttaagc-3' | Eurofins MWG (HPLC) | |||||||||||||
| HTP con largo G | 119 | 5'-ctgctagagattttccacactgactaaaagggtctgagggatctctagttaccagagtcacacaacagacgggcacacactactttgagcactcaaggcaagctttattgaggcttaag-3' | Eurofins MWG (HPLC) | |||||||||||||
| HTP con largo T | 116 | 5'-ctgctagagattttccacactgactaaaagggtctgagggatctctagttaccagagtcacacaacagacgggcacacactactttgagcactcaaggcaagctttattgaggctt-3' | Eurofins MWG (HPLC) | |||||||||||||
| HTP con largo A | 118 | 5'-ctgctagagattttccacactgactaaaagggtctgagggatctctagttaccagagtcacacaacagacgggcacacactactttgagcactcaaggcaagctttattgaggcttaa-3' | Eurofins MWG (HPLC) | |||||||||||||
| Con HTP mediados C | 76 | 5'-ctgctagagattttccacactgactaaaagggtctgagggatctctagttaccagagtcacacaacagacgggcac-3' | Eurofins MWG (HPLC) | |||||||||||||
| Con HTP mediados G (a) | 71 | 5'-ctgctagagattttccacactgactaaaagggtctgagggatctctagttaccagagtcacacaacagacg-3' | Eurofins MWG (HPLC) | |||||||||||||
| Con HTP mediados G (b) | 72 | 5'-ctgctagagattttccacactgactaaaagggtctgagggatctctagttaccagagtcacacaacagacgg-3' | Eurofins MWG (HPLC) | |||||||||||||
| Con HTP mediados A | 69 | 5'-ctgctagagattttccacactgactaaaagggtctgagggatctctagttaccagagtcacacaacaga-3' | Eurofins MWG (HPLC) | |||||||||||||
| Con HTP mediados T | 85 | 5'-ctgctagagattttccacactgactaaaagggtctgagggatctctagttaccagagtcacacaacagacgggcacacactactt-3' | Eurofins MWG (HPLC) | |||||||||||||
| HTP con cortocircuito A | 40 | 5'-ctgctagagattttccacactgactaaaagggtctgaggga-3' | Eurofins MWG (HPLC) | |||||||||||||
| HTP con corto | 33 | 5'-ctgctagagattttccacactgactaaaagggt-3' | Eurofins MWG (HPLC) | |||||||||||||
| HTP con corto G | 41 | 5'-ctgctagagattttccacactgactaaaagggtctgaggg-3' | Eurofins MWG (HPLC) | |||||||||||||
| HTP con corta C | 34 | 5'-ctgctagagattttccacactgactaaaagggtc-3' | Eurofins MWG (HPLC) | |||||||||||||
| HTP Con 46 (T) | 46 | 5'-ctgctagagattttccacactg actaaaagggtctgagggatctct-3' | Eurofins MWG (HPLC) | |||||||||||||
| HTP Con 83 (C) | 83 | 5'-ctgctagagattttccacactg actaaaagggtctgagggatctctagttaccagagtcacacaacagacgggcacacactac-3' | Eurofins MWG (HPLC) | |||||||||||||
| HTP Con 103 (C) | 103 | 5'-ctgctagagattttccacactg actaaaagggtctgagggatctctagttaccagagtcacacaacagacgggcacacactactttgagcactcaaggcaagc-3' | Eurofins MWG (HPLC) | |||||||||||||
| Con HTP 107 (A) | 107 | 5'-ctgctagagattttccacactg actaaaagggtctgagggatctctagttaccagagtcacacaacagacgggcacacactactttgagcactcaaggcaagcttta-3' | Eurofins MWG (HPLC) | |||||||||||||
Tabla 2: tabla de 17 oligonucleótidos control sintético utilizado como muestra control positivo. Los oligonucleótidos top 13 fueron elegidos según el tamaño [largo (116 a 120 nt), medio (69 a 85 nt), corto (33 a 41 nt)] así como sus 3'-termini. La tabla está adaptada de una anterior publicación6. Haga clic aquí para descargar esta tabla como un archivo de excel.
Discusión
La disponibilidad de secuenciación profunda rápida, confiable y rentable ha revolucionado muchos aspectos en el campo de Ciencias de la vida, permitiendo a gran profundidad en los análisis de secuenciación. Un desafío restante radica en el diseño innovador y la creación del representante de bibliotecas de la secuencia. Aquí se describe un protocolo para capturar las moléculas de ADNc viral naciente, específicamente los intermedios del proceso de transcripción inversa de VIH-1.
El paso más crítico de esta estrategia es la ligadura de un adaptador para el open 3'-termini de manera cuantitativa y objetiva. Eficiencias de trompas entre dos termini de ssDNA, tanto inter- y intramolecular, investigaron y optimizado para diversas aplicaciones11,26,27,28,29. La opción de utilizar un adaptador de horquilla con T4 ADN ligasa en las condiciones descritas en el paso 3.3 es el resultado de la optimización empírica en la que se evaluaron diferentes ligasas, adaptadores y reactivos para la ligación de oligonucleótidos sintéticos que representan Secuencias de HIV-1 (tabla 2) (datos no mostrados). En estas reacciones de la prueba en vitro , confirmamos que el ligase de la DNA de T4 mediada por la ligadura del adaptador de la horquilla, según lo descrito por Kwok et al. 11, tiene un sesgo muy bajo y logra cerca ligadura completa de moléculas aceptoras cuando el adaptador se usa en exceso. La eficacia de la ligadura era inafectada por la adición de la secuencia de nucleótidos para representar el adaptador compatible para el sistema de primer multiplex (ver figura 4). En comparación, encontramos que un termoestable 5' ADN/ARN ligasa («Ligasa A», véase Tabla de materiales para exactos ligasas comparado aquí), que es una ingeniería ligasa de RNA que se desarrolló en parte para mejorar la eficacia de la ligadura con ssDNA como aceptor 27, fue más efectivo que ligan dos moléculas de ssDNA de ligasa de RNA "(B de la ligasa del) pero tenía un sesgo importante, con fuertes diferencias en la eficacia de la ligadura entre oligonucleótidos con diferencias de longitud de base individual [tabla 2 ; HTP con media G (a) y (b)]. Además, encontramos sólo un sesgo mínimo en reacciones con "Ligasa C" combinado con un adaptador que lleva al azar 5'-termini (una estrategia utilizada para compensar el sesgo de nucleótidos conocido "C ligasa"; véase por ejemplo Ding et al. 30). sin embargo, la "C de ligasa"-mediadas trompas intermoleculares estaban incompletos, haciendo el sistema de T4 ADN ligasa la opción superior.
Varias medidas de control de calidad sobre el curso de protocolo y la inclusión de controles positivos y negativos permiten para la detección de problemas potenciales antes de continuación de ensayo y proporcionan orientación para la solución de problemas de esfuerzos. Las cuantificaciones de qPCR en pasos 2.2.2 2.3.12 aseguran de que la cantidad de material de entrada es suficiente. Números de copia cDNA típica en el rango de elución (del paso 2.1) 200 μL de alrededor de 10.000 a 300.000 por μl. El paso de captura de híbrido puede resultar en una pérdida general de VIH-1 cantidad de cDNA, pero dará lugar a un fuerte enriquecimiento de cDNA específico del VIH-1 sobre el ADN celular, que puede determinarse mediante el uso de primers adecuados para cuantificar ADN genómico antes y después de enriquecimiento por qPCR o midiendo la concentración de DNA total. CDNA de VIH-1 recuperado después de que el híbrido captura pasos debe ser al menos el 10% de la entrada. Baja a partir de material de lo contrario puede explicar un control positivo de éxito del oligonucleótido (ver paso 3.3.2) pero limitado sólo lee en las muestras. Bajo Lee números general también podrían explicarse por la sobreestimación de la concentración de la biblioteca debido a la presencia de especies de ADN irrelevantes sin adaptadores MiSeq. Esto daría lugar a densidad baja del cluster y puede mejorarse mediante la determinación de la concentración de secuencias de HIV-1 en la biblioteca por qPCR además de la cantidad de ADN total por análisis fluorométrico. Debido a la naturaleza altamente sensible del método, se debe tener especial cuidado para evitar la contaminación incluso de bajo nivel, tanto de otras muestras (en particular, de las existencias de oligonucleótidos de alta concentración control) así como de equipos de laboratorio. Trabajando en la estación de trabajo PCR de esterilización UV es beneficioso en este sentido. La electroforesis automatizada de la biblioteca final (paso 6.1.2) es otra medida de control de calidad. El rango de tamaño de ácidos nucleicos observado por lo general es entre 150 a 500 nt. imprimaciones que pueden detectarse en el control opcional después de la polimerización en cadena y antes de purificación (ver nota en el paso 5.2) ahora esté ausente. En un resultado representativo, la curva de intensidad de la muestra tiene un pico alrededor de 160 a 170 nt y un segundo más agudo pico alrededor de 320 a 350 nt. Esto probablemente refleja la abundancia superior ve a menudo en la transcripción inversa de relativamente corto (longitud de inserción de 1 a 20 nt) y larga duración fuerte parada (longitud de inserción de 180 a 182 nt) (figura 3b).
Mientras que el protocolo presentado y las cartillas son específicas para construcciones de transcripción reversa de HIV-1 tempranas, el método es generalmente aplicable a cualquier estudio con el objetivo de determinar abierto 3' termini de ADN. Las principales modificaciones en otros contextos será el método de captura híbrida y la estrategia de diseño de la cartilla. Por ejemplo, si el objetivo es adaptarse a las transcripciones finales de VIH-1, un mayor número de diferentes oligonucleótidos biotinilados captura recocido a lo largo del cDNA sería recomendable y probablemente disminuirá la pérdida en el paso de captura de híbridos. Como se mencionó en la introducción, es importante considerar las limitaciones en el diseño de la gama que 3'-termini está a detectar para evitar diversas fuentes de sesgo. En primer lugar, puede haber un sesgo en las reacciones de polimerización en cadena si las plantillas con el adaptador son de longitud muy variable. En segundo lugar, la plataforma la secuencia utilizada aquí (por ejemplo, MiSeq) tiene un rango de longitud de inserción preferido para el agrupamiento óptimo y significativamente productos más cortos y más largo no pueden ser ordenados con la misma eficiencia. En parte, esto puede abordarse computacionalmente, como se hizo mediante el cálculo de un factor de corrección por sesgo de longitud (ver figura 4, gráfico inferior). Sin embargo, si la región de donde se desea la 3'-termini mapeo es larga (> 1000 nt), es más aconsejable dividir las reacciones con las transcripciones ligadas y usar múltiples cebadores aguas arriba para evaluar 3'-termini en secciones.
El programa de análisis fue escrito en la empresa con el propósito específico de analizar ambos el último nucleótido de la secuencia del VIH-1 adyacente a la secuencia del adaptador fijo, así como la variación de la base de todas las bases para identificar cualquier mutaciones. Los pasos individuales comprenden las siguientes: en primer lugar, las secuencias de adaptador se recortan utilizando el toolkit de fastx-0.0.13; Luego, se eliminan cualquier secuencia que está duplicado (es decir, idénticas secuencias incluyendo el código de barras). Todas las lecturas únicas restantes entonces se alinean con la secuencia del VIH-1 con Bowtie (http://bowtie-bio.sourceforge.net/index.shtml) el desajuste máximo en tres bases. La secuencia de la plantilla está conformada por la primera 635 nt del cDNA de VIH-1 (cepa NL4.3), que incluye la secuencia - sss y el primer producto de transferencia de filamento hasta la pista de polypurine (U5-R-U3-PPT; ver figura 1). Por lo tanto, el software suministrado y plantillas aptas sólo directamente si el método se utiliza para la misma aplicación (detección de tempranas transcripciones inversas de la de VIH-1NL4.3). Ajustes tendrá que hacer para otras secuencias diana. Las posiciones de la 3'-termini para cada lectura se determinaron por la posición en la alineación. Se registran llamadas base para cada posición y se calculan tasas de mutación de la cobertura total de cada base, que varía, como Lee es de diferentes longitudes e insertos largos podrían no estar cubiertos completamente por la secuencia de la base de 125 en Read2.
Para concluir, creemos que el método descrito para ser una herramienta valiosa para muchos tipos de estudios. Aplicaciones obvias incluyen las investigaciones de los mecanismos de inhibición de reversa de la transcripción a través de medicamentos antirretrovirales o factores de restricción celular. Sin embargo, sólo relativamente pequeños ajustes deben ser necesarios adaptar el sistema a 3'-termini mapeo dentro de otros intermediarios virales monocatenario DNA, que están presentes, por ejemplo, en la replicación del parvovirus. Además, el principio del método, particularmente su paso ligadura optimizado, puede proporcionar una parte fundamental del diseño de preparación de biblioteca para la caracterización de las extensiones de ADN 3', incluyendo elongaciones catalizadas por ADN de doble cadena celular polimerasas.
Divulgaciones
Los autores declaran que no tienen nada que revelar.
Agradecimientos
Los autores reconocen el apoyo de los miembros de la laboratorio Malim, Luis Apolonia, Jernej Ule y Rebecca Oakey. Los autores agradecen a Matt Arno en genómica del centro de Londres de la Universidad del rey y Debbie Hughes en la Universidad College Londres (UCL), Instituto de Neurología siguiente secuencia planta de generación, para obtener ayuda con la secuencia MiSeq se ejecuta. El trabajo fue apoyado por el Consejo de investigación médica de Reino Unido (G1000196 y M001199/MR/1 a M.M.), la Wellcome Trust (106223/14/Z/Z a M.M.), séptimo programa de la Comisión Europea marco (FP7/2007-2013) en convenio de subvención no. PIIF-GA-2012-329679 (a D.P.) y el Departamento de salud vía un institutos nacionales en Premio salud investigación integral centro de investigación biomédica de Guy y St. Thomas' NHS Foundation Trust en asociación con College Londres de rey y del rey College Hospital NHS Foundation Trust.
Materiales
| Name | Company | Catalog Number | Comments |
| 293T cells | ATCC | CRL-3216 | |
| Dulbecco's Modified Eagle's Medium | Gibco | 31966-021 | |
| Penicillin/Streptomycin | Gibco | 15150-122 | |
| Fetal Bovine Serum | Gibco | 10270-106 | |
| HeraCell Vios 250i CO2 Incubator | Thermo Scientific | 51030966 | |
| Laminar flow hood - CAS BioMAT2 | Wolflabs | CAS001-C2R-1800 | |
| 10mm TC-treated culture dish | Corning | 430167 | |
| TrypLE™ Express (1x), Stable Trypsin Replacement Enzyme | Gibco | 12605-010 | |
| OptiMEM® (Minimal Essential Medium) | Gibco | 31985-047 | |
| HIV-1 NL4-3 Infectious Molecular Clone (pNL4-3) | NIH Aids reagent program | 114 | |
| Polyethylenimine (PEI) - MW:25000 | PolySciences Inc | 23966-2 | dissolved at 1mg/ml and adjusted to pH7 |
| RQ1- Rnase free Dnase | Promega | M6101 | |
| Filter 0.22 μm | Triple Red Limited | FPE404025 | |
| 15 mL polypropylene tubes | Corning | CLS430791 | |
| Sucrose | Calbiochem | 573113 | |
| Phosphate Buffered Saline (1x) | Gibco | 14190-094 | |
| Ultracentrifuge tubes | Beckman Coulter | 344060 | |
| Ultracentrifuge | Sorval | WX Ultra Series | Th-641 Rotor |
| Alliance HIV-1 p24 antigen ELISA kit | Perkin Elmer | NEK050001KT | |
| CEM-SS cells | NIH Aids reagent program | 776 | |
| Roswell Park Memorial Institute Medium | Gibco | 31870-025 | |
| CoStar® TC treated multiple well plates | Corning | CLS3513-50EA | |
| Benchtop centrifuge: Heraus™ Multifuge™ X3 FR | Thermo Scientific | 75004536 | |
| TX-1000 Swinging Bucket Rotor | Thermo Scientific | 75003017 | |
| Microcentrifuge: 5424R | Eppendorf | 5404000060 | |
| Total DNA extraction kit (DNeasy Blood and Tissue kit) | Qiagen | 69504 | |
| Nuclease free H2O | Ambion | AM9937 | |
| Cutsmart buffer | New England Biolabs (part of DpnI enzyme) | R0176S | |
| DpnI restriction enzyme | New England Biolabs | R0176S | |
| Oligonucleotides for qPCR | MWG Eurofins | N/A | HPSF purification |
| TaqMan PCR Universal Mastermix | Thermo | 4304437 | |
| LoBind Eppendorf® tubes | Eppendorf | 30108078 | |
| Axygen™ aerosol filter pipette tips, 1000 μL | Fisher Scientific | TF-000-R-S | |
| Axygen™ aerosol filter pipette tips, 200 μL | Fisher Scientific | TF-200-R-S | |
| Axygen™ aerosol filter pipette tips, 20 μL | Fisher Scientific | TF-20-R-S | |
| Axygen™ aerosol filter pipette tips, 10 μL | Fisher Scientific | TF-10-R-S | |
| PCR clean hood | LabCaire | Model PCR-62 | |
| DynaMag™2-magnet | Thermo | 12321D | |
| Streptavidin MagneSphere® paramagnetic particles | Promega | Z5481 | |
| Casein | Thermo Scientific | 37582 | |
| End over end rotator, Revolver™ 360° | Labnet | H5600 | |
| Tris-Base | Fisher Scientific | BP152-5 | |
| Hydrochloric Acid | Sigma | H1758-100ML | |
| EDTA disodium salt dihydrate | Electran (VWR) | 443885J | |
| Sodium Chloride | Sigma | S3014 | |
| Dri-Block® Analog Block Heater | Techne | UY-36620-13 | |
| PCR tubes and domed caps | Thermo Scientific | AB0266 | |
| PCR machine | Eppendorf | Mastercycler® series | |
| T4 DNA ligase | New England Biolabs | M0202M | |
| 40% Polyethylene glycol solution (PEG) in H2O, MW: 8000 | Sigma | P1458-25ML | |
| Betaine solution, 5M | Sigma | B0300-1VL | |
| Gel loading buffer II (formamide buffer) | Thermo Scientific | AM8546G | |
| Precast 6% TBE urea gels | Invitrogen | EC6865BOX | |
| Mini cell electrophoresis system | Invitrogen, Novex | XCell SureLock™ | |
| Tris/Borate/EDTA solution (10x) | Fisher Scientific | 10031223 | |
| Needle 21 G x1 1/2 | VWR | 613-2022 | |
| SYBR Gold nucleic acid stain (10000x) | Life Technologies | S11494 | |
| Dark Reader DR46B transilluminator | Fisher Scientific | NC9800797 | |
| Ammonium acetate | Merck | 101116 | |
| SDS solution 20% (w/v) | Biorad | 161-0418 | |
| Centrifuge tube filter | Appleton Woods | BC591 | |
| Filter Glass Fibre Gf/D 10mm | Whatman (VWR) | 512-0427 | |
| polyadenylic acid (polyA) RNA | Sigma | 10108626001 | |
| Glycogen, molecular biology grade | Thermo Scientific | R0561 | |
| Isopropanol (2-propanol) | Fisher Scientific | 15809665 | |
| Ethanol, molecular biology grade | Fisher Scientific | 10041814 | |
| Accuprime™ Supermix I (DNA polymerase premix) | Life Technologies | 12342-010 | |
| NEBNext® Multiplex Oligo for Illumina (Index Primer Set 1 and 2) | New England Biolabs | E7335S; E7500S | |
| Tapestation D1000 Screentape High sensitivity | Agilent Technologies | 5067- 5584 | |
| Tapestation D1000 Reagents | Agilent Technologies | 5067- 5585 | |
| 2200 Tapestation - automated gel electrophoresis system | Agilent Technologies | G2965AA | |
| Agencourt® AMPure® beads XP | Beckman Coulter | A63880 | |
| Qubit™ dsDNA HS Assay Kit | Invitrogen | Q32851 | |
| Qubit™ 2.0 Fluorometer | Invitrogen | Q32866 | |
| Topo™ TA cloning Kit | Invitrogen | 450071 | |
| Sequencing platform: MiSeq System | Illumina | ||
| Experiment Manager (Sample sheet software) | Illumina | Note: Use TruSeq LT as a template | |
| Miseq™ Reagent kit V3 (150 cycle) | Illumina | MS-102-3001 | |
| Sequencing hub: Basespace | Illumina | https://basespace.illumina.com | |
| Ligase A: Thermostable 5’ App DNA/RNA ligase | NEB | M0319S | Not used in this protocol, but tested in optimization process with results described in the discussion. |
| Ligase B: T4 RNA ligase 1 | NEB | M0204 | Not used in this protocol, but tested in optimization process with results described in the discussion. |
| Ligase C: CircLigase | Epicentre | CL4111K | Not used in this protocol, but tested in optimization process with results described in the discussion. |
Referencias
- Herschhorn, A., Hizi, A. Retroviral reverse transcriptases. Cellular and Molecular Life Sciences. 67 (16), 2717-2747 (2010).
- Hu, W. S., Hughes, S. H. HIV-1 reverse transcription. Cold Spring Harbor Perspectives in Medicine. 2 (10), (2012).
- Levin, J. G., Mitra, M., Mascarenhas, A., Musier-Forsyth, K. Role of HIV-1 nucleocapsid protein in HIV-1 reverse transcription. RNA Biology. 7 (6), 754-774 (2010).
- Menendez-Arias, L., Sebastian-Martin, A., Alvarez, M. Viral reverse transcriptases. Virus Research. , (2016).
- Telesnitsky, A., Goff, S. P., Coffin, J. M., Hughes, S. H., Varmus, H. E. . Retroviruses. , (1997).
- Pollpeter, D., et al. Deep sequencing of HIV-1 reverse transcripts reveals the multifaceted antiviral functions of APOBEC3G. Nature Microbiology. 3 (2), 220-233 (2018).
- Frohman, M. A., Dush, M. K., Martin, G. R. Rapid production of full-length cDNAs from rare transcripts: amplification using a single gene-specific oligonucleotide primer. Proceedings of the National Academy of Sciences of the United States of America. 85 (23), 8998-9002 (1988).
- Liu, X., Gorovsky, M. A. Mapping the 5' and 3' ends of Tetrahymena thermophila mRNAs using RNA ligase mediated amplification of cDNA ends (RLM-RACE). Nucleic Acids Research. 21 (21), 4954-4960 (1993).
- Ince, I. A., Ozcan, K., Vlak, J. M., van Oers, M. M. Temporal classification and mapping of non-polyadenylated transcripts of an invertebrate iridovirus. Journal of General Virology. 94, 187-192 (2013).
- Hafner, M., et al. RNA-ligase-dependent biases in miRNA representation in deep-sequenced small RNA cDNA libraries. RNA. 17 (9), 1697-1712 (2011).
- Kwok, C. K., Ding, Y., Sherlock, M. E., Assmann, S. M., Bevilacqua, P. C. A hybridization-based approach for quantitative and low-bias single-stranded DNA ligation. Analytical Biochemistry. 435 (2), 181-186 (2013).
- Abram, M. E., Tsiang, M., White, K. L., Callebaut, C., Miller, M. D. A cell-based strategy to assess intrinsic inhibition efficiencies of HIV-1 reverse transcriptase inhibitors. Antimicrobial Agents and Chemotherapy. 59 (2), 838-848 (2015).
- Bishop, K. N., Verma, M., Kim, E. Y., Wolinsky, S. M., Malim, M. H. APOBEC3G inhibits elongation of HIV-1 reverse transcripts. PLoS Pathogens. 4 (12), 1000231 (2008).
- Zack, J. A., Haislip, A. M., Krogstad, P., Chen, I. S. Incompletely reverse-transcribed human immunodeficiency virus type 1 genomes in quiescent cells can function as intermediates in the retroviral life cycle. Journal of Virology. 66 (3), 1717-1725 (1992).
- Adachi, A., et al. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. Journal of Virology. 59 (2), 284-291 (1986).
- Shah, V. B., Aiken, C. In vitro uncoating of HIV-1 cores. Journal of Visualized Experiments. (57), (2011).
- JoVE Science Education Database. Science Education Database: Basic Methods in Cellular and Molecular Biology: Passaging Cells. Journal of Visualized Experiments. , (2018).
- JoVE Science Education Database. JoVE Science Education Database: Basic Methods in Cellular and Molecular Biology: Using a Hemocytometer to Count Cells. Journal of Visualized Experiments. , (2018).
- Zhou, M. Y., Gomez-Sanchez, C. E. Universal TA cloning. Current Issues in Molecular Biology. 2 (1), 1-7 (2000).
- Zhang, S., Cahalan, M. D. Purifying plasmid DNA from bacterial colonies using the QIAGEN Miniprep Kit. Journal of Visualized Experiments. (6), 247 (2007).
- Mangeat, B., et al. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature. 424 (6944), 99-103 (2003).
- Gillick, K., et al. Suppression of HIV-1 infection by APOBEC3 proteins in primary human CD4(+) T cells is associated with inhibition of processive reverse transcription as well as excessive cytidine deamination. Journal of Virology. 87 (3), 1508-1517 (2013).
- Harris, R. S., et al. DNA deamination mediates innate immunity to retroviral infection. Cell. 113 (6), 803-809 (2003).
- Zhang, H., et al. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature. 424 (6944), 94-98 (2003).
- Konig, J., et al. iCLIP--transcriptome-wide mapping of protein-RNA interactions with individual nucleotide resolution. Journal of Visualized Experiments. (50), (2011).
- Troutt, A. B., McHeyzer-Williams, M. G., Pulendran, B., Nossal, G. J. Ligation-anchored PCR: a simple amplification technique with single-sided specificity. Proceedings of the National Academy of Sciences of the United States of America. 89 (20), 9823-9825 (1992).
- Zhelkovsky, A. M., McReynolds, L. A. Structure-function analysis of Methanobacterium thermoautotrophicum RNA ligase - engineering a thermostable ATP independent enzyme. BMC Molecular Biology. 13 (24), (2012).
- Li, T. W., Weeks, K. M. Structure-independent and quantitative ligation of single-stranded DNA. Analytical Biochemistry. 349 (2), 242-246 (2006).
- Gansauge, M. T., et al. Single-stranded DNA library preparation from highly degraded DNA using T4 DNA ligase. Nucleic Acids Research. 45 (10), 79 (2017).
- Ding, Y., Kwok, C. K., Tang, Y., Bevilacqua, P. C., Assmann, S. M. Genome-wide profiling of in vivo RNA structure at single-nucleotide resolution using structure-seq. Nature Protocols. 10 (7), 1050-1066 (2015).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados