
Method Article
Investigation of the Transcriptional Role of a RUNX1 Intronic Silencer par CRISPR/Cas9 Ribonucleoprotein in Acute Myeloid Leukemia Cells
Dans cet article
Résumé
La livraison directe des complexes préassemblés de ribonucleoprotein d'ARN de Cas9/guide est un moyen rapide et efficace pour l'édition de génome dans les cellules hématopoïétiques. Ici, nous utilisons cette approche pour supprimer un silencieux intronique RUNX1 et examiner les réponses transcriptionnelles dans les cellules leucémiques OCI-AML3.
Résumé
La majeure partie du génome humain (98 %) est composé de séquences non codantes. Leséléments cis-régulateurs (CEs) sont des séquences d'ADN non codantes qui contiennent des sites de liaison pour les régulateurs transcriptionnels afin de moduler l'expression des gènes. Des modifications des CRE ont été impliquées dans diverses maladies, y compris le cancer. Alors que les promoteurs et les exhausteurs ont été les principaux CRE pour l'étude de la régulation des gènes, très peu est connu sur le rôle du silencieux, qui est un autre type de CRE qui assure la médiation de la répression des gènes. Initialement identifié comme un système d'immunité adaptative chez les procaryotes, CRISPR/Cas9 a été exploité pour être un outil puissant pour l'édition du génome eucaryotique. Ici, nous présentons l'utilisation de cette technique pour supprimer un silencieux intronique dans le gène humain RUNX1 et étudier les impacts sur l'expression des gènes dans les cellules leucémiques OCI-AML3. Notre approche repose sur la livraison par électroporation de deux complexes préassemblés de ribonucleoprotéine (RNP) de Cas9/guide RNA (RNP) pour créer deux ruptures à double brin (DSB) qui flanquent le silencieux. Les suppressions peuvent être facilement examinées par analyse fragmentaire. Les analyses d'expression de différents ARNm transcrites par d'autres promoteurs aident à évaluer les effets dépendants des promoteurs. Cette stratégie peut être utilisée pour étudier d'autres CRE et convient particulièrement aux cellules hématopoïétiques, qui sont souvent difficiles à transfect avec des méthodes à base de plasmide. L'utilisation d'une stratégie sans plasmide et sans virus permet d'évaluer rapidement et simple les fonctions de régulation des gènes.
Introduction
Leséléments Cis-régulateurs (CEs) sont des séquences d'ADN non codantes qui contiennent des sites de liaison pour les régulateurs transcriptionnels pour contrôler l'expression des gènes1,2. Ces éléments sont généralement de 100 à 1 000 paires de base (bp). Les promoteurs et les exhausteurs sont les deux types les plus caractérisés d'ERC. Les promoteurs sont présents à proximité des sites de démarrage de transcription et constituent l'unité de base de la transcription. Beaucoup de gènes ont plus d'un promoteur et leur utilisation alternative contribue à la diversité transcriptome et la spécificité des tissus3,4. D'autre part, les exhausteurs activent la transcription et peuvent être situés en amont, en aval ou à l'intérieur des introns des gènes cibles. Les exhausteurs peuvent agir de loin (sur une mégabase) et indépendamment de l'orientation1,2. Les CRE comprennent également des silencieux et des isolants5,6. Le premier agit à l'opposé pour améliorer l'expression des gènes en se liant aux répresseurs transcriptionnels, tandis que le second divise le génome en domaines topologiquement discrets pour isoler les gènes d'autres CRE des domaines voisins. Ces éléments agissent de concert les uns avec les autres par le biais d'interactions de chromatine à courte et/ou longue portée et sont organisés en plaques de régulation pour diriger l'expression spatiotemporale appropriée des gènes. Les progrès récents dans les techniques de séquençage à haut débit ont accéléré l'identification et l'annotation fonctionnelle de nombreux CRE qui ont grandement facilité notre compréhension des réseaux transcriptionnels qui dictent le gène spécifique à la lignée expression dans différents types de cellules et de tissus7,8,9,10,11,12.
Étant donné les rôles fondamentaux des CRE dans la régulation de la transcription, leurs altérations peuvent conduire à une expression génique aberrante. Il a été démontré que les CRE sont fréquemment perturbés par des changements génétiques et épigénétiques dans différents types de cancers humains, contribuant ainsi à l'initiation, à la progression et à l'agressivité tumorales13,14. En outre, les facteurs contraignants CRE sont souvent mutés et/ou mal exprimés dans divers types de cancer, ce qui souligne encore plus l'importance de la déréglementation de la CRE dans l'oncogenèse15. Les CRE peuvent également être affectés par des aberrations structurelles, comme en témoignent les réarrangements chromosomiques fréquents de l'améliorateur de gène lourd d'immunoglobuline (IgH) qui ont comme conséquence l'activation anormale des oncogènes voisins dans les lymphomes de B-cellule16. Dans la leucémie myéloïde aigue (LAM), le repositionnement d'un seul exhausteur par les réarrangements de chromosome 3q provoque la régulation cofixe et l'activation d'EVI1, qui peuvent être potentiellement ciblées par l'inhibition bet des fonctions d'amélioration 17. Récemment, nous avons caractérisé une nouvelle translocation chromosomique impliquant la perturbation d'un silencieux intronique RUNX1 qui pourrait contribuer à la progression de la LAM chez un patient pédiatrique18. Ainsi, déchiffrer le génome du cancer non codant fournit des avenues fructueuses pour élucider la pathogénie de la maladie, la découverte de biomarqueurs et les interventions thérapeutiques, qui finissent par améliorer les résultats pour les patients.
La voie de nucléane CRISPR/Cas9, identifiée à l'origine comme système immunitaire adaptatif dans les cellules procaryotes, a été exploitée comme un moyen rapide et rentable pour l'édition génomique spécifique au site dans les cellules vivantes et les organismes19,20 ,21,22. Le système CRISPR/Cas9 comporte deux composantes principales : le gRNA et le streptocoque pyogenes-dérivéde cas9 nucléase. Le gRNA contient une séquence spécifique appelée protospacer qui reconnaît la région cible et dirige Cas9 pour l'édition. Le gRNA est composé de deux parties : l'ARN CRISPR (crRNA), généralement une séquence de nucléotide de 20 mer s'agence à l'ADN cible, et un crRNA trans-activant (tracrRNA), qui sert d'échafaudage liant pour la nucléase. Un motif adjacent protospacer (PAM) (5'-NGG) immédiatement adjacent au site cible est nécessaire pour le clivage Cas9 et le site de clivage est situé 3 nucléotides en amont du PAM. CRISPR/Cas9-mediated gene editing is commonly performed by transfecting cells avec un plasmide qui code Cas9 et le gRNA cloné23. Cependant, cette approche est difficile pour les cellules hématopoïétiques, qui sont souvent difficiles à transfect et nécessitent de longues méthodes de transduction à base de virus. Une autre approche est la livraison cellulaire directe des complexes RNP Cas9/gRNA préassemblés24. Une méthode courante pour la livraison de RNP est l'électroporation, qui génère des pores temporaires dans la membrane cellulaire, permettant ainsi l'entrée des complexes RNP dans les cellules25,26. Les avantages de cette approche comprennent la facilité d'utilisation, la réduction des effets hors cible et la stabilité des complexes RNP. Ici, nous décrivons un protocole d'utilisation de la méthode d'administration de RNP pour étudier le rôle transcriptionnel d'un silencieux intronique RUNX1 dans la ligne de cellules de leucémie OCI-AML318, qui a été établie à partir du sang périphérique d'un patient de LAM diagnostiqué avec le sous-type Français-américain-britannique M427. Le protocole comprend la conception de crRNA, la préparation des complexes RNP, l'électroporation ainsi que le dépistage et la caractérisation ultérieure des clones souhaités.
Protocole
1. Conception de crRNA
- Concevoir deux crRNAs, l'un 5' et l'autre 3' de la CRE cible à l'aide d'un outil de conception CHIPSR basé sur le Web28,29,30,31. Assurez-vous qu'un PAM de NGG est situé immédiatement en aval de la séquence cible pour la reconnaissance Cas9. La conception des deux CRRNAs (crRNA-1 et crRNA-2) pour la suppression du silencieux RUNX1 18 est présentée dans la figure 1.
REMARQUE: Un crRNA contient généralement un protospacer de 20 mers qui est complémentaire à la séquence cible. - Vérifiez la présence de polymorphismes nucléotides simples (SNP) /indels dans les séquences de PAM cibles et adjacentes en entrant les emplacements génomiques des séquences dans la boîte de recherche d'un navigateur de génome en ligne (p. ex., NCBI 1000 Genomes ou UCSC Genome Browser).
REMARQUE: Les SNP/indels communs ont une fréquence d'allèle mineure d'au moins 1 % dans la population générale. - Soumettez les séquences crRNA sélectionnées pour synthèse avec un fournisseur commercial. Aussi, acheter l'ARN tracr pour la formation duplex gRNA.
2. Conception des amorces de criblage de suppression
- Concevoir une paire d'amorces qui flanquent la région de suppression prévue. Assurez-vous que les amorces sont au moins 50 bp des sites de clivage Cas9 de sorte que l'amplification PCR est minimalement affectée par les indels formés sur les sites de clivage.
- Assurez-vous que l'amplicon est plus petit que 1200 bp (de préférence plus petit que 600 bp pour une meilleure résolution de taille). En outre, étiquetez l'un des amorces avec un colorant fluorescent (par exemple, 6-carboxyfluorescein (6-FAM)) à la 5'-extrémité pour la détection. Les amorces utilisées pour le criblage de la suppression du silencieux RUNX1 18 sont indiquées à la figure 1.
3. Préparation des complexes Cas9/gRNA RNP
- Resuspendre les crARN et tracrRNA dans un tampon 1x TE (10 mM Tris, 0,1 mM EDTA, pH 7,5) à une concentration finale de 200 M.
- Pour chaque crRNA, mélanger 2,2 L de 200 m crRNA, 2,2 L de 200 'M tracrRNA et 5,6 'L de 1x TE tampon (total 10 'L) dans un tube de 0,2 mL pour obtenir une concentration finale en duplex de 44 'M.
- Incuber à 95 oC pendant 5 min dans un thermocycleur. Laisser refroidir les tubes à température ambiante pour la formation complexe de gRNA.
- Diluer 10,4 L de 62 nucléanes Cas9 recombinantes avec 7,6 L de 1x salin tamponné par phosphate (PBS) pour obtenir une concentration finale de nucléane de 36 M.
REMARQUE: Ce montant est suffisant pour la préparation de deux complexes Cas9/gRNA. - Mélanger des volumes égaux (8,5 L) de la nucléase diluée avec chacun des duplex gRNA obtenus à partir de l'étape 3.3.
- Incuber à température ambiante pendant 20 min pour permettre la formation du complexe RNP. Conserver les mélanges sur la glace jusqu'à l'électroporation.
4. Électroporation des complexes RNP en cellules OCI-AML3
- Culture OCI-AML3 cellules dans RPMI 1640 milieu complété par 10 % de sérum bovin fœtal inactivé par la chaleur (FBS), 1x GlutaMAX, 100 unités/mL de pénicilline et 100 g/mL de streptomycine (ci-après appelé RPMI complet 1640 milieu) à 37 oC avec 5 % CO2.
- Compter les cellules à l'aide de taches bleues trypan. Assurer la viabilité de la cellule au moment de l'électroporation est de plus de 90%.
- Centrifugeuse 2,5 x 106 cellules dans un tube de 1,5 ml à 500 x g pendant 5 min à température ambiante. Retirer le supernatant et laver les cellules avec 1 ml de 1x PBS. Faites tourner à nouveau les cellules et retirez tous les supernatants résiduels.
- Resuspendre les cellules dans 163 'L de RPMI 1640 milieu sans phenol rouge. Ajouter 16,7 L de chaque complexe RNP (à partir de l'étape 3.6) et 3,6 'L de 100 'M Electroporation Enhancer aux cellules (volume total de 200 'L). Mélanger délicatement par pipetting.
REMARQUE: L'Améliorateur D'Electroporation est un ADN porteur pour améliorer l'efficacité d'édition. - Transférer le mélange dans une cuvette d'électroporation de 0,2 cm sans bulles. Effectuer l'électroporation avec un système d'électroporation (Mode: exponentiel; Tension : 150 V; Capacité de 700 'F; Résistance à 50 euros).
- Transférer les cellules dans un flacon de culture tissulaire T-25 contenant 6 ml de milieu rpMI 1640 complet et incuber à 37 oC avec 5 % de CO2.
5. Dépistage et sélection de clones cellulaires avec suppressions biallelic
- Diluer les cellules à 5 x 103/mL dans le rpMI complet 1640 moyen un jour après l'électroporation. Ajouter 100 L de la suspension cellulaire diluée dans chaque puits de plaques de culture tissulaire de 96 puits et permettre aux cellules de se développer pendant 7-14 jours.
- Extraire l'ADN génomique des cellules à l'aide d'un système de purification à haut débit.
- Ajouter 100 l de fixation de plaque et tampon de lyse à chaque puits d'une plaque d'extraction de 96 puits. Ajouter 5 x 104 cellules resuspendues dans 10 'L de 1x PBS dans le tampon et les mélanger par pipetting.
- Incuber à température ambiante pendant 30 min pour permettre la liaison de l'ADN génomique aux puits.
- Aspirez la solution des puits sans gratter les surfaces du puits. Laver les puits à l'œil de 120 l'amorti de lavage.
- Sécher à l'air les puits contenant l'ADN lié.
- Préparer 20 l de mélange PCR (2 'L de 10x high Fidelity buffer, 0.8 'L de 50 mM MgSO4, 0.4 'L de 10 mM dNTP, 0.4 'L de 10 'M FAM-étiqueté amorce avant (étape 2.2), 0.4 'L de 10 'M inversé non étiqueté et 0,4 U de Taq'
REMARQUE: Préparer un mélange maître pour s'assurer de l'ajout de quantités normalisées de réactifs dans chaque échantillon. - Ajouter le mélange de PCR à chaque puits de la plaque d'extraction et faire fonctionner les réactions dans un thermocycleur (Conditions : initiale de 94 oC pendant 2 min, suivie de 35 cycles de 94 oC pour 15 s, 56 oC pour 30 s et 68 oC pendant 1 min).
- Estimer la quantité des produits en mesurant leurs concentrations dans un certain nombre d'échantillons à l'aide d'un fluoromètre. Diluer tous les échantillons avec H2O sans nauséalence à 0,5 ng/L.
- Mélanger 1 l des produits PCR dilués avec 8,5 l de formamide déionisé et 0,5 l de norme de taille fluorescente étiquetée colorant dans une plaque de 96 puits compatible avec l'analyseur génétique.
REMARQUE: Préparer un mélange maître contenant du formamide déionisé et la norme de taille. - Couvrir la plaque d'une plaque septa et dénaturer les échantillons à 95 oC pendant 3 min dans un thermocycleur. Ne fermez pas le couvercle de la machine.
- Effectuer l'électrophoresis de gel capillaire pour séparer les produits étiquetés PCR comme précédemment décrit32.
- Après l'électrophoresis, ouvrez le logiciel d'analyse pour analyser les résultats.
- Cliquez sur Nouveau Projet et sélectionnez Microsatellite. Ensuite, cliquez sur OK.
- Cliquez sur Ajouter des échantillons au projet et sélectionnez les fichiers de résultats (contiennent l'extension .fsa). Cliquez ensuite sur Ajouter à la liste pour importer les fichiers.
- Dans le tableau montrant les fichiers de résultats sélectionnés, choisissez Microsatellite Par défaut dans la colonne Méthode d'analyse. En outre, sélectionnez la norme de taille utilisée dans la colonne Taille standard. Cliquez ensuite sur l'icône Analyse, entrez le nom de l'expérience et enregistrez l'expérience.
- Cliquez sur Afficher les parcelles pour afficher les résultats et choisir l'analyse des fragments dans le paramètre de l'intrigue.
- Choisissez les canaux colorés appropriés pour l'analyse. Vérifiez l'icône orange pour afficher les fragments étiquetés dans la norme de taille pour évaluer la qualité des appels de taille.
- Vérifiez l'icône bleue pour afficher les produits PCR étiquetés. Identifier les pics qui correspondent aux produits de type sauvage et mutant (c.-à-d. porteurs des suppressions attendues). Estimer le niveau de mutant dans chaque échantillon en divisant la zone sous le pic mutant par la somme de la zone sous les pics sauvages et mutants.
- Sélectionnez plusieurs pools cellulaires avec des niveaux élevés de suppressions attendues pour d'autres dilutions en série.
- Répétez l'extraction de l'ADN, le PCR fluorescent et les étapes d'électrophorèse capillaire. Sélectionnez les clones cellulaires avec des niveaux mutants :95 % représentant des suppressions bialleliques pour des analyses ultérieures.
- Vérifier l'identité des suppressions dans les clones sélectionnés par séquençage Sanger.
6. Analyses fonctionnelles de la suppression du silencieux par analyse quantitative RT-PCR en temps réel
- Extraire l'ARN total des clones sélectionnés et effectuer la synthèse complémentaire de l'ADN (ADNc).
- Mélanger 1 g d'ARN avec 1 oL de 50 oM oligo (dT)20 apprêt et 1 l de mélange de 10 ml de dNTP dans un volume total de 10 oL. Inc. Inc. pendant 5 min, puis placer sur la glace pendant au moins 1 min.
- Ajouter 10 l de mélange de synthèse de cDNA contenant 2 l de tampon de RT 10x, 4 oL de 25 mMg MgCl2, 2 l de 0,1 M DTT, 1 l d'inhibiteur de la RNase (40 U/L) et 1 l de transcriptase inversée (200 U/L). Incuber à 50 oC pendant 50 min, puis 85 oC pendant 5 min dans un thermocycleur.
REMARQUE: Préparer un mix maître pour la transcription inversée. - Refroidir les échantillons sur la glace. Ajouter 1 oL de RNase H et incuber à 37 oC pendant 20 min. Conserver l'ADNc à -20 oC.
- Amorces de conception et sondes TaqMan qui reconnaissent spécifiquement les variantes de transcription individuelles générées par des promoteurs alternatifs.
REMARQUE: Des ensembles d'amorce/sonde spécifiques à la transcription préconçus sont disponibles dans le commerce. - Clone fragments d'ADN contenant les séquences de transcription spécifiques dans l'ADN plasmide. Préparer une série de dilution supplicées (106 à 10 copies) des plasmides recombinants en tant que courbes standard pour la quantification de la transcription.
- Préparer 20 l de mélange PCR (0,5 l de modèle d'ADN, 1 L de 20x d'analyse de sonde/primer TaqMan préconçue et 10 l de 2x TaqMan PCR Master Mix) pour chaque échantillon (normes cDNA et plasmide). Mesurer chaque échantillon en triple.
REMARQUE: Préparer un master mix pour le PCR en temps réel. - Exécuter les réactions dans une machine PCR en temps réel (Conditions : initiale de 50 oC pour 2 min et 95 oC pendant 10 min, suivie de 40 cycles de 94 oC pour 15 s et 60 oC pour 1 min).
- Après l'amplification, cliquez sur l'icône Analyze dans le logiciel pour analyser les données. Vérifier la pente et le coefficient de corrélation des courbes standard pour évaluer l'efficacité et la linéarité des réactions. Assurez-vous que les pentes se sitrouvent entre -3,1 et -3,6 et que les coefficients de corrélation sont supérieurs à 0,99.
- Normaliser le numéro de copie des transcriptions cibles de chaque échantillon avec un gène d'entretien ménager (p. ex. GAPDH).
Résultats
Le but de cette expérience est de supprimer un silencieux intronique dans le gène RUNX1 et d'examiner les impacts sur la transcription RUNX1 dans les cellules OCI-AML3. Le silencieux a été identifié par une approche moléculaire combinatoire et s'est avéré contenir un élément de base de 209 pb18. Afin de permettre une évaluation plus précise de cet élément central dans le contrôle de l'expression RUNX1, les CRRNAs (crRNA-1 et crRNA-2) ont été conçus pour cibler étroitement cette région18 (figure 1). Les sites de clivage Cas9 prévus apportés par crRNA-1 et crRNA-2 étaient respectivement de 29 pb et 35 pb de l'élément central (figure 1). Il convient de noter que, bien que les sites PAM des deux CRRNAs résident sur des brins opposés, les ORD se produiront indépendamment de l'emplacement de la séquence PAM. Ainsi, l'introduction concomitante de deux complexes Cas9/gRNA RNP guidés par crRNA-1 et crRNA-2 devrait exciser l'élément silencieux du locus RUNX1.
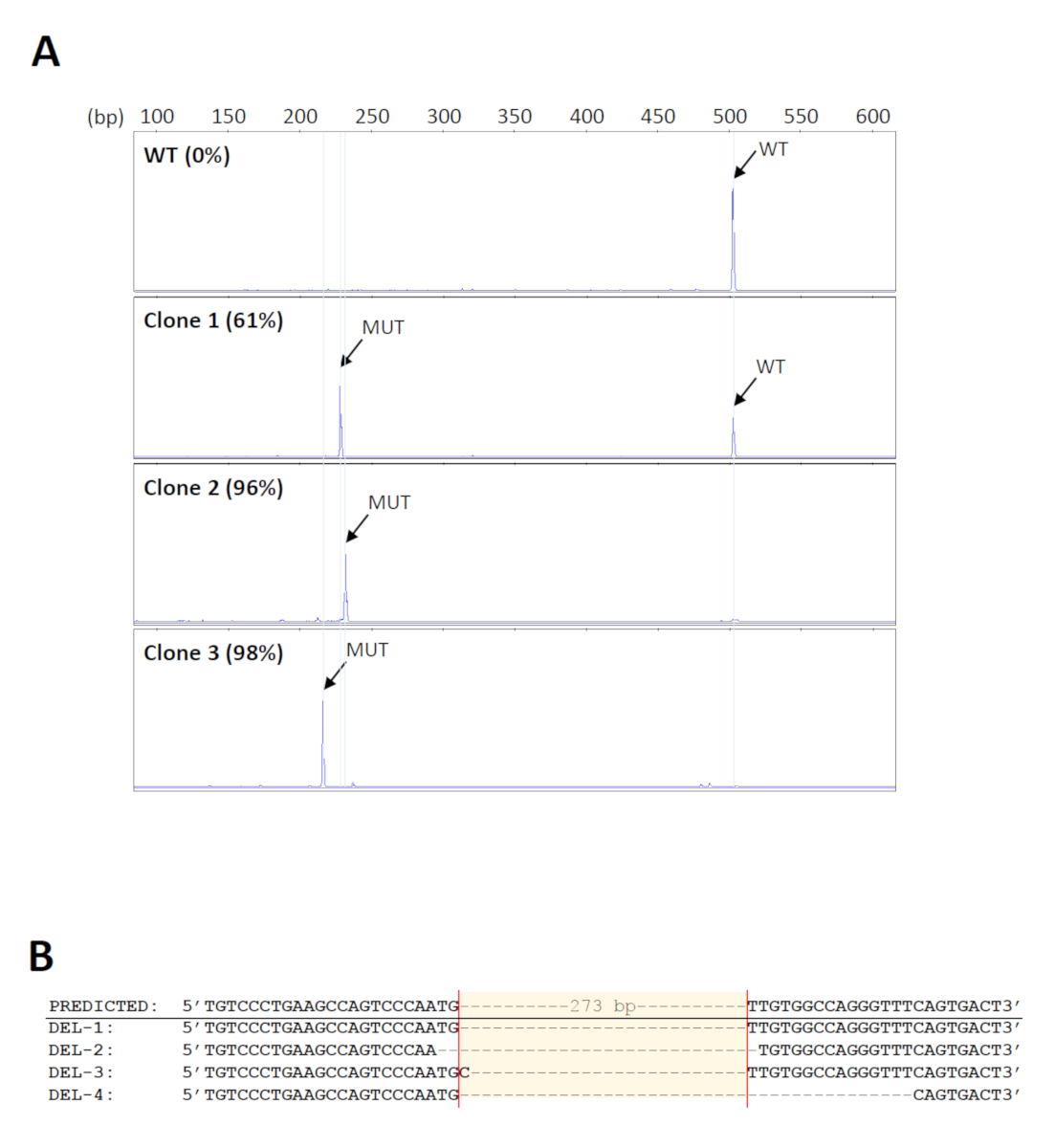
Pour dépister les suppressions souhaitées dans un grand nombre d'échantillons, un format de 96 puits de kit d'extraction d'ADN génomique a été utilisé pour la purification à haut débit. En outre, l'ADN lié sur les puits des plaques d'extraction peut être soumis à une amplification directe, minimisant ainsi les erreurs ou la contamination dues au transfert répété d'échantillons. Les amorces utilisées pour le criblage des suppressions18 sont indiquées à la figure 1. La taille prévue du produit PCR de type sauvage est d'environ 500 pb. Étant donné que la suppression prévue s'étend sur 273 pb, le produit mutant devrait être d'environ 230 pb. Cette gamme de taille permet une analyse de fragments simple et rapide par électrophorèse de gel capillaire. Un total de 160 pools cellulaires initiaux ont été examinés et 14 ont été trouvés pour porter les suppressions attendues avec des niveaux mutants d'au moins 70%. Cinq pools ont ensuite été sélectionnés pour d'autres dilutions en série afin d'identifier les clones portant des suppressions bialleliques. Les électrophétographies représentatives provenant de clones cellulaires ayant différents niveaux de produits mutants sont indiquées à la figure 2A. L'identité des suppressions a été vérifiée par séquençage De Sanger (Figure 2B). Comme prévu, des indels formés par la réparation non-homologue de jointure des DSB s'est produit33 dans les sites de clivage prévus dans les clones de suppression. Il en est résulté l'amplification de produits mutants de différentes tailles, qui pourraient également être détectés par électrophoresis capillaire (Figure 2A).
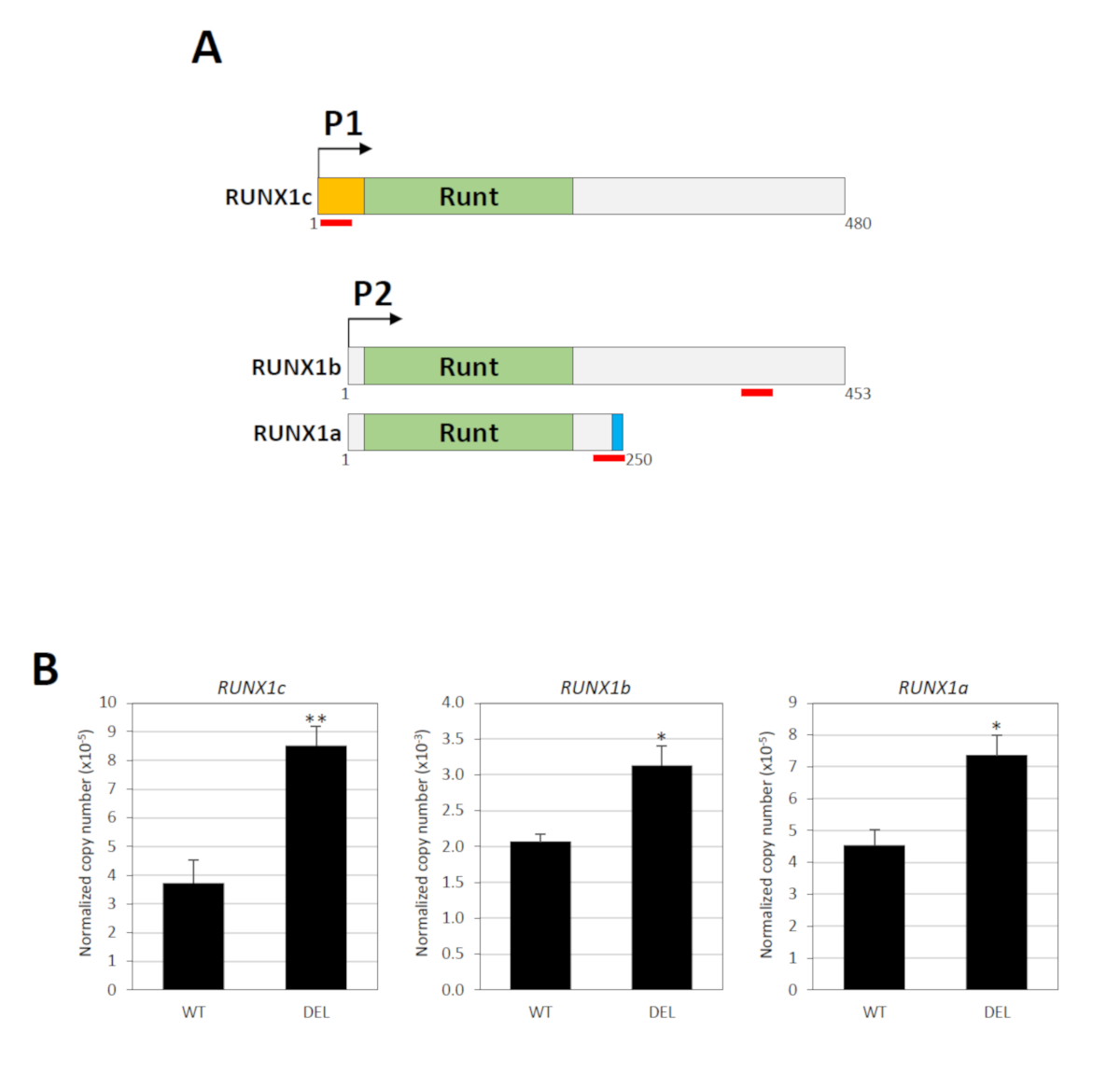
Le gène RUNX1 contient deux promoteurs, à savoir le P1 distal et proximal P2, qui sont séparés par un grand intron abritant l'élément silencieux34. Trois transcriptions majeures de l'ARNm sont produites par ces promoteurs : RUNX1c par P1 et RUNX1a et RUNX1b par P234. La séquence de nucléotide de RUNX1c et RUNX1b sont identiques sauf que le premier a un terminus N unique, à partir duquel un essai spécifique d'expression génique TaqMan peut être conçu (Figure 3A). Pour mesurer RUNX1b, un essai TaqMan reconnaissant à la fois RUNX1b et RUNX1c a été utilisé (Figure 3A). Les niveaux de RUNX1b ont ensuite été déterminés en soustrayant le total RUNX1b/RUNX1c de RUNX1c. RUNX1a est un isoforme nettement plus court en raison de l'épissage alternatif et un essai spécifique TaqMan est disponible pour cette variante (Figure 3A). Ainsi, l'activité des promoteurs P1 et P2 peut être déterminée individuellement. Le RT-PCR quantitatif en temps réel a montré que la suppression de l'élément silencieux a considérablement modifié les niveaux d'expression des transcriptions dérivées de P1 et P2 (figure3B).

Figure 1 : Stratégie de suppression du silencieux intronic RUNX1. Le silencieux (boîte rouge) est situé dans le premier intron du gène RUNX1 séparant les deux promoteurs P1 et P2 (les coordonnées hg19 sont montrées). Deux CRRNAs (crRNA-1 et crRNA-2) ont été conçus pour introduire des DSB flanquant l'élément silencieux. Les sites de clivage Cas9 prévus sont indiqués par des lignes rouges verticales et les sites PAM (NGG) sont en violet. Les deux amorces utilisées pour le criblage des suppressions sont représentées par des flèches ouvertes. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 2 : Identification des clones de suppression. (A) Électrophéromies représentatifs de clones cellulaires montrant différents niveaux de produits MUTANTs PCR (MUT). La taille des produits mutants varie d'un clone à l'autre en raison des indels formés sur les sites de clivage. WT - type sauvage. (B) Vérification des suppressions de clones représentatifs par séquençage Sanger. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 3 : Conséquences fonctionnelles de la suppression du silencieux. (A) Les trois principaux isoformes RUNX1 (RUNX1a, RUNX1b et RUNX1c) sont présentés. Ces variantes contiennent le même domaine de liaison d'ADN Runt mais différents N- (orange) ou C-terminus (bleu). L'emplacement des paires de sondes/amorces TaqMan est indiqué par des lignes rouges. Les chiffres indiquent les résidus d'acides aminés. (B) Analyse quantitative RT-PCR en temps réel des transcriptions dérivées de RUNX1 P1- et P2 dans les populations cellulaires avec (DEL) ou sans (WT) les suppressions bialleliques18. GAPDH a été utilisé pour la normalisation. et l'indique P 'lt; 0.05 et P 'lt; 0.01, respectivement par le test de Mann-Whitney. Ce chiffre a été modifié à partir de Cheng et coll.18. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}
Discussion
Le système CRISPR/Cas9 a été utilisé dans un large éventail d'applications d'édition du génome telles que les études de gène knock-in et knock-in35,36, régulation transcriptionnelle37,38, génie génétique de divers organismes modèles39,40,41,42,43,44 et thérapie génique45,46. Ici, nous démontrons l'utilisation de CRISPR/Cas9 pour étudier les conséquences fonctionnelles de la suppression d'un silencieux intronique sur le gène RUNX1. La livraison des composants CRISPR dans notre approche ne s'appuyait pas sur l'ADN plasmide, le clonage de gRNA ou de virus, mais sur l'électroporation des complexes RNP Cas9/gRNA préassemblés. Il a été démontré que l'utilisation de l'ADN exogène peut être associée à l'intégration indésirable de séquences vectorielles étrangères dans le génome de l'hôte, à une toxicité accrue et à une faible efficacité25,47,48, alors que les méthodes de transduction du virus prennent beaucoup de temps. En outre, l'expression prolongée de Cas9 à partir de l'ADN plasmide peut augmenter les effets hors cible48. Au contraire, l'approche de livraison directe basée sur le RNP a été établie comme la méthode préférée car elle est rapide et directe avec une efficacité d'édition améliorée, la sélectivité et la viabilité cellulaire. En effet, une variété de méthodes telles que lipofection49,50, électroporation25,51, nanoparticules52, peptides pénétrant les cellules53, iTOP54 et TRIAMF 55 ont été développés pour une livraison efficace de CRISPR/Cas9 dans divers types de cellules ainsi que sur des espèces animales et végétales24,25,26,56,57 , 58 Annonces , 59 Annonces , 60 Annonces , 61 Annonces , 62 Annonces , 63. Étant donné que les séquences d'ADN non codantes sont des points chauds des variations génétiques64, la vérification de la présence de SNP/indels communs dans les séquences de PAM cibles et voisines est particulièrement pertinente lors de la conception de l'ARNc qui cible les rudiments.
Un goulot d'étranglement dans l'édition du génome CRISPR/Cas9 implique le dépistage des clones mutants désirés dans un grand nombre d'échantillons. Nous avons employé le PCR fluorescent couplé avec l'électrophoresis capillaire de gel pour le criblage car la mutation cible est une petite suppression génomique d'environ 300 bp. Cette méthode est rapide et sensible et peut être effectuée d'une manière à haut débit. En outre, cette méthode permet une estimation précise des niveaux de mutants et des tailles de suppression simultanément. En outre, l'analyse multiplexe des fragments de PCR étiquetés avec différents colorants fluorescents est prise en charge. Nous avons été couramment en utilisant cette technique pour génotype de petites insertions / suppressions dans les néoplasmes myéloïdes65,66. D'après notre expérience, nous pouvons constamment détecter des tailles de fragments qui diffèrent de 4 pb avec une grande précision et un fardeau mutant jusqu'à 3 %. Cependant, il convient de noter que cette méthode a une limite de taille fragmentaire de 1200 bp, et donc il n'est pas approprié pour le criblage des grandes suppressions. De plus, les substitutions de base (résultant en des fragments inchangés) et les événements hors cible potentiels dans d'autres régions génomiques ne peuvent pas être détectés. Pour ce dernier, le séquençage du génome entier coûteux est nécessaire pour dresser un profil complet des changements indésirables globaux dans les clones cibles. Adopter notre approche actuelle pour l'étude de grandes séquences réglementaires non codantes (1 000 pb), une suppression détaillée et des analyses mutagénagénagénagénagénagénèse des sites de liaison des facteurs de transcription putatifs à l'aide d'essais de gènes reporter in vitro peuvent être effectuées à l'avance de délimiter la région fonctionnelle minimale pour CRISPR/Cas9 édition18.
Comme de nombreux gènes contiennent plus d'un promoteur3,4, il est important d'être conscient de l'existence de promoteurs alternatifs dans le locus gène cible que la manipulation des éléments réglementaires peuvent affecter les promoteurs différentiellement. Ainsi, les variantes de transcription dérivées de différents promoteurs doivent être mesurées individuellement pour évaluer les réponses spécifiques aux promoteurs. L'utilisation d'essais basés sur la sonde TaqMan est préférée à SYBR Green en raison d'une meilleure spécificité et reproductibilité. Si le système PCR numérique plus avancé est disponible, la quantification de transcription peut être exécutée plus précisément sans avoir besoin de construction de courbe standard.
Une considération importante dans l'exécution des expériences CRISPR/Cas9 dans les lignées de cellules cancéreuses est le numéro de copie de ploidy et de gène cible dans les cellules utilisées comme pratiquement toutes les lignées de cellules cancéreuses hébergent des altérations génétiques comprenant des variations structurales et de nombre de copie. Dans notre cas, OCI-AML3 a un karyotype hyperdiploïde avec 45 à 50 chromosomes. En outre, la lignée cellulaire s'est avérée porter un numéro de copie RUNX1 normal tel que révélé à partir de l'Encyclopédie67 de la lignée cellulaire du cancer et des études d'hybridation in situ18. Lorsque vous ciblez un gène avec un gain de numéro de copie, la méthode d'administration peut avoir besoin d'être optimisée pour fournir des niveaux suffisants des composants CRISPR pour l'édition. En outre, plus de clones peuvent avoir besoin d'être examinés afin d'identifier les KO complets. Fait important, il a été démontré que le ciblage dans les régions génomiques amplifiées, en particulier celles causées par des réarrangements structurels, peut déclencher des réponses antiprolimiques indépendantes des gènes dans les cellules cancéreuses, conduisant à des résultats faussement positifs dans les gènes études fonctionnelles68,69,70. À cet égard, d'autres approches comme l'interférence de l'ARN (ARNi) knockdown et / ou cDNA surexpression devrait être utilisé pour vérifier les résultats CRISPR. En outre, plusieurs lignées cellulaires devraient être utilisées pour éviter une mauvaise interprétation des effets d'édition CRISPR spécifiques à la ligne cellulaire mais indépendants des gènes.
Le système CRISPR/Cas9 a révolutionné la recherche fondamentale et translationnelle en fournissant un moyen simple et efficace d'édition du génome. Ici nous démontrons la facilité d'utiliser CRISPR/Cas9 pour perturber un silencieux intronique pour des études transcriptionnelles dans une lignée de cellules cancéreuses. Cette technique permet l'étude des CRE au niveau de l'ADN et offre la possibilité d'examiner les fonctions CRE dans le contexte endogène plutôt que les gènes traditionnels de reporter hétérologue. Récemment, un système d'édition d'ARN basé sur CRISPR a également été identifié71 et peut servir de nouvel outil pour étudier les CRE en ciblant l'ARN transcrit à partir des éléments réglementaires. En combinant avec des techniques de capture de conformation chromosomique, CRISPR/Cas9 aidera certainement à déchiffrer les participations des CRE dans l'organisation altérée du génome et l'expression des gènes liées à divers problèmes de santé.
Déclarations de divulgation
Les auteurs n'ont rien à révéler.
Remerciements
Les auteurs aimeraient remercier le professeur M.D. Minden (Princess Margaret Cancer Centre, University Health Network, Toronto, Canada) d'avoir fourni la lignée cellulaire OCI-AML3. En outre, les auteurs tiens à remercier les services publics de base de la génomique du cancer et de la pathobiologie (Université chinoise de Hong Kong) pour fournir les installations et l'aide à l'appui de cette recherche.
matériels
| Name | Company | Catalog Number | Comments |
| 0.2 cm gap electroporation cuvette | Bio-Rad | 1652086 | |
| 1×TE buffer, pH 7.5 | Integrated DNA Technologies | 11-01-02-02 | |
| 10 mM dNTP mix | Thermo Fisher Scientific | 18427013 | |
| 3500 Genetic Analyzer | Thermo Fisher Scientific | 4405673 | |
| 6-FAM-labeled fluorescent PCR forward primer | Thermo Fisher Scientific | None | |
| 7300 Real-Time PCR System | Thermo Fisher Scientific | None | |
| 7300 System SDS Software | Thermo Fisher Scientific | None | Version 1.3.1 |
| Bio-Rad Gene Pulser Xcell system | Bio-Rad | 1652660 | |
| ChargeSwitch Direct gDNA Purification Kit, 96-well | Thermo Fisher Scientific | CS11205 | |
| crRNA-1 | Integrated DNA Technologies | Alt-R CRISPR-Cas9 crRNA | Components of the Cas9/gRNA complex |
| crRNA-2 | Integrated DNA Technologies | Alt-R CRISPR-Cas9 crRNA | Components of the Cas9/gRNA complex |
| Deionized formamide | Thermo Fisher Scientific | 4311320 | |
| Electroporation Enhancer | Integrated DNA Technologies | 1075915 | |
| Fetal bovine serum | Thermo Fisher Scientific | 10270098 | |
| Fluorometer | Thermo Fisher Scientific | Q32857 | |
| GeneMapper Software 5 | Thermo Fisher Scientific | 4475073 | |
| GeneScan 600 LIZ Size Standard | Thermo Fisher Scientific | 4408399 | |
| GlutaMAX | Thermo Fisher Scientific | 35050061 | |
| PBS, 10x Solution, pH 7.4 | Affymetrix | 75889 | |
| Penicillin and streptomycin | Thermo Fisher Scientific | 15140122 | |
| Platinum Taq DNA Polymerase High Fidelity | Thermo Fisher Scientific | 11304029 | |
| Qubit dsDNA HS Assay Kit | Thermo Fisher Scientific | Q32854 | |
| Recombinant S. pyogenes Cas9 nuclease | Integrated DNA Technologies | 1081058 | Components of the Cas9/gRNA complex |
| RPMI 1640 medium | Thermo Fisher Scientific | 31800-022 | |
| RPMI 1640 medium without phenol red | Thermo Fisher Scientific | 11835030 | |
| RUNX1a TaqMan gene expression assays | Thermo Fisher Scientific | 4331182 | Hs04186042_m1 |
| RUNX1b/c TaqMan gene expression assays | Thermo Fisher Scientific | 4331182 | Hs00231079_m1 |
| RUNX1c TaqMan gene expression assays | Thermo Fisher Scientific | 4331182 | Hs01021966_m1 |
| SuperScript III First-Strand Synthesis System | Thermo Fisher Scientific | 18080051 | |
| TaqMan Universal PCR Master Mix | Thermo Fisher Scientific | 4304437 | |
| tracrRNA | Integrated DNA Technologies | 1072533 | Components of the Cas9/gRNA complex |
| TRIzol Reagent | Thermo Fisher Scientific | 15596018 | |
| Unlabeled PCR reverse primer | Thermo Fisher Scientific | None |
Références
- Maston, G. A., Evans, S. K., Green, M. R. Transcriptional regulatory elements in the human genome. Annual Review of Genomics and Human Genetics. 7, 29-59 (2006).
- Shlyueva, D., Stampfel, G., Stark, A. Transcriptional enhancers: from properties to genome-wide predictions. Nature Reviews Genetics. 15 (4), 272-286 (2014).
- Kimura, K., et al. Diversification of transcriptional modulation: large-scale identification and characterization of putative alternative promoters of human genes. Genome Research. 16 (1), 55-65 (2006).
- Landry, J. R., Mager, D. L., Wilhelm, B. T. Complex controls: the role of alternative promoters in mammalian genomes. Trends in Genetics. 19 (11), 640-648 (2003).
- West, A. G., Gaszner, M., Felsenfeld, G. Insulators: many functions, many mechanisms. Genes & Development. 16 (3), 271-288 (2002).
- Riethoven, J. J. Regulatory regions in DNA: promoters, enhancers, silencers, and insulators. Methods in Molecular Biology. 674, 33-42 (2010).
- Thurman, R. E., et al. The accessible chromatin landscape of the human genome. Nature. 489 (7414), 75-82 (2012).
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 489 (7414), 57-74 (2012).
- Roadmap Epigenomics Consortium. Integrative analysis of 111 reference human epigenomes. Nature. 518 (7539), 317-330 (2015).
- Andersson, R., et al. An atlas of active enhancers across human cell types and tissues. Nature. 507 (7493), 455-461 (2014).
- Carninci, P., et al. The transcriptional landscape of the mammalian genome. Science. 309 (5740), 1559-1563 (2005).
- Bernstein, B. E., et al. The NIH roadmap epigenomics mapping consortium. Nature Biotechnology. 28 (10), 1045-1048 (2010).
- Zhou, S., Treloar, A. E., Lupien, M. Emergence of the Noncoding Cancer Genome: A Target of Genetic and Epigenetic Alterations. Cancer Discovery. 6 (11), 1215-1229 (2016).
- Khurana, E., et al. Role of non-coding sequence variants in cancer. Nature Reviews Genetics. 17 (2), 93-108 (2016).
- Herz, H. M., Hu, D., Shilatifard, A. Enhancer malfunction in cancer. Molecular Cell. 53 (6), 859-866 (2014).
- Willis, T. G., Dyer, M. J. The role of immunoglobulin translocations in the pathogenesis of B-cell malignancies. Blood. 96 (3), 808-822 (2000).
- Gröschel, S., et al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell. 157 (2), 369-381 (2014).
- Cheng, C. K., et al. RUNX1 upregulation via disruption of long-range transcriptional control by a novel t(5;21)(q13;q22) translocation in acute myeloid leukemia. Molecular Cancer. 17 (1), 133 (2018).
- Wiedenheft, B., Sternberg, S. H., Doudna, J. A. RNA-guided genetic silencing systems in bacteria and archaea. Nature. 482 (7385), 331-338 (2012).
- Bhaya, D., Davison, M., Barrangou, R. CRISPR-Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation. Annual Review of Genetics. 45, 273-297 (2011).
- Mali, P., et al. RNA-guided human genome engineering via Cas9. Science. 339 (6121), 823-826 (2013).
- Jinek, M., et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337 (6096), 816-821 (2012).
- Ran, F. A., et al. Genome engineering using the CRISPR-Cas9 system. Nature Protocols. 8 (11), 2281-2308 (2013).
- Cho, S. W., Lee, J., Carroll, D., Kim, J. S., Lee, J. Heritable gene knockout in Caenorhabditis elegans by direct injection of Cas9-sgRNA ribonucleoproteins. Genetics. 195 (3), 1177-1180 (2013).
- Kim, S., Kim, D., Cho, S. W., Kim, J., Kim, J. S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Research. 24 (6), 1012-1019 (2014).
- Lin, S., Staahl, B. T., Alla, R. K., Doudna, J. A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife. 3, 04766 (2014).
- Quentmeier, H., et al. Cell line OCI/AML3 bears exon-12 NPM gene mutation-A and cytoplasmic expression of nucleophosmin. Leukemia. 19 (10), 1760-1767 (2005).
- Heigwer, F., Kerr, G., Boutros, M. E-CRISP: fast CRISPR target site identification. Nature Methods. 11 (2), 122-123 (2014).
- Montague, T. G., Cruz, J. M., Gagnon, J. A., Church, G. M., Valen, E. CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Research. 42, 401-407 (2014).
- Doench, J. G., et al. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nature Biotechnology. 32 (12), 1262-1267 (2014).
- Haeussler, M., et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biology. 17 (1), 148 (2016).
- Ramlee, M. K., Wang, J., Cheung, A. M., Li, S. Using a Fluorescent PCR-capillary Gel Electrophoresis Technique to Genotype CRISPR/Cas9-mediated Knockout Mutants in a High-throughput Format. Journal of Visualized Experiments. (122), e55586 (2017).
- Canver, M. C., et al. Characterization of genomic deletion efficiency mediated by clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 nuclease system in mammalian cells. Journal of Biological Chemistry. 289 (31), 21312-21324 (2014).
- Sood, R., Kamikubo, Y., Liu, P. Role of RUNX1 in hematological malignancies. Blood. 129 (15), 2070-2082 (2017).
- Wang, H., et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 153 (4), 910-918 (2013).
- Yang, H., et al. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell. 154 (6), 1370-1379 (2013).
- Wright, J. B., Sanjana, N. E. CRISPR Screens to Discover Functional Noncoding Elements. Trends in Genetics. 32 (9), 526-529 (2016).
- Korkmaz, G., et al. Functional genetic screens for enhancer elements in the human genome using CRISPR-Cas9. Nature Biotechnology. 34 (2), 192-198 (2016).
- Cong, L., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Hwang, W. Y., et al. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nature Biotechnology. 31 (3), 227-229 (2013).
- Gratz, S. J., et al. Genome engineering of Drosophila with the CRISPR RNA-guided Cas9 nuclease. Genetics. 194 (4), 1029-1035 (2013).
- Chen, C., Fenk, L. A., de Bono, M. Efficient genome editing in Caenorhabditis elegans by CRISPR-targeted homologous recombination. Nucleic Acids Research. 41 (20), 193 (2013).
- Jacobs, T. B., LaFayette, P. R., Schmitz, R. J., Parrott, W. A. Targeted genome modifications in soybean with CRISPR/Cas9. BMC Biotechnology. 15, 16 (2015).
- Svitashev, S., et al. Targeted Mutagenesis, Precise Gene Editing, and Site-Specific Gene Insertion in Maize Using Cas9 and Guide RNA. Plant Physiology. 169 (2), 931-945 (2015).
- Schwank, G., et al. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell. 13 (6), 653-658 (2013).
- Ye, L., et al. Genome editing using CRISPR-Cas9 to create the HPFH genotype in HSPCs: An approach for treating sickle cell disease and β-thalassemia. Proceedings of the National Academy of Sciences of the United States of America. 113 (38), 10661-10665 (2016).
- Gabriel, R. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nature Biotechnology. 29 (9), 816-823 (2011).
- Gaj, T., Guo, J., Kato, Y., Sirk, S. J., Barbas, C. F. Targeted gene knockout by direct delivery of zinc-finger nuclease proteins. Nature Methods. 9 (8), 805-807 (2012).
- Zuris, J. A., et al. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nature Biotechnology. 33 (1), 73-80 (2015).
- Wang, M., et al. Efficient delivery of genome-editing proteins using bioreducible lipid nanoparticles. Proceedings of the National Academy of Sciences of the United States of America. 113 (11), 2868-2873 (2016).
- Gundry, M. C., et al. Highly Efficient Genome Editing of Murine and Human Hematopoietic Progenitor Cells by CRISPR/Cas9. Cell Reports. 17 (5), 1453-1461 (2016).
- Mout, R., et al. Direct Cytosolic Delivery of CRISPR/Cas9-Ribonucleoprotein for Efficient Gene Editing. ACS Nano. 11 (3), 2452-2458 (2017).
- Ramakrishna, S., et al. Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Research. 24 (6), 1020-1027 (2014).
- D'Astolfo, D. S., et al. Efficient intracellular delivery of native proteins. Cell. 161 (3), 674-690 (2015).
- Yen, J., et al. TRIAMF: A New Method for Delivery of Cas9 Ribonucleoprotein Complex to Human Hematopoietic Stem Cells. Scientific Reports. 8 (1), 16304 (2018).
- Sung, Y. H., et al. Highly efficient gene knockout in mice and zebrafish with RNA-guided endonucleases. Genome Research. 24 (1), 125-131 (2014).
- Martin, A., et al. CRISPR/Cas9 mutagenesis reveals versatile roles of Hox genes in crustacean limb specification and evolution. Current Biology. 26 (1), 14-26 (2016).
- Menoret, S., et al. Homology-directed repair in rodent zygotes using Cas9 and TALEN engineered proteins. Scientific Reports. 5, 14410 (2015).
- Woo, J. W., et al. DNA-free genome editing in plants with preassembled CRISPR-Cas9 ribonucleoproteins. Nature Biotechnology. 33 (11), 1162-1164 (2015).
- Malnoy, M., et al. DNA-free genetically edited grapevine and apple protoplast using CRISPR/Cas9 ribonucleoproteins. Frontiers in Plant Science. 7, 1904 (2016).
- Liang, Z., et al. Efficient DNA-free genome editing of bread wheat using CRISPR/Cas9 ribonucleoprotein complexes. Nature Communications. 8, 14261 (2017).
- Svitashev, S., Schwartz, C., Lenderts, B., Young, J. K., Mark Cigan, A. Genome editing in maize directed by CRISPR-Cas9 ribonucleoprotein complexes. Nature Communications. 7, 13274 (2016).
- Shin, S. E., et al. CRISPR/Cas9-induced knockout and knock-in mutations in Chlamydomonas reinhardtii. Scientific Reports. 6, 27810 (2016).
- 1000 Genomes Project Consortium et al. A global reference for human genetic variation. Nature. 526 (7571), 68-74 (2015).
- Klampfl, T., et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. The New England Journal of Medicine. 369 (25), 2379-2390 (2013).
- Döhner, H., et al. Diagnosis and management of AML in adults: 2017 ELN recommendations fro an international expert panel. Blood. 129 (4), 424-447 (2017).
- Barretina, J., et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 483 (7391), 603-607 (2012).
- Aguirre, A. J., et al. Genomic Copy Number Dictates a Gene-Independent Cell Response to CRISPR/Cas9 Targeting. Cancer Discovery. 6 (8), 914-929 (2016).
- Munoz, D. M., et al. CRISPR Screens Provide a Comprehensive Assessment of Cancer Vulnerabilities but Generate False-Positive Hits for Highly Amplified Genomic Regions. Cancer Discovery. 6 (8), 900-913 (2016).
- Gonçalves, E., et al. Structural rearrangements generate cell-specific, gene-independent CRISPR-Cas9 loss of fitness effects. Genome Biology. 20 (1), 27 (2019).
- Abudayyeh, O. O., et al. RNA targeting with CRISPR-Cas13. Nature. 550 (7675), 280-284 (2017).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.