
Method Article
Investigación del papel transcripcional de un silenciador intrónico RUNX1 por RIBonucleoprotein CRISPR/Cas9 en células de leucemia mieloide aguda
En este artículo
Resumen
La entrega directa de complejos de ribonucleoproteínacas Cas9/ARN guía premontados es un medio rápido y eficiente para la edición del genoma en células hematopoyéticas. Aquí, utilizamos este enfoque para eliminar un silenciador intrónico RUNX1 y examinar las respuestas transcripcionales en las células leucémicas OCI-AML3.
Resumen
La mayor parte del genoma humano (98%) se compone de secuencias no codificante. Los elementos reguladores DeCis (CRU) son secuencias de ADN no codificante que contienen sitios de unión para que los reguladores transcripcionales modulan la expresión génica. Se han implicado alteraciones de las Enclos en diversas enfermedades, incluido el cáncer. Si bien los promotores y potenciadores han sido las principales CRED para estudiar la regulación genética, se sabe muy poco sobre el papel del silenciador, que es otro tipo de CRE que media la represión génica. Originalmente identificado como un sistema de inmunidad adaptativa en prokaryotes, CRISPR/Cas9 ha sido explotado como una poderosa herramienta para la edición del genoma eucariota. Aquí, presentamos el uso de esta técnica para eliminar un silenciador intrónico en el gen RUNX1 humano e investigar los impactos en la expresión génica en las células leucemias OCI-AML3. Nuestro enfoque se basa en la entrega mediada por electroporación de dos complejos de ribonucleoproteína (RNP) Cas9/ARN guía (GRNA) premontados para crear dos roturas de doble cadena (DSB) que flanquean el silenciador. Las eliminaciones se pueden examinar fácilmente mediante el análisis de fragmentos. Los análisis de expresiones de diferentes ARNm transcritos de promotores alternativos ayudan a evaluar los efectos dependientes del promotor. Esta estrategia se puede utilizar para estudiar otras CREs y es particularmente adecuada para células hematopoyéticas, que a menudo son difíciles de transfectar con métodos basados en plásmidos. El uso de una estrategia de plásmido y libre de virus permite evaluaciones simples y rápidas de las funciones reguladoras de genes.
Introducción
Loselementos reguladores Cis (CES) son secuencias de ADN no codificanteque contienen sitios de unión para reguladores transcripcionales para controlar la expresión génica 1,2. Estos elementos suelen tener entre 100 y 1.000 pares de base (bp) de largo. Los promotores y potenciadores son los dos tipos más caracterizados de CREs. Los promotores están presentes muy cerca de los sitios de inicio de la transcripción y constituyen la unidad básica de transcripción. Muchos genes tienen más de un promotor y su uso alternativo contribuyea la diversidad de transcriptoma y la especificidad de los tejidos 3,4. Por otro lado, los potenciadores activan la transcripción y se pueden ubicar aguas arriba, aguas abajo o dentro de los intrones de los genes diana. Los potenciadores pueden actuar desde lejos (sobre unamegabase) e independientes de la orientación 1,2. Las CLE también incluyen silenciadoresy aislantes 5,6. El primero actúa opuestamente a potenciadores para inhibir la expresión génica mediante la unión a represores transcripcionales, mientras que el segundo divide el genoma en dominios topológicamente discretos para aislar genes de otras CREs de dominios vecinos. Estos elementos actúan en conjunto entre sí a través de interacciones de cromatina de corto y/o largo alcance y se organizan en centros reguladores para dirigir la expresión génica espaciotemporal adecuada. Los recientes avances en las técnicas de secuenciación de alto rendimiento han acelerado la identificación y la anotación funcional de muchas CTE que han facilitado en gran medida nuestra comprensión de las redes transcripcionales que dictan genes específicos del linaje expresión en diferentes tipos de células y tejidos7,8,9,10,11,12.
Dadas las funciones fundamentales de las RTE en la regulación de la transcripción, sus alteraciones pueden conducir a una expresión génica aberrante. Se ha demostrado que las ENC se ven frecuentemente interrumpidas por cambios genéticos y epigenéticos en diferentes tipos de cáncer humano, contribuyendo así a la iniciación, progresión y agresividad del tumor13,14. Además, los factores de unión a la CRE a menudo se mutan y/o se expresan erróneamente en varios tipos de cáncer, lo que pone de relieve aún más la importancia de la desregulación de la CRE en la oncogénesis15. Las ENGEN también pueden verse afectadas por aberraciones estructurales, como lo ejemplifican los frecuentes reordenamientos cromosómicos del potenciador del gen de la inmunoglobulina pesada (IgH) que dan lugar a la activación anormal de los oncogenes vecinos en los linfomas de células B16. En la leucemia mieloide aguda (LMA), el reposicionamiento de un solo potenciador por reordenamientos del cromosoma 3q provoca una regulación descendente de GATA2 concomitante y activación de EVI1, que puede ser potencialmente dirigida por la inhibición de BET de las funciones del potenciador 17. Recientemente, caracterizamos una novedosa translocación cromosómica que implica perturbación de un silenciador intrónico RUNX1 que podría contribuir a la progresión de la LMA en un paciente pediátrico18. Por lo tanto, descifrar el genoma del cáncer no codificante proporciona vías fructíferas para dilucidar la patogénesis de la enfermedad, el descubrimiento de biomarcadores y las intervenciones terapéuticas, que en última instancia mejoran los resultados de los pacientes.
La vía de nucleasa CRISPR/Cas9, originalmente identificada como un sistema inmune adaptativo en células procakóticas, ha sido explotada como un medio rápido y rentable para la edición genómica específica del sitio en células y organismos vivos19,20 ,21,22. El sistema CRISPR/Cas9 implica dos componentes principales: el ARB y el Streptococcus pyogenes-derivadosegún la nucleasa Cas9. El gRNA contiene una secuencia específica llamada protoespaciador que reconoce la región de destino y dirige Cas9 para su edición. El ARNm se compone de dos partes: ARN CRISPR (crRNA), típicamente una secuencia de nucleótidos de 20 mers complementaria al ADN objetivo, y un crRNA activador trans(trarRNA), que sirve como un andamio de unión para la nucleasa. Se requiere un motivo adyacente protoespacial (PAM) (5'-NGG) inmediatamente adyacente al sitio de destino para el escisión Cas9 y el sitio de escisión se encuentra 3 nucleótidos aguas arriba del PAM. con un plásmido que codifica Cas9 y el gRNA clonado23. Sin embargo, este enfoque es difícil para las células hematopoyéticas, que a menudo son difíciles de transfectar y requieren largos métodos de transducción basados en virus. Un enfoque alternativo es la entrega celular directa de los complejos Cas9/gRNA RNP premontados24. Un método común para la entrega de RNP es la electroporación, que genera poros temporales en la membrana celular, permitiendo así la entrada de los complejos RNP en las células25,26. Las ventajas de este enfoque incluyen la facilidad de uso, la reducción de los efectos fuera del objetivo y la estabilidad de los complejos RNP. Aquí, describimos un protocolo de uso del método de administración de RNP para investigar el papel transcripcional de un silenciador intrónico RUNX1 en la línea18de la célula de leucemia OCI-AML3, que se estableció a partir de la sangre periférica de un paciente con LMA diagnosticado con el subtipo27francés-estadounidense-británico M4. El protocolo incluye el diseño de crRNA, la preparación de complejos RNP, electroporación, así como la detección y posterior caracterización de los clones deseados.
Protocolo
1. Diseño de crRNA
- Diseñar dos crRNAs, uno 5' y el otro 3' del CRE objetivo utilizando una herramienta de diseño CRISPR basada en web28,29,30,31. Asegúrese de que un PAM de NGG esté situado inmediatamente aguas abajo de la secuencia de destino para el reconocimiento Cas9. El diseño de los dos crRNAs (crRNA-1 y crRNA-2) para la eliminación del silenciador RUNX1 18 se muestra en la Figura1.
NOTA: Un crRNA normalmente contiene un protoespaciador de 20 mer que es complementario a la secuencia de destino. - Compruebe la presencia de polimorfismos de nucleótido único (SNP)/indels en las secuencias de PAM objetivo y adyacentes introduciendo las ubicaciones genómicas de las secuencias en el cuadro de búsqueda de un navegador de genoma en línea (por ejemplo, genomas NCBI 1000 o navegador genoma UCSC).
NOTA: Los SNP/indels comunes tienen una frecuencia de alelos menor de al menos el 1% en la población general. - Envíe las secuencias crRNA seleccionadas para su síntesis con un proveedor comercial. Además, compre el tracrRNA para la formación dúplex de ARNm.
2. Diseño de Primers de Detección de Eliminación
- Diseñe un par de imprimaciones que flanqueen la región de eliminación prevista. Asegúrese de que las imprimaciones estén al menos a 50 bp de los sitios de escisión Cas9 para que la amplificación de PCR se vea mínimamente afectada por las indels formadas en los sitios de escisión.
- Asegúrese de que el amplificador es menor que 1.200 bp (preferiblemente menor que 600 bp para una mejor resolución de tamaño). Además, etiquete una de las imprimaciones con un tinte fluorescente (por ejemplo, 6-carboxifluoresceína (6-FAM)) en el extremo de 5'para la detección. Las imprimaciones utilizadas para la selección del silenciador RUNX1 18 se muestran en la Figura1.
3. Preparación de complejos Cas9/gRNA RNP
- Resuspenda los crRNAs y tracrRNA en 1búfer TE (10 mM Tris, 0,1 mM EDTA, pH 7,5) a una concentración final de 200 m.
- Para cada CRRNA, mezcle 2,2 ml de crRNA de 200 m, 2,2 ml de armero de 200 oM y 5,6 ml de 1 tampón te (total 10 ml) en un tubo de 0,2 ml para obtener una concentración dúplex final de 44 oM.
- Incubar a 95oC durante 5 min en un termociclador. Deje que los tubos se enfríen a temperatura ambiente para la formación compleja de ARNm.
- Diluir 10,4 l de 62 m de nucleasa recombinante Cas9 con 7,6 l de solución salina tamponada de fosfato 1x (PBS) para obtener una concentración final de nucleasa de 36 m.
NOTA: Esta cantidad es suficiente para la preparación de dos complejos Cas9/gRNA. - Mezclar volúmenes iguales (8,5 l) de la nucleasa diluida con cada uno de los dúplex de ARNm obtenidos del paso 3.3.
- Incubar a temperatura ambiente durante 20 min para permitir la formación compleja de RNP. Mantenga las mezclas sobre hielo hasta la electroporación.
4. Electroporación de los Complejos RNP en Células OCI-AML3
- Cultivo de células OCI-AML3 en RPMI 1640 medio suplementado con 10 % suero bovino fetal inactivado por calor (FBS), 1x GlutaMAX, 100 unidades/ml de penicilina y 100 g/ml de estreptomicina (en adelante, medio RPMI 1640 completo) a 37 oC con 5 % de CO2.
- Cuente las células usando la tinción azul de trypan. Asegúrese de que la viabilidad celular en el momento de la electroporación sea superior al 90%.
- Centrífuga 2,5 x 106 células en un tubo de 1,5 ml a 500 x g durante 5 min a temperatura ambiente. Retire el sobrenadante y lave las células con 1 ml de PBS 1x. Vuelva a girar las células y retire todo sobrenadante residual.
- Resuspenda las células en 163 ml de rpm 1640 de medio sin rojo fenol. Añadir 16,7 l de cada complejo de RNP (a partir del paso 3.6) y 3,6 l de potenciador de electroporación de 100 oM a las células (volumen total de 200 ol). Mezcle suavemente pipeteando.
NOTA: El Electroporation Enhancer es un ADN portador para mejorar la eficiencia de edición. - Transfiera la mezcla a una cubeta de electroporación de 0,2 cm de brecha sin burbujas. Realizar electroporación con un sistema de electroporación (Modo: exponencial; Voltaje: 150 V; Capacitancia a 700 oF; Resistencia a 50o).
- Transfiera las células a un matraz de cultivo tisular T-25 que contenga 6 ml de medioRPMI 1640 completo e incubar a 37 oC con un 5 % de CO2.
5. Detección y selección de clones celulares con eliminaciones bialélílicas
- Diluir las células a 5 x 103/mL en el medio RPMI 1640 completo un día después de la electroporación. Añadir 100 l de la suspensión celular diluida en cada pozo de placas de cultivo de tejido de 96 pocillos y permitir que las células crezcan durante 7-14 días.
- Extraiga ADN genómico de las células utilizando un sistema de purificación de alto rendimiento.
- Añadir 100 l de encuadernación de placas y tampón de lisis a cada pocal de una placa de extracción de 96 pocillos. Agregue 5 x 104 células resuspendidas en 10 l de 1x PBS al búfer y mezcle con pipeteo.
- Incubar a temperatura ambiente durante 30 min para permitir la unión del ADN genómico a los pozos.
- Aspirar la solución de los pozos sin raspar las superficies del pozo. Lave los pozos con 120 ml de tampón de lavado.
- Seque al aire los pozos que contienen el ADN enlazado.
- Preparar 20 ml de mezcla de PCR (2 l de tampón de alta fidelidad de 10x, 0,8 ml de 50 mM MgSO4, 0,4 ml de dNTPs de 10 mM, 0,4 ml de 10 m de fAM con etiqueta de imprimación delantera (paso 2.2), 0,4 ml de imprimación inversa sin etiquetar de 10 m y 0,4 U de polimerada de ADN Taq para cada muestra).
NOTA: Prepare una mezcla maestra para garantizar la adición de cantidades estandarizadas de reactivos en cada muestra. - Añadir la mezcla de PCR a cada pocal de la placa de extracción y ejecutar las reacciones en un termociclador (Condiciones: inicial 94 oC durante 2 min, seguido de 35 ciclos de 94oC para 15 s, 56oC para 30 s y 68oC durante 1 min).
- Estimar la cantidad de productos midiendo sus concentraciones en un número seleccionado de muestras utilizando un fluorómetro. Diluir todas las muestras con sin nucleasas H2O a 0,5 ng/L.
- Mezclar 1 l de los productos de PCR diluidos con 8,5 ml de formación desionizada y 0,5 ml de tamaño con etiqueta de tinte fluorescente estándar en una placa de 96 pocillos compatible con el analizador genético.
NOTA: Prepare una mezcla maestra que contenga formamida desionizada y el tamaño estándar. - Cubrir la placa con un septo de placa y desnaturalizar las muestras a 95 oC durante 3 min en un termociclador. No cierre la tapa de la máquina.
- Realice la electroforesis de gel capilar para separar los productos pcR etiquetados como se describió anteriormente32.
- Después de la electroforesis, abra el software de análisis para analizar los resultados.
- Haga clic en Nuevo proyecto y seleccione Microsatélite. A continuación, haga clic en Aceptar.
- Haga clic en Agregar ejemplos al proyecto y seleccione los archivos de resultados (contienen la extensión .fsa). A continuación, haga clic en Agregar a la lista para importar los archivos.
- En la tabla que muestra los archivos de resultados seleccionados, elija Microsatélite predeterminado en la columna Método de análisis. Además, seleccione el estándar de tamaño utilizado en la columna Tamaño estándar. A continuación, haga clic en el icono Analizar, escriba el nombre del experimento y guarde el experimento.
- Haga clic en Visualizar trazados para ver los resultados y elija Análisis de fragmentos en la configuración de trazado.
- Elija los canales de color adecuados para el análisis. Compruebe el icono naranja para ver los fragmentos etiquetados en el estándar de tamaño para evaluar la calidad de las llamadas de tamaño.
- Compruebe el icono azul para ver los productos PCR etiquetados. Identificar los picos que corresponden a los productos de tipo salvaje y mutante (es decir, llevar las eliminaciones esperadas). Estimar el nivel de mutante en cada muestra dividiendo el área bajo el pico mutante por la suma del área bajo los picos de tipo salvaje y mutante.
- Seleccione varias agrupaciones de celdas con altos niveles de las eliminaciones esperadas para diluciones en serie adicionales.
- Repita los pasos de extracción de ADN, PCR fluorescente y electroforesis capilar. Seleccione clones celulares con niveles mutantes >95% que representen eliminaciones biloélicas para análisis posteriores.
- Compruebe la identidad de las eliminaciones en los clones seleccionados mediante la secuenciación de Sanger.
6. Análisis funcionales de la eliminación del silenciador mediante análisis cuantitativos RT-PCR en tiempo real
- Extraiga el ARN total de los clones seleccionados y realice la síntesis complementaria de ADN (ADNc).
- Mezclar 1 g de ARN con 1 l de 50 oM de oligo(dT)20 imprimación y 1 l de mezcla dNTP de 10 mM en un volumen total de 10 l. Incubar a 65 oC durante 5 min y luego colocar en hielo durante al menos 1 min.
- Añadir 10 ml de mezcla de síntesis de ADN que contenga 2 ml de tampón RT de 10x, 4 ml de MgCl2, 2 ml de TDT de 0,1 M, 1 l de inhibidor de la RNase (40 U/L) y 1 l de transcriptasa inversa (200 U/L). Incubar a 50oC durante 50 min y luego 85oC durante 5 min en un termociclador.
NOTA: Prepare una mezcla maestra para la transcripción inversa. - Enfríe las muestras en hielo. Añadir 1 l de RNase H e incubar a 37oC durante 20 min. Almacenar el ADNc a -20oC.
- Imprimadores de diseño y sondas TaqMan que reconocen específicamente variantes de transcripción individuales generadas a partir de promotores alternativos.
NOTA: Los conjuntos de imprimación/sonda específicos para transcripción prediseñados están disponibles comercialmente. - Clonar fragmentos de ADN que contienen las secuencias de transcripción específicas en el ADN plásmido. Preparar una serie de dilución de 10 veces (10de 6 a 10 copias) de los plásmidos recombinantes como curvas estándar para la cuantificación de transcripciones.
- Preparar 20 ml de mezcla de PCR (0,5 l de plantilla de ADN, 1 l de ensayo de sonda/imprimación TaqMan prediseñado de 20x y 10 l de 2x TaqMan PCR Master Mix) para cada muestra (estándares de ADN y plásmido). Mida cada muestra en triplicado.
NOTA: Prepare una mezcla maestra para la PCR en tiempo real. - Ejecuta las reacciones en una máquina de PCR en tiempo real (Condiciones: inicial 50oC durante 2 min y 95oC durante 10 min, seguida de 40 ciclos de 94oC para 15 s y 60oC durante 1 min).
- Después de la amplificación, haga clic en el icono Analizar del software para analizar los datos. Compruebe la pendiente y el coeficiente de correlación de las curvas estándar para evaluar la eficiencia y linealidad de las reacciones. Asegúrese de que las pendientes estén entre -3.1 y -3.6 y que los coeficientes de correlación sean mayores que 0.99.
- Normalizar el número de copia de las transcripciones de destino en cada muestra con un gen de limpieza (por ejemplo, GAPDH).
Resultados
El objetivo de este experimento es eliminar un silenciador intrónico en el gen RUNX1 y examinar los impactos en la transcripción de RUNX1 en células OCI-AML3. El silenciador fue identificado por un enfoque molecular combinatorio y se encontró que contenía un elemento central de 209 bp18. Para permitir una evaluación más precisa de este elemento principal en el control de la expresión RUNX1, los crRNAs (crRNA-1 y crRNA-2) se diseñaron para orientarse estrechamente a esta región18 (Figura1). Los sitios de escote Cas9 previstos traídos por crRNA-1 y crRNA-2 fueron 29 bp y 35 bp del elemento central, respectivamente (Figura1). Cabe señalar que mientras que los sitios PAM de los dos crRNAs residen en hebras opuestas, los DSB ocurrirán independientemente de la ubicación de la secuencia PAM. Por lo tanto, se espera que la introducción concomitante de dos complejos Cas9/gRNA RNP guiados por crRNA-1 y crRNA-2 expuse el elemento silenciador del locus RUNX1.
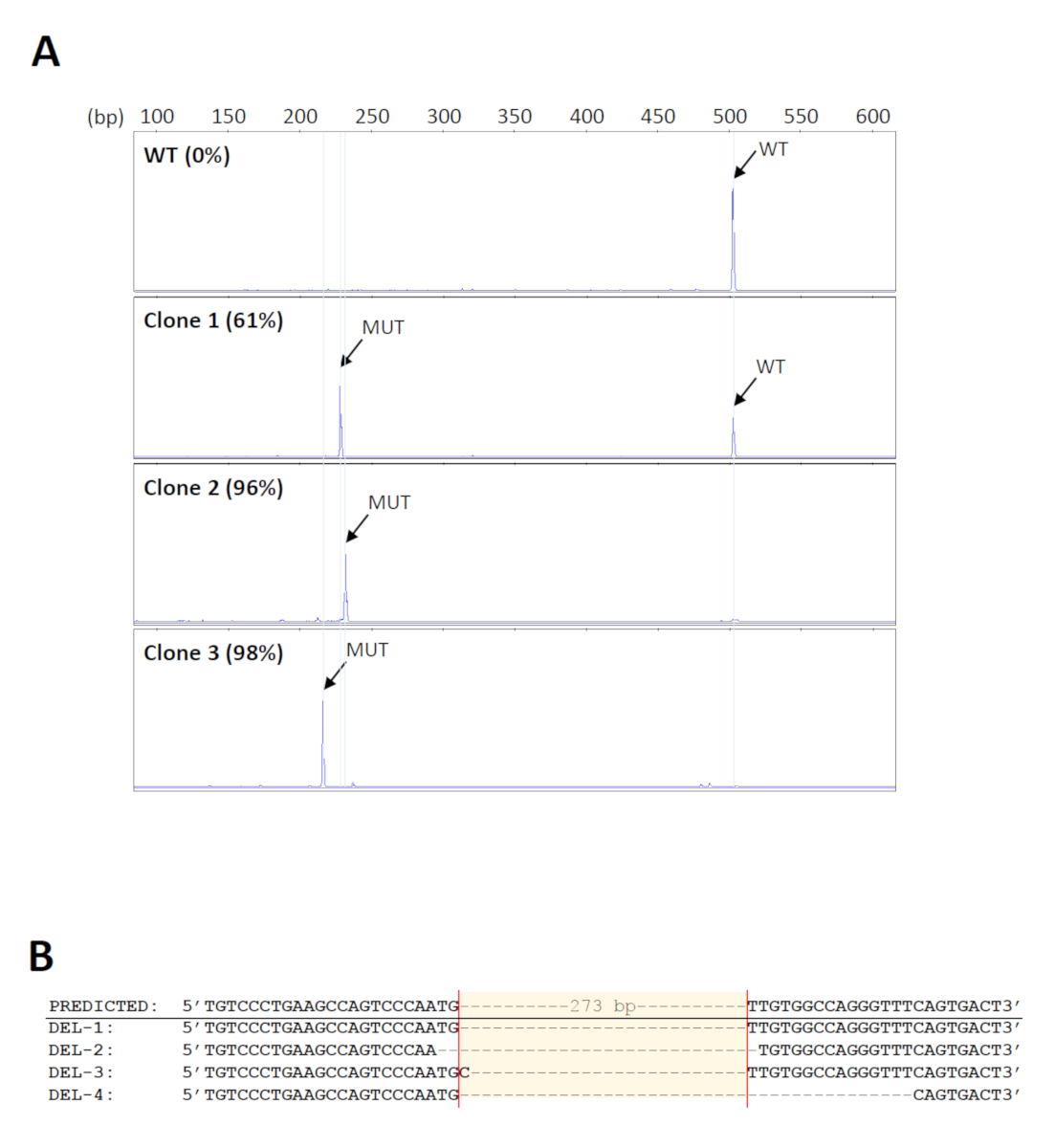
Para detectar las eliminaciones deseadas en un gran número de muestras, se utilizó un formato de 96 polos de kit de extracción de ADN genómico para la purificación de alto rendimiento. Además, el ADN unido en los pozos de las placas de extracción puede ser sometido a amplificación directa, minimizando así errores o contaminación debido a la transferencia repetida de muestras. Las imprimaciones utilizadas para el cribado de las eliminaciones18 se muestran en la Figura1. El tamaño esperado del producto PCR de tipo salvaje es de aproximadamente 500 bp. Dado que la eliminación prevista abarca 273 bp, se espera que el producto mutante sea de aproximadamente 230 bp. Este rango de tamaño permite un análisis de fragmentos simple y rápido mediante electroforesis de gel capilar. Un total de 160 grupos de células iniciales fueron examinados y 14 fueron encontrados para llevar las eliminaciones esperadas con niveles mutantes de al menos 70%. A continuación, se seleccionaron cinco grupos para realizar más diluciones en serie para identificar clones que llevaban eliminaciones biallélicas. Los electroferogramas representativos de clones celulares con diferentes niveles de productos mutantes se muestran en la Figura 2A. La identidad de las eliminaciones fue verificada por la secuenciación de Sanger (Figura2B). Como era de esperar, se observaron indels formados por reparaciones de unión final no homólogas de DSBs33 en los sitios de escisión previstos en los clones de eliminación. Esto dio lugar a la amplificación de productos mutantes de diferentes tamaños, que también podrían ser detectados por electroforesis capilar (Figura2A).
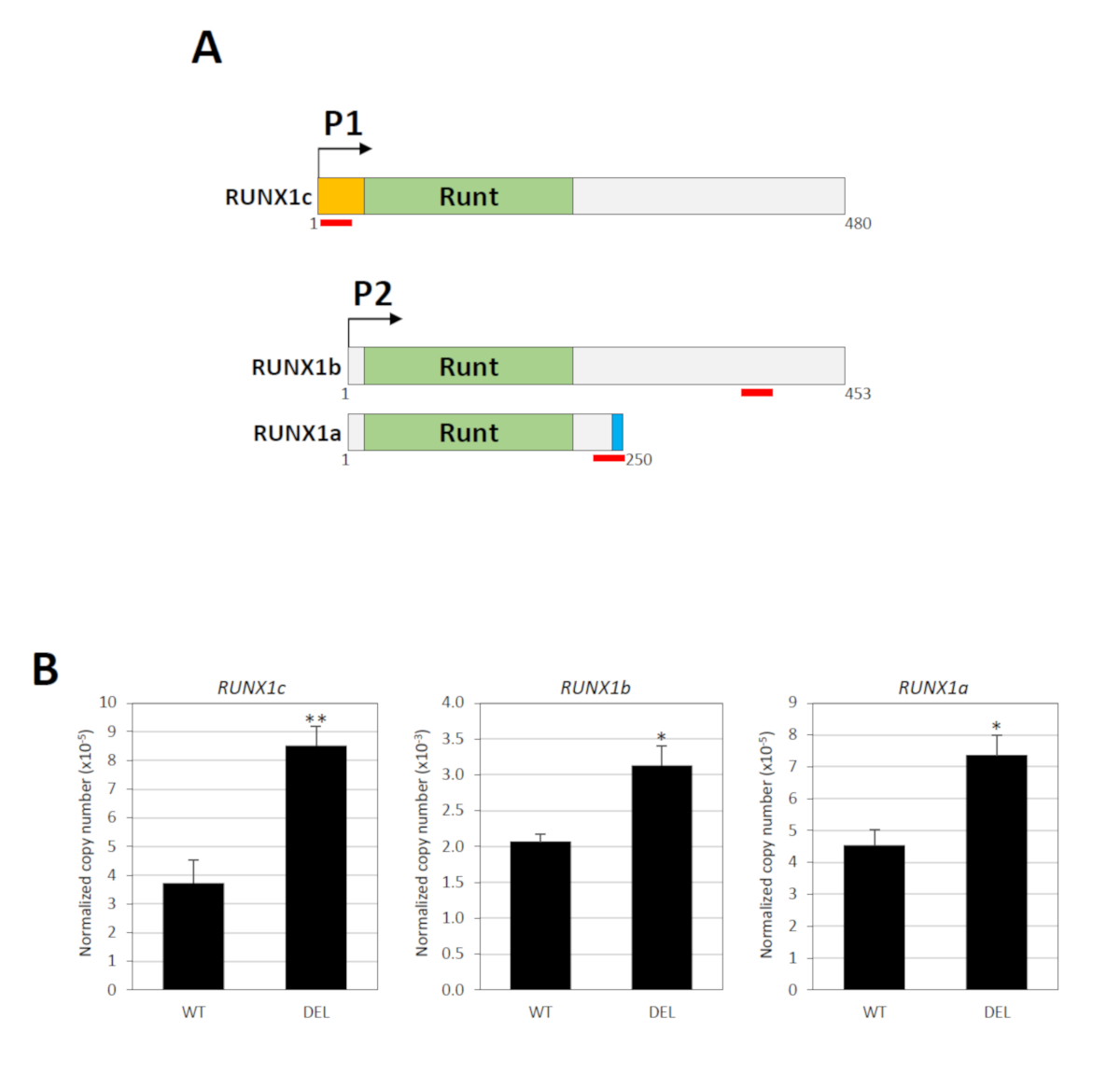
El gen RUNX1 contiene dos promotores, a saber, el P1 distal y el P2 proximal, que están separados por un gran intrón que alberga el elemento silenciador34. Tres transcripciones principales de ARNm son producidas por estos promotores: RUNX1c por P1 y RUNX1a y RUNX1b por P234. La secuencia de nucleótidos de RUNX1c y RUNX1b son idénticas, excepto que la primera tiene un N-terminus único, a partir del cual se puede diseñar un ensayo específico de expresión génica TaqMan (Figura3A). Para medir RUNX1b, se utilizó un ensayo TaqMan que reconoce tanto RUNX1b como RUNX1c (Figura 3A). Los niveles runX1b se determinaron entonces restando el total runX1b/RUNX1c de RUNX1c. RUNX1a es una isoforma claramente más corta debido al empalme alternativo y un ensayo TaqMan específico está disponible para esta variante (Figura3A). Por lo tanto, la actividad de los promotores P1 y P2 se puede determinar individualmente. RT-PCR cuantitativo en tiempo real mostró que la eliminación del elemento silenciador reguló significativamente los niveles de expresión de las transcripciones derivadas de P1 y P2 (Figura3B).

Figura 1 : Estrategia para eliminar el silenciador intrónico RUNX1. El silenciador (caja roja) se encuentra en el primer intrón del gen RUNX1 que separa los dos promotores P1 y P2 (se muestran las coordenadas hg19). Dos crRNAs (crRNA-1 y crRNA-2) fueron diseñados para introducir DSB flanqueando el elemento silenciador. Los sitios de escote Cas9 previstos están indicados por líneas rojas verticales y los sitios PAM (NGG) están en color púrpura. Las dos imprimaciones utilizadas para el cribado de las eliminaciones están representadas por flechas abiertas. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2 : Identificación de los clones de eliminación. (A) Electroferogramas representativos de clones celulares que muestran diferentes niveles de productos mutantes de PCR (MUT). El tamaño de los productos mutantes varía entre los clones debido a los indels formados en los sitios de escote. WT - tipo salvaje. (B) Verificación de las eliminaciones de clones representativos por secuenciación de Sanger. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3 : Consecuencias funcionales de la eliminación del silenciador. (A) Se muestran las tres isoformas principales de RUNX1 (RUNX1a, RUNX1b y RUNX1c). Estas variantes contienen el mismo dominio de unión de ADN Runt pero diferente N- (naranja) o C-terminus (azul). La ubicación de los pares de sonda/imprimación TaqMan se indica mediante líneas rojas. Los números indican los residuos de aminoácidos. (B) Análisis cuantitativo en tiempo real de RT-PCR de transcripciones derivadas de RUNX1 P1 y P2 en poblaciones celulares con (DEL) o sin (WT) las eliminaciones bialélíticas18. GAPDH se utilizó para la normalización. * y ** indican P < 0.05 y P < 0.01, respectivamente por la prueba Mann-Whitney. Esta cifra ha sido modificada de Cheng et al.18. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Discusión
El sistema CRISPR/Cas9 se ha utilizado en una amplia gama de aplicaciones de edición del genoma, como los estudios de knockout y knock-in35,36,regulación transcripcional37,38,ingeniería genética de organismos modelo39,40,41,42,43,44 y terapia génica45,46. Aquí, demostramos el uso de CRISPR/Cas9 para investigar las consecuencias funcionales de la eliminación de un silenciador intrónico en el gen RUNX1. La entrega de los componentes CRISPR en nuestro enfoque no se basó en el ADN plásmido, clonación de ARNm o virus, sino en la electroporación de complejos Cas9/gRNA RNP premontados. Se ha demostrado que el uso de ADN exógeno puede asociarse con la integración indeseable de secuencias vectoriales extrañas en el genoma del huésped, aumento de la toxicidad y baja eficiencia25,47,48,mientras que los métodos de transducción de virus consumen mucho tiempo. Además, la expresión prolongada de Cas9 a partir de ADN plásmido puede aumentar los efectos fuera del objetivo48. Por el contrario, el enfoque de entrega directa basado en RNP se ha establecido como el método preferido, ya que es rápido y directo con una eficiencia de edición mejorada, selectividad y viabilidad celular. De hecho, una variedad de métodos como lipofección49,50, electroporación25,51, nanopartículas52, péptidos de penetración celular53, iTOP54 y TRIAMF 55 han sido desarrollados para la entrega eficiente de CRISPR/Cas9 en diversos tipos de células, así como especies animales y vegetales24,25,26,56,57 , 58 , 59 , 60 , 61 , 62 , 63. Dado que las secuencias de ADN no codificantes son puntos críticos de las variaciones genéticas64, comprobar la presencia de SNP/indels comunes en las secuencias de PAM objetivo y vecina es particularmente relevante al diseñar gRNA que se dirige a las regulaciones Elementos.
Un cuello de botella en la edición del genoma de CRISPR/Cas9 implica la detección de clones mutantes deseados en un gran número de muestras. Empleamos PCR fluorescente junto con electroforesis de gel capilar para el cribado, ya que la mutación objetivo es una pequeña eliminación genómica de aproximadamente 300 bp. Este método es rápido y sensible y se puede realizar de una manera de alto rendimiento. Además, este método permite una estimación precisa de los niveles mutantes y los tamaños de eliminación simultáneamente. Además, se admite el análisis multiplex de fragmentos de PCR etiquetados con diferentes colorantes fluorescentes. Hemos estado utilizando rutinariamente esta técnica para genotipo pequeñas inserciones/eliminaciones en neoplasias mieloides65,66. En nuestra experiencia, podemos detectar consistentemente tamaños de fragmentos que difieren en 4 bp con alta precisión y carga mutante hasta 3 %. Sin embargo, cabe señalar que este método tiene un límite de tamaño de fragmento de 1.200 bp, y por lo tanto no es adecuado para el cribado de grandes eliminaciones. Además, no se pueden detectar sustituciones de base (que dan como resultado un tamaño de fragmento sin cambios) y posibles eventos fuera de destino en otras regiones genómicas. Para este último, la costosa secuenciación del genoma completo es necesaria para perfilar exhaustivamente los cambios globales indeseables en los clones de destino. Para adoptar nuestro enfoque actual para la investigación de grandes secuencias reguladoras no codificativas (>1.000 bp), se pueden realizar análisis detallados de eliminación y mutagénesis de sitios de unión al factor de transcripción putativa utilizando ensayos genéticos de reporteroin vitro previamente para delinear la región funcional mínima para la edición CRISPR/Cas918.
Dado que muchos genes contienen más de un promotor3,4, es importante ser conscientes de la existencia de promotores alternativos en el locus gen objetivo ya que la manipulación de elementos reguladores puede afectar diferencialmente a los promotores. Por lo tanto, las variantes de transcripción derivadas de diferentes promotores deben medirse individualmente para evaluar cualquier respuesta específica del promotor. Se prefiere el uso de ensayos basados en sondas TaqMan sobre SYBR Green debido a una mejor especificidad y reproducibilidad. Si el sistema de PCR digital más avanzado está disponible, la cuantificación de transcripciones se puede realizar con mayor precisión sin necesidad de construcción de curvas estándar.
Una consideración importante en la realización de experimentos CRISPR/Cas9 en líneas celulares cancerosas es el número de copia del gen objetivo y ploidy en las células utilizadas como prácticamente todas las líneas celulares cancerosas albergan alteraciones genéticas, incluidas las variaciones estructurales y de números de copia. En nuestro caso, OCI-AML3 tiene un cariotipo hiperdiloide con 45 a 50 cromosomas. Además, se encontró que la línea celular llevaba un número de copia RUNX1 normal, como se reveló en la Enciclopedia de la Línea Celular de Cáncer67 y los estudios de hibridación in situ de fluorescencia18. Cuando se dirige a un gen con ganancia de número de copia, es posible que sea necesario optimizar el método de entrega para proporcionar niveles suficientes de los componentes CRISPR para la edición. Además, es posible que sea necesario realizar más clones para identificar los knockouts completos. Es importante destacar que se ha demostrado que la focalización en regiones genómicas amplificadas, en particular las causadas por reordenamientos estructurales, puede desencadenar respuestas antiproliferativas independientes de los genes en las células cancerosas, lo que conduce a resultados falsos positivos en el gen estudios funcionales68,69,70. A este respecto, deben emplearse enfoques alternativos como el derribo de la interferencia del ARN (ARN) y/o la sobreexpresión del ADNr para verificar los hallazgos de CRISPR. Además, se deben utilizar varias líneas celulares para evitar la interpretación errónea de los efectos de edición CRISPR específicos de la línea celular pero independientes de los genes.
El sistema CRISPR/Cas9 ha revolucionado la investigación básica y traslacional al proporcionar un medio sencillo y eficiente para la edición del genoma. Aquí demostramos la facilidad de usar CRISPR/Cas9 para interrumpir un silenciador intrónico para estudios transcripcionales en una línea celular de cáncer. Esta técnica permite el estudio de las CRU a nivel de ADN y ofrece la oportunidad de examinar las funciones de la CRE en el contexto endógeno en lugar de los genes tradicionales de reporteros hetólogos. Recientemente, un sistema de edición de ARN basado en CRISPR también se ha identificado71 y puede servir como una herramienta novedosa para estudiar las RCA mediante el ARN cruzado de los elementos reglamentarios. Al combinarcon técnicas de captura de conformación cromosómica, CRISPR/Cas9 sin duda ayudará a descifrar las implicaciones de las CLE en la organización del genoma alterado y la expresión génica vinculada a diversos problemas de salud.
Divulgaciones
Los autores no tienen nada que revelar.
Agradecimientos
Los autores desean agradecer al Prof. M.D. Minden (Princess Margaret Cancer Centre, University Health Network, Toronto, Canadá) por proporcionar la línea celular OCI-AML3. Además, los autores quieren agradecer a los principales servicios de enseñanza de la genómica y la patobiología del cáncer (Universidad China de Hong Kong) por proporcionar las facilidades y la asistencia en apoyo de esta investigación.
Materiales
| Name | Company | Catalog Number | Comments |
| 0.2 cm gap electroporation cuvette | Bio-Rad | 1652086 | |
| 1×TE buffer, pH 7.5 | Integrated DNA Technologies | 11-01-02-02 | |
| 10 mM dNTP mix | Thermo Fisher Scientific | 18427013 | |
| 3500 Genetic Analyzer | Thermo Fisher Scientific | 4405673 | |
| 6-FAM-labeled fluorescent PCR forward primer | Thermo Fisher Scientific | None | |
| 7300 Real-Time PCR System | Thermo Fisher Scientific | None | |
| 7300 System SDS Software | Thermo Fisher Scientific | None | Version 1.3.1 |
| Bio-Rad Gene Pulser Xcell system | Bio-Rad | 1652660 | |
| ChargeSwitch Direct gDNA Purification Kit, 96-well | Thermo Fisher Scientific | CS11205 | |
| crRNA-1 | Integrated DNA Technologies | Alt-R CRISPR-Cas9 crRNA | Components of the Cas9/gRNA complex |
| crRNA-2 | Integrated DNA Technologies | Alt-R CRISPR-Cas9 crRNA | Components of the Cas9/gRNA complex |
| Deionized formamide | Thermo Fisher Scientific | 4311320 | |
| Electroporation Enhancer | Integrated DNA Technologies | 1075915 | |
| Fetal bovine serum | Thermo Fisher Scientific | 10270098 | |
| Fluorometer | Thermo Fisher Scientific | Q32857 | |
| GeneMapper Software 5 | Thermo Fisher Scientific | 4475073 | |
| GeneScan 600 LIZ Size Standard | Thermo Fisher Scientific | 4408399 | |
| GlutaMAX | Thermo Fisher Scientific | 35050061 | |
| PBS, 10x Solution, pH 7.4 | Affymetrix | 75889 | |
| Penicillin and streptomycin | Thermo Fisher Scientific | 15140122 | |
| Platinum Taq DNA Polymerase High Fidelity | Thermo Fisher Scientific | 11304029 | |
| Qubit dsDNA HS Assay Kit | Thermo Fisher Scientific | Q32854 | |
| Recombinant S. pyogenes Cas9 nuclease | Integrated DNA Technologies | 1081058 | Components of the Cas9/gRNA complex |
| RPMI 1640 medium | Thermo Fisher Scientific | 31800-022 | |
| RPMI 1640 medium without phenol red | Thermo Fisher Scientific | 11835030 | |
| RUNX1a TaqMan gene expression assays | Thermo Fisher Scientific | 4331182 | Hs04186042_m1 |
| RUNX1b/c TaqMan gene expression assays | Thermo Fisher Scientific | 4331182 | Hs00231079_m1 |
| RUNX1c TaqMan gene expression assays | Thermo Fisher Scientific | 4331182 | Hs01021966_m1 |
| SuperScript III First-Strand Synthesis System | Thermo Fisher Scientific | 18080051 | |
| TaqMan Universal PCR Master Mix | Thermo Fisher Scientific | 4304437 | |
| tracrRNA | Integrated DNA Technologies | 1072533 | Components of the Cas9/gRNA complex |
| TRIzol Reagent | Thermo Fisher Scientific | 15596018 | |
| Unlabeled PCR reverse primer | Thermo Fisher Scientific | None |
Referencias
- Maston, G. A., Evans, S. K., Green, M. R. Transcriptional regulatory elements in the human genome. Annual Review of Genomics and Human Genetics. 7, 29-59 (2006).
- Shlyueva, D., Stampfel, G., Stark, A. Transcriptional enhancers: from properties to genome-wide predictions. Nature Reviews Genetics. 15 (4), 272-286 (2014).
- Kimura, K., et al. Diversification of transcriptional modulation: large-scale identification and characterization of putative alternative promoters of human genes. Genome Research. 16 (1), 55-65 (2006).
- Landry, J. R., Mager, D. L., Wilhelm, B. T. Complex controls: the role of alternative promoters in mammalian genomes. Trends in Genetics. 19 (11), 640-648 (2003).
- West, A. G., Gaszner, M., Felsenfeld, G. Insulators: many functions, many mechanisms. Genes & Development. 16 (3), 271-288 (2002).
- Riethoven, J. J. Regulatory regions in DNA: promoters, enhancers, silencers, and insulators. Methods in Molecular Biology. 674, 33-42 (2010).
- Thurman, R. E., et al. The accessible chromatin landscape of the human genome. Nature. 489 (7414), 75-82 (2012).
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 489 (7414), 57-74 (2012).
- Roadmap Epigenomics Consortium. Integrative analysis of 111 reference human epigenomes. Nature. 518 (7539), 317-330 (2015).
- Andersson, R., et al. An atlas of active enhancers across human cell types and tissues. Nature. 507 (7493), 455-461 (2014).
- Carninci, P., et al. The transcriptional landscape of the mammalian genome. Science. 309 (5740), 1559-1563 (2005).
- Bernstein, B. E., et al. The NIH roadmap epigenomics mapping consortium. Nature Biotechnology. 28 (10), 1045-1048 (2010).
- Zhou, S., Treloar, A. E., Lupien, M. Emergence of the Noncoding Cancer Genome: A Target of Genetic and Epigenetic Alterations. Cancer Discovery. 6 (11), 1215-1229 (2016).
- Khurana, E., et al. Role of non-coding sequence variants in cancer. Nature Reviews Genetics. 17 (2), 93-108 (2016).
- Herz, H. M., Hu, D., Shilatifard, A. Enhancer malfunction in cancer. Molecular Cell. 53 (6), 859-866 (2014).
- Willis, T. G., Dyer, M. J. The role of immunoglobulin translocations in the pathogenesis of B-cell malignancies. Blood. 96 (3), 808-822 (2000).
- Gröschel, S., et al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell. 157 (2), 369-381 (2014).
- Cheng, C. K., et al. RUNX1 upregulation via disruption of long-range transcriptional control by a novel t(5;21)(q13;q22) translocation in acute myeloid leukemia. Molecular Cancer. 17 (1), 133 (2018).
- Wiedenheft, B., Sternberg, S. H., Doudna, J. A. RNA-guided genetic silencing systems in bacteria and archaea. Nature. 482 (7385), 331-338 (2012).
- Bhaya, D., Davison, M., Barrangou, R. CRISPR-Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation. Annual Review of Genetics. 45, 273-297 (2011).
- Mali, P., et al. RNA-guided human genome engineering via Cas9. Science. 339 (6121), 823-826 (2013).
- Jinek, M., et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337 (6096), 816-821 (2012).
- Ran, F. A., et al. Genome engineering using the CRISPR-Cas9 system. Nature Protocols. 8 (11), 2281-2308 (2013).
- Cho, S. W., Lee, J., Carroll, D., Kim, J. S., Lee, J. Heritable gene knockout in Caenorhabditis elegans by direct injection of Cas9-sgRNA ribonucleoproteins. Genetics. 195 (3), 1177-1180 (2013).
- Kim, S., Kim, D., Cho, S. W., Kim, J., Kim, J. S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Research. 24 (6), 1012-1019 (2014).
- Lin, S., Staahl, B. T., Alla, R. K., Doudna, J. A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife. 3, 04766 (2014).
- Quentmeier, H., et al. Cell line OCI/AML3 bears exon-12 NPM gene mutation-A and cytoplasmic expression of nucleophosmin. Leukemia. 19 (10), 1760-1767 (2005).
- Heigwer, F., Kerr, G., Boutros, M. E-CRISP: fast CRISPR target site identification. Nature Methods. 11 (2), 122-123 (2014).
- Montague, T. G., Cruz, J. M., Gagnon, J. A., Church, G. M., Valen, E. CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Research. 42, 401-407 (2014).
- Doench, J. G., et al. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nature Biotechnology. 32 (12), 1262-1267 (2014).
- Haeussler, M., et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biology. 17 (1), 148 (2016).
- Ramlee, M. K., Wang, J., Cheung, A. M., Li, S. Using a Fluorescent PCR-capillary Gel Electrophoresis Technique to Genotype CRISPR/Cas9-mediated Knockout Mutants in a High-throughput Format. Journal of Visualized Experiments. (122), e55586 (2017).
- Canver, M. C., et al. Characterization of genomic deletion efficiency mediated by clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 nuclease system in mammalian cells. Journal of Biological Chemistry. 289 (31), 21312-21324 (2014).
- Sood, R., Kamikubo, Y., Liu, P. Role of RUNX1 in hematological malignancies. Blood. 129 (15), 2070-2082 (2017).
- Wang, H., et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 153 (4), 910-918 (2013).
- Yang, H., et al. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell. 154 (6), 1370-1379 (2013).
- Wright, J. B., Sanjana, N. E. CRISPR Screens to Discover Functional Noncoding Elements. Trends in Genetics. 32 (9), 526-529 (2016).
- Korkmaz, G., et al. Functional genetic screens for enhancer elements in the human genome using CRISPR-Cas9. Nature Biotechnology. 34 (2), 192-198 (2016).
- Cong, L., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Hwang, W. Y., et al. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nature Biotechnology. 31 (3), 227-229 (2013).
- Gratz, S. J., et al. Genome engineering of Drosophila with the CRISPR RNA-guided Cas9 nuclease. Genetics. 194 (4), 1029-1035 (2013).
- Chen, C., Fenk, L. A., de Bono, M. Efficient genome editing in Caenorhabditis elegans by CRISPR-targeted homologous recombination. Nucleic Acids Research. 41 (20), 193 (2013).
- Jacobs, T. B., LaFayette, P. R., Schmitz, R. J., Parrott, W. A. Targeted genome modifications in soybean with CRISPR/Cas9. BMC Biotechnology. 15, 16 (2015).
- Svitashev, S., et al. Targeted Mutagenesis, Precise Gene Editing, and Site-Specific Gene Insertion in Maize Using Cas9 and Guide RNA. Plant Physiology. 169 (2), 931-945 (2015).
- Schwank, G., et al. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell. 13 (6), 653-658 (2013).
- Ye, L., et al. Genome editing using CRISPR-Cas9 to create the HPFH genotype in HSPCs: An approach for treating sickle cell disease and β-thalassemia. Proceedings of the National Academy of Sciences of the United States of America. 113 (38), 10661-10665 (2016).
- Gabriel, R. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nature Biotechnology. 29 (9), 816-823 (2011).
- Gaj, T., Guo, J., Kato, Y., Sirk, S. J., Barbas, C. F. Targeted gene knockout by direct delivery of zinc-finger nuclease proteins. Nature Methods. 9 (8), 805-807 (2012).
- Zuris, J. A., et al. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nature Biotechnology. 33 (1), 73-80 (2015).
- Wang, M., et al. Efficient delivery of genome-editing proteins using bioreducible lipid nanoparticles. Proceedings of the National Academy of Sciences of the United States of America. 113 (11), 2868-2873 (2016).
- Gundry, M. C., et al. Highly Efficient Genome Editing of Murine and Human Hematopoietic Progenitor Cells by CRISPR/Cas9. Cell Reports. 17 (5), 1453-1461 (2016).
- Mout, R., et al. Direct Cytosolic Delivery of CRISPR/Cas9-Ribonucleoprotein for Efficient Gene Editing. ACS Nano. 11 (3), 2452-2458 (2017).
- Ramakrishna, S., et al. Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Research. 24 (6), 1020-1027 (2014).
- D'Astolfo, D. S., et al. Efficient intracellular delivery of native proteins. Cell. 161 (3), 674-690 (2015).
- Yen, J., et al. TRIAMF: A New Method for Delivery of Cas9 Ribonucleoprotein Complex to Human Hematopoietic Stem Cells. Scientific Reports. 8 (1), 16304 (2018).
- Sung, Y. H., et al. Highly efficient gene knockout in mice and zebrafish with RNA-guided endonucleases. Genome Research. 24 (1), 125-131 (2014).
- Martin, A., et al. CRISPR/Cas9 mutagenesis reveals versatile roles of Hox genes in crustacean limb specification and evolution. Current Biology. 26 (1), 14-26 (2016).
- Menoret, S., et al. Homology-directed repair in rodent zygotes using Cas9 and TALEN engineered proteins. Scientific Reports. 5, 14410 (2015).
- Woo, J. W., et al. DNA-free genome editing in plants with preassembled CRISPR-Cas9 ribonucleoproteins. Nature Biotechnology. 33 (11), 1162-1164 (2015).
- Malnoy, M., et al. DNA-free genetically edited grapevine and apple protoplast using CRISPR/Cas9 ribonucleoproteins. Frontiers in Plant Science. 7, 1904 (2016).
- Liang, Z., et al. Efficient DNA-free genome editing of bread wheat using CRISPR/Cas9 ribonucleoprotein complexes. Nature Communications. 8, 14261 (2017).
- Svitashev, S., Schwartz, C., Lenderts, B., Young, J. K., Mark Cigan, A. Genome editing in maize directed by CRISPR-Cas9 ribonucleoprotein complexes. Nature Communications. 7, 13274 (2016).
- Shin, S. E., et al. CRISPR/Cas9-induced knockout and knock-in mutations in Chlamydomonas reinhardtii. Scientific Reports. 6, 27810 (2016).
- 1000 Genomes Project Consortium et al. A global reference for human genetic variation. Nature. 526 (7571), 68-74 (2015).
- Klampfl, T., et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. The New England Journal of Medicine. 369 (25), 2379-2390 (2013).
- Döhner, H., et al. Diagnosis and management of AML in adults: 2017 ELN recommendations fro an international expert panel. Blood. 129 (4), 424-447 (2017).
- Barretina, J., et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 483 (7391), 603-607 (2012).
- Aguirre, A. J., et al. Genomic Copy Number Dictates a Gene-Independent Cell Response to CRISPR/Cas9 Targeting. Cancer Discovery. 6 (8), 914-929 (2016).
- Munoz, D. M., et al. CRISPR Screens Provide a Comprehensive Assessment of Cancer Vulnerabilities but Generate False-Positive Hits for Highly Amplified Genomic Regions. Cancer Discovery. 6 (8), 900-913 (2016).
- Gonçalves, E., et al. Structural rearrangements generate cell-specific, gene-independent CRISPR-Cas9 loss of fitness effects. Genome Biology. 20 (1), 27 (2019).
- Abudayyeh, O. O., et al. RNA targeting with CRISPR-Cas13. Nature. 550 (7675), 280-284 (2017).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados