
Method Article
Investigação do papel transcricional de um silenciador intrônico RUNX1 por crispr/Cas9 Ribonucleoproteína em células de leucemia mielóide aguda
Neste Artigo
Resumo
A entrega direta de complexos de ribonucleoproteína de RNA de Cas9/guia pré-montados é um meio rápido e eficiente para a edição de genoma em células hematopoiéticas. Aqui, nós utilizamos esta aproximação para suprimir de um silenciador intronic RUNX1 e para examinar as respostas transcricional em pilhas leucêmicas OCI-AML3.
Resumo
A maior parte do genoma humano (~ 98%) é composto de seqüências de não-codificação. Os elementos cis-Regulatory (cres) são seqüências do ADN da não-codificação que contêm locais obrigatórios para reguladores do transcricional para modular a expressão de Gene. As alterações de CREs foram implicadas em várias doenças que incluem o cancro. Quando os promotores e os potenciadores forem o cres preliminar para estudar a regulação genética, muito pouco se sabe sobre o papel do silenciador, que é um outro tipo de cre que medeia a repressão genética. Originalmente identificado como um sistema de imunidade adaptativa em procariontes, CRISPR/Cas9 tem sido explorado para ser uma ferramenta poderosa para a edição do genoma eucariótico. Aqui, nós apresentamos o uso desta técnica para suprimir de um silenciador intronic no gene RUNX1 humano e para investigar os impactos na expressão de gene em pilhas LEUCÊMICAS OCI-AML3. A nossa abordagem baseia-se na entrega mediada por electroporação de dois complexos de ribonucleoproteína (RNP) de RNA de Cas9/guia (gRNA) pré-montados para criar duas quebras de dupla vertente (DSBs) que flanqueiam o silenciador. As eliminações podem ser prontamente rastreadas por análise de fragmento. As análises de expressão de diferentes mRNAs transcritas de promotores alternativos ajudam a avaliar os efeitos dependentes do promotor. Esta estratégia pode ser usada para estudar outros CRE e é particular apropriada para pilhas hematopoietic, que são frequentemente difíceis de transfecção com os métodos Plasmid-baseados. O uso de uma estratégia de plasmídeo e livre de vírus permite avaliações simples e rápidas das funções regulatórias do gene.
Introdução
Os elementos cis-Regulatory (cres) são seqüências do ADN da não-codificação que contêm locais obrigatórios para reguladores do transcricional para controlar a expressão de gene1,2. Esses elementos são tipicamente 100 a 1.000 pares de base (BP) de comprimento. Promotores e potenciadores são os dois tipos mais caracterizados de CREs. Os promotores estão presentes em estreita proximidade com os sítios de início da transcrição e constituem a unidade básica de transcrição. Muitos genes têm mais de um promotor e seu uso alternativo contribui para a diversidade do transcriptoma e a especificidade do tecido3,4. Por outro lado, potenciadores ativam a transcrição e podem ser localizados a montante, a jusante ou dentro de íntrons dos genes alvo. Potenciadores podem atuar de longe distância (sobre uma megabase) e independente da orientação1,2. CREs também incluem silenciadores e isoladores5,6. Os atos anteriores oposta aos potenciadores para inibir a expressão de gene ligando aos repressores transcricional, visto que o último divide o genoma em domínios topologicamente discretos para isolar genes de outros cres dos domínios vizinhos. Esses elementos atuam em conjunto uns com os outros através de interações de cromatina de curto e/ou longo alcance e são organizados em hubs regulatórios para direcionar a expressão genética espaciotemporal adequada. Os avanços recentes em técnicas de sequenciamento de alta taxa de transferência aceleraram a identificação e a anotação funcional de muitos cre que facilitaram muito nossos entendimentos das redes transcricional que ditam o gene linhagem-específico expressão em diferentes tipos de células e tecidos7,8,9,10,11,12.
Dado os papéis fundamentais de CREs em regular a transcrição, suas alterações podem conduzir à expressão de gene aberrante. Tem sido demonstrado que os CRE são freqüentemente interrompidos por alterações genéticas e epigenéticas em diferentes tipos de cânceres humanos, contribuindo assim para a iniciação tumoral, progressão e agressividade13,14. Além disso, os fatores de ligação CRE são freqüentemente mutantes e/ou misexpressionados em vários tipos de câncer, destacando ainda mais o significado da desregulamentação CRE na oncogênese15. CREs pode igualmente ser afetado por aberrações estruturais, como exemplificado por rearranjos cromossomáticos freqüentes do potenciador de gene pesado da imunoglobulina (igh) que resultam na ativação anormal dos oncogenes vizinhos em linfomas da B-pilha16. Na leucemia mielóide aguda (LMA), o reposicionamento de um único potenciador por retrações do cromossomo 3Q provoca a GATA2 concomitante de downregulation e EVI1 , o que pode ser potencialmente direcionado pela inibição Bet das funções do potenciador 17. recentemente, nós caracterizamos um translocação cromossomático novo que envolve o perturbação de um silenciador intronic RUNX1 que possa contribuir à progressão de AML em um paciente pediatra18. Assim, decifrar o genoma não-codificante do cancro fornece avenidas frutíferas para elucidar a patogénese da doença, a descoberta do biomarcador e as intervenções terapêuticas, que melhoram finalmente resultados pacientes.
A via de nuclease crispr/Cas9, originalmente identificada como um sistema imunológico adaptativo em células procarióticas, tem sido explorada como um meio rápido e rentável para a edição genômica específica do local em células vivas e organismos19,20 ,21,22. O sistema CRISPR/Cas9 envolve dois componentes principais: o gRNA e o Streptococcus nuclease de Cas9 derivado de pyogenes. O gRNA contém uma seqüência específica chamada protospacer que reconhece a região de destino e direciona Cas9 para edição. O gRNA é compor de duas porções: RNA de CRISPR (crRNA), tipicamente uma seqüência do nucleotide de 20 Mer complementar ao ADN do alvo, e um crRNA trans-ativando (tracrRNA), que sirva como um andaime obrigatório para o nuclease. Um motivo adjacente ao protoespaçador (PAM) (5 '-NGG) imediatamente adjacente ao local de destino é necessário para a clivagem de Cas9 e o local de clivagem está localizado a 3 nucleotídeos a montante do PAM. a edição de genes mediados por CRISPR/Cas9 é comumente realizada por células transfecting com um plasmídeo que codifica Cas9 e o gRNA clonado23. Entretanto, esta aproximação é desafiante para pilhas hematopoietic, que são frequentemente duras ao transfecção e exigem métodos Virus-based longos da transdução. Uma aproximação alternativa é entrega celular direta de complexos pré-montados de Cas9/gRNA RNP24. Um método comum para a entrega da RNP é a eletroporação, que gera poros temporários na membrana celular, permitindo assim a entrada dos complexos RNP nas células25,26. As vantagens desta abordagem incluem a facilidade de uso, efeitos off-Target reduzidos e estabilidade dos complexos RNP. Aqui, nós descrevemos um protocolo de usar o método da entrega de RNP para investigar o papel transcricional de um silenciador intronic RUNX1 na linha de pilha18da leucemia OCI-AML3, que foi estabelecida do sangue periférico de um paciente de AML diagnosticado com o subtipo francês-americano-britânico M427. O protocolo inclui o delineamento de crRNA, preparo de complexos RNP, eletroporação, bem como triagem e posterior caracterização dos clones desejados.
Protocolo
1. projeto de crRNA
- Projete dois crrnas, um 5 ' e os outros 3 ' do CRE do alvo usando uma ferramenta de projeto Web-based de crispr28,29,30,31. Assegure-se de que um PAM de NGG esteja localizado imediatamente a jusante da sequência de destino para o reconhecimento de Cas9. O delineamento das duas crRNAs (crRNA-1 e crRNA-2) para a deleção do silenciador RUNX1 18 é mostrado na Figura 1.
Nota: Um crRNA geralmente contém um protoespaçador de 20 Mer que é complementar à sequência de destino. - Verifique a presença de polimorfismos de nucleotídeo único (SNPs)/indels no alvo e seqüências adjacentes PAM inserindo os locais genômicas das sequências na caixa de pesquisa de um navegador de genoma on-line (por exemplo, NCBI 1000 genomes ou UCSC genoma browser).
Nota: SNPs/indels comuns têm uma frequência de alelo menor de pelo menos 1% na população geral. - Envie as sequências de crRNA selecionadas para síntese com um fornecedor comercial. Além disso, comprar o tracrRNA para gRNA formação duplex.
2. projeto de eliminação de primers de triagem
- Projete um par de primers que flanqueiam a região de exclusão pretendida. Assegure-se de que os primers sejam pelo menos 50 BP dos sítios de clivagem de Cas9 para que a amplificação do PCR seja minimamente afetada pelos indelos formados nos sítios de clivagem.
- Assegure-se de que o amplicon seja menor do que 1.200 BP (preferivelmente menor do que 600 BP para a melhor definição do tamanho). Também, rotule um dos primers com um corante fluorescente (por exemplo, 6-carboxyfluorescein (6-FAM)) no 5 '-End para a detecção. Os primers utilizados para a triagem do apagamento do silenciador RUNX1 18 são mostrados na Figura 1.
3. preparação de complexos de Cas9/gRNA RNP
- Ressuscitar as crRNAs e tracrRNA em 1x TE buffer (10 mM Tris, 0,1 mM EDTA, pH 7,5) para uma concentração final de 200 μM.
- Para cada crRNA, misture 2,2 μL de crRNA de 200 μM, 2,2 μL de tracrRNA de 200 μM e 5,6 μL de tampão de 1x TE (total 10 μL) em um tubo de 0,2 mL para obter uma concentração duplex final de 44 μM.
- Incubar a 95 ° c durante 5 min num termocicer. Permita que os tubos esfriem à temperatura ambiente para a formação complexa do gRNA.
- Diluir 10,4 μL de 62 μM de nuclease de Cas9 recombinante com 7,6 μL de soro fisiológico com tampão de fosfato 1x (PBS) para obter uma concentração de nuclease final de 36 μM.
Nota: Este montante é suficiente para a preparação de dois complexos de Cas9/gRNA. - Misture volumes iguais (8,5 μL) da nuclease diluída com cada um dos duplex grna obtidos a partir do passo 3,3.
- Incubar à temperatura ambiente por 20 min para permitir a formação complexa de RNP. Mantenha as misturas no gelo até a electroporação.
4. electroporação dos complexos RNP em células OCI-AML3
- Cultura OCI-AML3 células em RPMI 1640 meio suplementado com 10% de soro bovino fetal inativado por calor (FBS), 1x GlutaMAX, 100 unidades/mL de penicilina e 100 μg/mL de estreptomicina (doravante referido como completo RPMI 1640 médio) a 37 ° c com 5% CO2.
- Contagem de células usando Tripan coloração azul. Assegure-se de que a viabilidade celular no momento da eletroporação seja acima de 90%.
- Centrifugador 2,5 x 106 pilhas em um tubo de 1,5 mL em 500 x g por 5 minutos na temperatura ambiente. Retire o sobrenadante e lave as células com 1 mL de 1X PBS. Gire para baixo as pilhas outra vez e remova todo o sobrenadante residual.
- Ressuscitem as células em 163 μL de RPMI 1640 médio sem fenol vermelho. Adicionar 16,7 μL de cada complexo RNP (a partir do passo 3,6) e 3,6 μL de 100 μM Electroporation Enhancer para as células (volume total = 200 μL). Misture suavemente com pipetagem.
Nota: O electroporation Enhancer é um DNA portador para melhorar a eficiência de edição. - Transfira a mistura a uma cubeta do Electroporation da Gap de 0,2 cm sem nenhumas bolhas. Realize o electroporation com um sistema do Electroporation (modalidade: exponencial; Tensão =: 150 V; Capacitância = 700 μF; Resistência = 50 Ω).
- Transfira as células para um balão de cultura de tecido T-25 contendo 6 mL de meio completo de RPMI 1640 e incubar a 37 ° c com 5% de CO2.
5. triagem e seleção de clones de células com deleções Bialélicas
- Diluir as células para 5 x 103/ml em completo rpmi 1640 meio um dia após o electroporation. Adicione 100 μL da suspensão da pilha diluída em cada poço de placas da cultura do tecido de 96 poços e permita que as pilhas cresçam por 7-14 dias.
- Extraia o DNA genómico das células usando um sistema de purificação de alta taxa de transferência.
- Adicionar 100 μL de ligação de placa e tampão de Lise a cada poço de uma placa de extração de 96 poços. Adicionar 5 x 104 células ressuscipended em 10 μL de 1X PBS para o tampão e misturá-los por pipetagem.
- Incubar à temperatura ambiente durante 30 min para permitir a ligação do ADN genómico aos poços.
- Aspirar a solução dos poços sem raspar as superfícies do poço. Lave os poços com 120 μL de tampão de lavagem.
- O ar seco os poços que contêm o ADN encadernado.
- Prepare 20 μL de mistura de PCR (2 μL de tampão de alta fidelidade 10x, 0,8 μL de 50 mM MgSO4, 0,4 μL de dNTPs de 10 mm, 0,4 μL de primer de 10 μm para a frente com etiqueta fam (passo 2,2), 0,4 μL de 10 μm de primer reverso sem rótulo e 0,4 U de Taq DNA polymerase para cada amostra).
Nota: Prepare uma mistura mestra para garantir a adição de quantidades padronizadas de reagentes em cada amostra. - Adicione a mistura do PCR a cada poço da placa da extração e funcione as reações em um termociclador (circunstâncias: ° c 94 inicial por 2 minutos, seguidos por 35 ciclos de 94 ° c para 15 s, de 56 ° c para 30 s e de 68 ° c por 1 minuto).
- Estimar a quantidade de produtos medindo suas concentrações em um número selecionado de amostras usando um fluorômetro. Diluir todas as amostras com H2o de nuclease-livre a 0,5 ng/μl.
- Misture 1 μL dos produtos de PCR diluídos com 8,5 μL de formamida deionizada e 0,5 μL de padrão de tamanho rotulado fluorescente em uma placa de 96 poços compatível com o analisador genético.
Nota: Prepare uma mistura mestra contendo formamida deionizada e o padrão de tamanho. - Cubra a placa com uma placa septos e desnaturem as amostras a 95 ° c por 3 min em um termocicer. Não feche a tampa da máquina.
- Realize a electroforese capilar do gel para separar os produtos etiquetados do PCR como descrito previamente32.
- Após a eletroforese, abra o software de análise para analisar os resultados.
- Clique em novo projeto e selecione microssatélite. Em seguida, clique em OK.
- Clique em Adicionar amostras ao projeto e selecione os arquivos de resultado (contêm a extensão. FSA). Em seguida, clique em Adicionar à lista para importar os arquivos.
- Na tabela que mostra os arquivos de resultados selecionados, escolha padrão de microssatélites na coluna método de análise . Além disso, selecione o padrão de tamanho usado na coluna tamanho padrão . Em seguida, clique no ícone analisar , insira o nome do experimento e salve o experimento.
- Clique em Exibir plotagens para exibir os resultados e escolha análise de fragmento na configuração de plotagem.
- Escolha os canais coloridos apropriados para análise. Verifique o ícone laranja para visualizar os fragmentos rotulados no padrão de tamanho para avaliar a qualidade da chamada de tamanho.
- Verifique o ícone azul para ver os produtos etiquetados do PCR. Identifique os picos que correspondem ao tipo selvagem e mutante (ou seja, carregando os apagamentos esperados) produtos. Estimar o nível mutante em cada amostra, dividindo a área o pico mutante pela soma da área o selvagem-tipo e picos mutantes.
- Selecione vários pools de células com altos níveis das exclusões esperadas para outras diluições seriais.
- Repita a extração do ADN, PCR fluorescente e etapas capilares da electroforese. Selecione clones de células com níveis mutantes > 95% representando deleções biallélicas para análises subsequentes.
- Verifique a identidade das exclusões nos clones selecionados por sequenciamento de Sanger.
6. análises funcionais do apagamento do silenciador pela análise quantitativa real-time do RT-PCR
- Extraia o RNA total dos clones selecionados e realize a síntese complementar do DNA (cDNA).
- Misture 1 μg de RNA com 1 μL de 50 μM de oligo (dT)20 primer e 1 μL de mistura dNTP de 10 mm num volume total de 10 μL. incubar a 65 ° c durante 5 min e depois coloque no gelo durante pelo menos 1 min.
- Adicionar 10 μL de mistura de síntese de cDNA contendo 2 μL de tampão 10x RT, 4 μL de MgCl de 25 mM2, 2 μl de 0,1 M DTT, 1 μL de inibidor de rnase (40 u/μL) e 1 μL de transcriptase reversa (200 u/μL). Incubar a 50 ° c por 50 min e, em seguida, 85 ° c durante 5 min num termocicer.
Nota: Prepare um Mix mestre para a transcrição reversa. - Resfrie as amostras no gelo. Adicionar 1 μL de RNase H e incubar a 37 ° c durante 20 min. armazene o cDNA a-20 ° c.
- Projete primers e sondas TaqMan que reconhecem especificamente variantes de transcrição individuais geradas a partir de promotores alternativos.
Nota: Os conjuntos de primer/sonda específicos de transcrição pré-projetados estão disponíveis comercialmente. - Clone fragmentos do ADN que contêm as seqüências específicas do transcrito no ADN do plasmídeo. Prepare uma série de diluição de 10 vezes (106 a 10 cópias) dos plasmídos recombinantes como curvas padrão para a quantificação do transcrito.
- Prepare 20 μL de mistura de PCR (0,5 μL de modelo de DNA, 1 μL de 20x de teste de sonda/primer TaqMan pré-concebidos e 10 μL de 2x TaqMan PCR Master Mix) para cada amostra (ambos os padrões de cDNA e plasmídeo). Meça cada amostra em Triplicate.
Nota: Prepare uma mistura mestra para o PCR em tempo real. - Execute as reações em uma máquina de PCR em tempo real (condições: 50 ° c inicial para 2 min e 95 ° c por 10 min, seguido por 40 ciclos de 94 ° c para 15 s e 60 ° c por 1 min).
- Após a amplificação, clique no ícone Analyze no software para analisar os dados. Verifique a inclinação e o coeficiente de correlação das curvas padrão para avaliar a eficiência e a linearidade das reações. Assegure-se de que as inclinações estejam entre-3,1 e-3,6 e os coeficientes de correlação sejam maiores de 0,99.
- Normalize o número de cópias das transcrições de destino em cada amostra com um gene de limpeza (por exemplo, GAPDH).
Resultados
O objetivo deste experimento é eliminar um silenciador intrônico no gene RUNX1 e examinar os impactos na transcrição RUNX1 em células OCI-AML3. O silenciador foi identificado por uma aproximação molecular combinatória e encontrado para conter um elemento do núcleo18de 209 BP. Para possibilitar uma avaliação mais acurada desse elemento central no controle da expressão RUNX1 , as crRNAs (crrna-1 e crrna-2) foram projetadas para direcionar de perto para esta região18 (Figura 1). Os sítios de clivagem de Cas9 preditos trazidos por crRNA-1 e crRNA-2 foram 29 BP e 35 BP do elemento central, respectivamente (Figura 1). Deve-se notar que, enquanto os sites PAM dos dois crRNAs residem em vertentes opostas, DSBs ocorrerá independentemente do local da seqüência PAM. Assim, a introdução concomitante de dois complexos de Cas9/gRNA RNP guiados por crRNA-1 e crRNA-2 é esperado para extirpar o elemento do silenciador do locus RUNX1 .
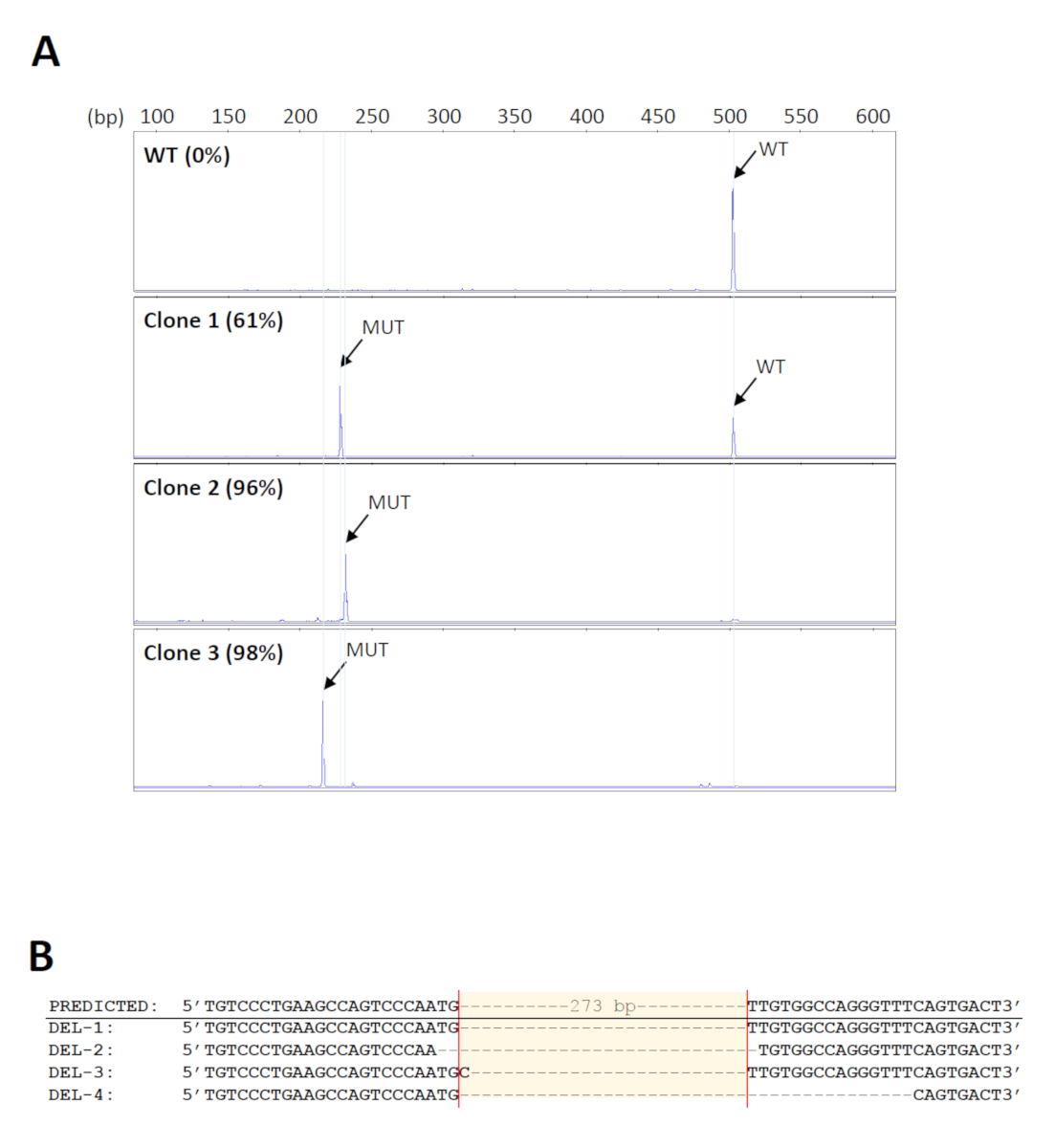
Para a tela para as eliminações desejadas em um grande número amostras, um formato 96-well do jogo genomic da extração do ADN foi usado para a purificação da elevado-produção. Além disso, o DNA vinculado aos poços das placas de extração pode ser submetido à amplificação direta, minimizando assim erros ou contaminação devido à transferência repetida da amostra. Os primers utilizados para a triagem das eliminações18 são mostrados na Figura 1. O tamanho esperado do produto do PCR do selvagem-tipo é aproximadamente 500 BP. Desde que o apagamento pretendido se estende 273 BP, o produto do mutante é esperado ser aproximadamente 230 BP. Esta escala do tamanho permite a análise simples e rápida do fragmento pela electroforese capilar do gel. Um total de 160 pools de células iniciais foram rastreados e 14 foram encontrados para transportar as exclusões esperadas com níveis mutantes de pelo menos 70%. Cinco pools foram selecionados então para umas diluições seriais mais adicionais para identificar os clones que carregam deleções bialélica. Os eletroferogramas representativos dos clones da pilha com níveis diferentes de produtos do mutante são mostrados em Figura 2A. A identidade das eliminações foi verificada pelo sequenciamento de Sanger (Figura 2B). Como esperado, os indels formados pelo reparo de junção final não-homérbio de DSBs33 foram observados nos locais de clivagem previstos nos clones de deleção. Estes resultaram na amplificação de produtos mutantes de tamanhos variados, o que também pode ser detectado por eletroforese capilar (Figura 2A).
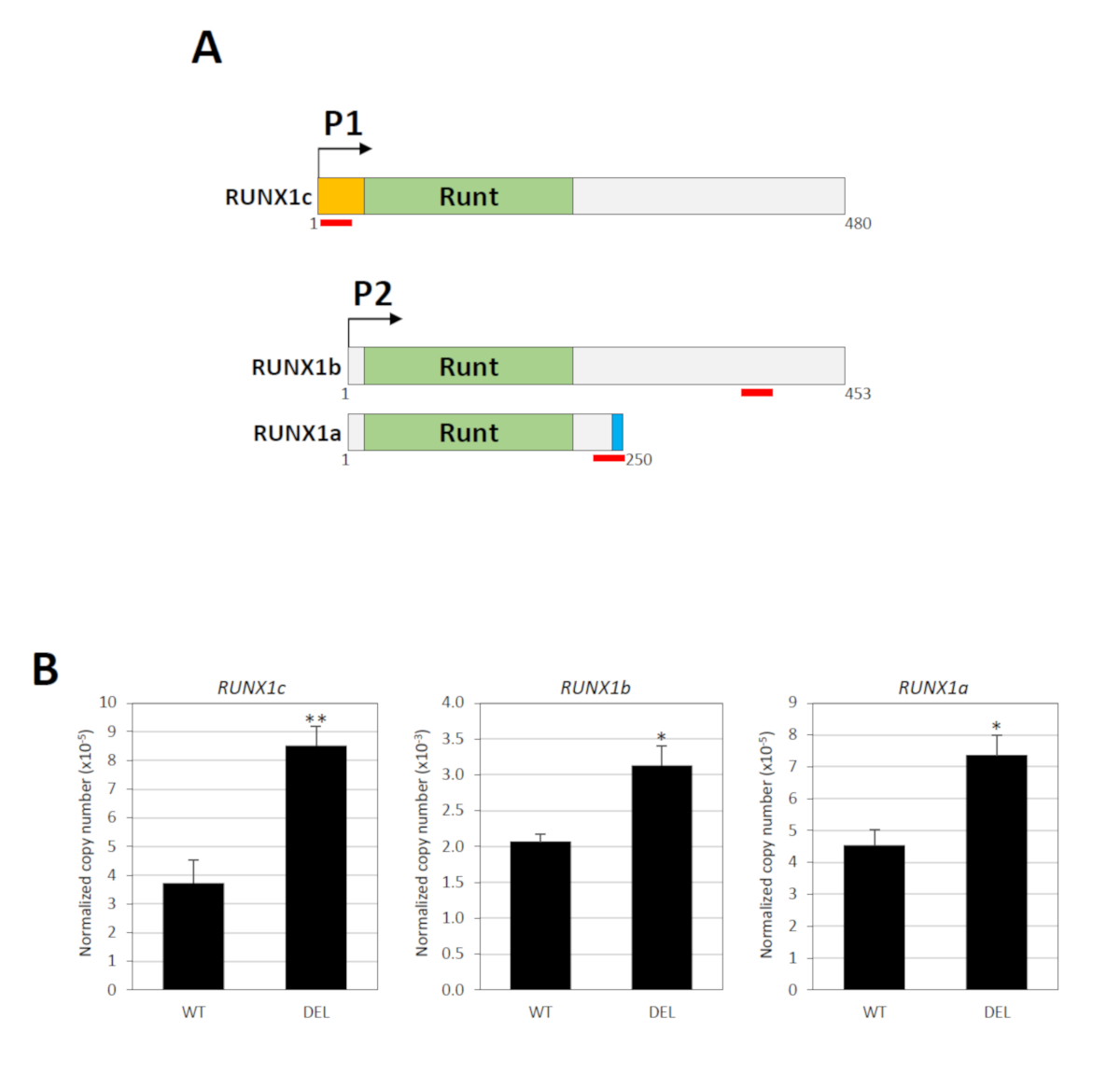
O gene RUNX1 contém dois promotores, a saber, o P1 distal e o P2 proximal, que são separados por um grande intron que abrigando o elemento silenciador34. Três transcrições principais do mRNA são produzidas por estes promotores: RUNX1c por P1 e por RUNX1a e por RUNX1b por P234. A seqüência do nucleotide de RUNX1c e de RUNX1b é idêntica exceto o anterior tem um N-Terminus original, de que um ensaio específico da expressão de gene de TaqMan pode ser projetado (Figura 3a). Para a mensuração do RUNX1b, utilizou-se o teste de TaqMan, reconhecendo tanto RUNX1b quanto RUNX1c (Figura 3a). Os níveis de RUNX1b foram então determinados subtraindo-se o total RUNX1b/RUNX1c de RUNX1c. RUNX1a é uma isoforma distintamente mais curta devido à emenda alternativa e um ensaio específico de TaqMan está disponível para esta variação (Figura 3a). Assim, a atividade dos promotores P1 e P2 pode ser determinada individualmente. O RT-PCR quantitativo em tempo real mostrou que o apagamento do elemento do silenciador upregulated significativamente os níveis da expressão dos transcritos P1-e P2-derivados (Figura 3B).

Figura 1 : Estratégia para eliminar o silenciador intrônico RUNX1 . O silenciador (caixa vermelha) está localizado no primeiro intron do gene RUNX1 separando os dois promotores P1 e P2 (hg19 coordenadas são mostradas). Dois crRNAs (crRNA-1 e crRNA-2) foram projetados introduzir DSBs que flanqueiam o elemento do silenciador. Os sítios de clivagem de Cas9 previstos são indicados por linhas vermelhas verticais e os sítios PAM (NGG) estão em roxo. Os dois primers utilizados para a triagem das eliminações são representados por setas abertas. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 2 : Identificação dos clones de deleção. (A) eletroferogramas representativos de clones da pilha que mostram níveis diferentes de produtos do PCR do mutante (MUT). O tamanho dos produtos mutantes varia entre os clones por causa dos indelos formados nos locais de clivagem. WT = Wild-Type. (B) verificação das eliminações de clones representativos por sequenciamento de Sanger. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 3 : Conseqüências funcionais do apagamento do silenciador. (A) sãomostradas as três principais isoformas RUNX1 (RUNX1a, RUNX1b e RUNX1c). Essas variantes contêm o mesmo domínio de ligação de DNA Runt, mas diferentes N-(laranja) ou C-Terminus (azul). A localização dos pares de sonda/primer TaqMan é indicada por linhas vermelhas. Os números indicam os resíduos do aminoácido. (B) análise quantitativa de RT-PCR em tempo real de transcrições derivadas de RUNX1 P1 e P2 em populações celulares com (del) ou sem (WT) as eliminações bialélicas18. GAPDH foi utilizado para normalização. * e * * indicam p < 0, 5 e p < 0, 1, respectivamente pelo teste de Mann-Whitney. Este número foi modificado de Cheng et al.18. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}
Discussão
O sistema crispr/Cas9 tem sido usado em uma ampla gama de aplicações de edição de genoma, como nocaute genético e knock-in Studies35,36, regulamento transcripcional37,38, engenharia genética de vários organismos modelo39,40,41,42,43,44 e terapia genética45,46. Aqui, demonstramos o uso de CRISPR/Cas9 para investigar as conseqüências funcionais da exclusão de um silenciador intrônico no gene RUNX1 . A entrega dos componentes de CRISPR em nossa aproximação não confiou no ADN do plasmídeo, na clonagem do gRNA ou no vírus mas no Electroporation de complexos pré-montados de Cas9/gRNA RNP. Demonstrou-se que o uso de DNA exógeno pode ser associado à integração indesejável de sequências vetoriais estrangeiras no genoma hospedeiro, aumento da toxicidade e baixa eficiência25,47,48, enquanto métodos de transdução de vírus são demorados. Além disso, a expressão prolongada de Cas9 do DNA plasmídeo pode aumentar os efeitos fora do alvo48. Pelo contrário, a abordagem de entrega direta baseada em RNP foi estabelecida como o método preferencial, pois é rápido e direto, com melhor eficiência de edição, seletividade e viabilidade celular. De fato, uma variedade de métodos como lipofecção49,50, Electroporation25,51, nanopartículas52, peptídeos penetrantes de células53, iTop54 e triamf 55 foram desenvolvidos para a entrega eficiente de crispr/Cas9 em tipos diferentes da pilha assim como a espécie animal e de planta24,25,26,56,57 , 58 , 59 , 60 , 61 , 62 , 63. uma vez que as sequências de ADN não codificantes são hotspots de variações genéticas64, a verificação da presença de SNPs/indels comuns no alvo e das sequências vizinhas do Pam é particularmente relevante na conceção do grna que visa a regulamentação Elementos.
Um gargalo na edição do genoma CRISPR/Cas9 envolve a triagem de clones mutantes desejados em um grande número de amostras. Nós empregamos o PCR fluorescente acoplado com a electroforese capilar do gel para a seleção porque a mutação do alvo é um apagamento genomic pequeno de aproximadamente 300 BP. Este método é rápido e sensível e pode ser realizado em uma forma de alta taxa de transferência. Também, este método permite a estimativa exata de níveis do mutante e de tamanhos do apagamento simultaneamente. Além disso, a análise multiplex de fragmentos de PCR rotulados com diferentes corantes fluorescentes é suportada. Nós temos usado rotineiramente esta técnica para genótipo pequenas inserções/deleções em neoplasma Myeloid65,66. Em nossa experiência, nós podemos consistentemente detectar tamanhos de fragmento que são diferentes por 4 BP com alta precisão e carga mutante para baixo para ~ 3%. No entanto, deve-se notar que este método tem um limite de tamanho de fragmento de 1.200 BP, e, portanto, não é adequado para a triagem de grandes deleções. Além disso, as substituições de base (resultando em tamanho de fragmento inalterado) e potenciais eventos fora do destino em outras regiões genômica não podem ser detectadas. Para este último, o sequenciamento de genoma todo caro é necessário para analisar globalmente as mudanças indesejáveis globais nos clones-alvo. Para adotar nossa abordagem atual para investigação de grandes seqüências reguladoras não codificantes (> 1000 BP), uma análise detalhada de deleção e mutagenese de sítios de ligação de fatores de transcrição putativos usando ensaios de genes in vitro Reporter pode ser realizada de antemão para delinear a região funcional mínima para a edição de CRISPR/Cas918.
Como muitos genes contêm mais de um promotores3,4, éimportante estar ciente da existência de promotores alternativos no locus do gene alvo como manipular elementos regulatórios pode afetar os promotores diferencialmente. Assim, as variantes de transcrição derivadas de diferentes promotores precisam ser medidas individualmente para avaliar quaisquer respostas específicas do promotor. O uso de ensaios à base de sonda TaqMan é preferível ao SYBR Green devido à melhor especificidade e reprodutibilidade. Se o sistema digital mais avançado do PCR está disponível, a quantificação do transcrito pode ser executada mais precisamente sem a necessidade da construção padrão da curva.
Uma consideração importante na realização de experimentos crispr/Cas9 em linhagens de células cancerosas é o número de cópia do gene ploidia e alvo nas células utilizadas como virtualmente todas as linhas de células cancerosas que abrigam alterações genéticas, incluindo variações estruturais e de número de cópias. Em nosso caso, OCI-AML3 tem um karyotype celular com 45 a 50 cromossomas. Também, a linha de pilha foi encontrada para carreg um número normal da cópia de RUNX1 como revelado do cancer Cell line enciclopédia67 e estudos in situ da hibridação da fluorescência18. Ao segmentar um gene com ganho de número de cópia, o método de entrega pode precisar ser otimizado para fornecer níveis suficientes dos componentes CRISPR para a edição. Também, mais clones podem precisar de ser selecionados a fim identificar os Knockouts completos. É importante ressaltar que a segmentação em regiões genómicas amplificadas, particularmente aquelas causadas por rearranjos estruturais, pode desencadear respostas antiproliferativas de genes independentes em células cancerosas, levando a resultados falsos positivos no gene estudos funcionais68,69,70. Neste sentido, as aproximações alternativas como o knockdown da interferência do RNA (RNAi) e/ou o superexpressão do cDNA devem ser empregadas para verificar os resultados de crispr. Além disso, várias linhas de células devem ser usadas para evitar interpretações incorretas de efeitos de edição de CRISPR específicos da linha de célula, mas independentes de genes.
O sistema CRISPR/Cas9 revolucionou a pesquisa básica e translacional, proporcionando um meio simples e eficiente para a edição do genoma. Aqui nós Demonstramos a facilidade de usar CRISPR/Cas9 para interromper um silenciador intrônico para estudos transcripcionais em uma linha de células cancerosas. Esta técnica permite o estudo de Cres no nível do ADN e oferece as oportunidades de examinar funções de CRE no contexto endógeno um pouco do que os genes heterólogo tradicionais do repórter. Recentemente, um sistema de edição de RNA baseado em CRISPR também foi identificado71 e pode servir como uma nova ferramenta para estudar Cres, visando o RNA transcrito a partir dos elementos regulatórios. Combinando com técnicas da captação do conformação do cromossoma, crispr/Cas9 ajudará certamente a decifrar os envolvimentos de Cres na organização alterada do genoma e na expressão de gene lig aos vários problemas de saúde.
Divulgações
Os autores não têm nada a revelar.
Agradecimentos
Os autores gostariam de agradecer ao Prof. M.D. Minden (Princess Margaret Cancer Centre, Universidade de saúde da rede, Toronto, Canadá) por fornecer a linha celular OCI-AML3. Além disso, os autores gostariam de agradecer as principais utilidades do cancer Genomics e Pathobiology (a universidade chinesa de Hong Kong) para fornecer as facilidades e o auxílio no apoio desta pesquisa.
Materiais
| Name | Company | Catalog Number | Comments |
| 0.2 cm gap electroporation cuvette | Bio-Rad | 1652086 | |
| 1×TE buffer, pH 7.5 | Integrated DNA Technologies | 11-01-02-02 | |
| 10 mM dNTP mix | Thermo Fisher Scientific | 18427013 | |
| 3500 Genetic Analyzer | Thermo Fisher Scientific | 4405673 | |
| 6-FAM-labeled fluorescent PCR forward primer | Thermo Fisher Scientific | None | |
| 7300 Real-Time PCR System | Thermo Fisher Scientific | None | |
| 7300 System SDS Software | Thermo Fisher Scientific | None | Version 1.3.1 |
| Bio-Rad Gene Pulser Xcell system | Bio-Rad | 1652660 | |
| ChargeSwitch Direct gDNA Purification Kit, 96-well | Thermo Fisher Scientific | CS11205 | |
| crRNA-1 | Integrated DNA Technologies | Alt-R CRISPR-Cas9 crRNA | Components of the Cas9/gRNA complex |
| crRNA-2 | Integrated DNA Technologies | Alt-R CRISPR-Cas9 crRNA | Components of the Cas9/gRNA complex |
| Deionized formamide | Thermo Fisher Scientific | 4311320 | |
| Electroporation Enhancer | Integrated DNA Technologies | 1075915 | |
| Fetal bovine serum | Thermo Fisher Scientific | 10270098 | |
| Fluorometer | Thermo Fisher Scientific | Q32857 | |
| GeneMapper Software 5 | Thermo Fisher Scientific | 4475073 | |
| GeneScan 600 LIZ Size Standard | Thermo Fisher Scientific | 4408399 | |
| GlutaMAX | Thermo Fisher Scientific | 35050061 | |
| PBS, 10x Solution, pH 7.4 | Affymetrix | 75889 | |
| Penicillin and streptomycin | Thermo Fisher Scientific | 15140122 | |
| Platinum Taq DNA Polymerase High Fidelity | Thermo Fisher Scientific | 11304029 | |
| Qubit dsDNA HS Assay Kit | Thermo Fisher Scientific | Q32854 | |
| Recombinant S. pyogenes Cas9 nuclease | Integrated DNA Technologies | 1081058 | Components of the Cas9/gRNA complex |
| RPMI 1640 medium | Thermo Fisher Scientific | 31800-022 | |
| RPMI 1640 medium without phenol red | Thermo Fisher Scientific | 11835030 | |
| RUNX1a TaqMan gene expression assays | Thermo Fisher Scientific | 4331182 | Hs04186042_m1 |
| RUNX1b/c TaqMan gene expression assays | Thermo Fisher Scientific | 4331182 | Hs00231079_m1 |
| RUNX1c TaqMan gene expression assays | Thermo Fisher Scientific | 4331182 | Hs01021966_m1 |
| SuperScript III First-Strand Synthesis System | Thermo Fisher Scientific | 18080051 | |
| TaqMan Universal PCR Master Mix | Thermo Fisher Scientific | 4304437 | |
| tracrRNA | Integrated DNA Technologies | 1072533 | Components of the Cas9/gRNA complex |
| TRIzol Reagent | Thermo Fisher Scientific | 15596018 | |
| Unlabeled PCR reverse primer | Thermo Fisher Scientific | None |
Referências
- Maston, G. A., Evans, S. K., Green, M. R. Transcriptional regulatory elements in the human genome. Annual Review of Genomics and Human Genetics. 7, 29-59 (2006).
- Shlyueva, D., Stampfel, G., Stark, A. Transcriptional enhancers: from properties to genome-wide predictions. Nature Reviews Genetics. 15 (4), 272-286 (2014).
- Kimura, K., et al. Diversification of transcriptional modulation: large-scale identification and characterization of putative alternative promoters of human genes. Genome Research. 16 (1), 55-65 (2006).
- Landry, J. R., Mager, D. L., Wilhelm, B. T. Complex controls: the role of alternative promoters in mammalian genomes. Trends in Genetics. 19 (11), 640-648 (2003).
- West, A. G., Gaszner, M., Felsenfeld, G. Insulators: many functions, many mechanisms. Genes & Development. 16 (3), 271-288 (2002).
- Riethoven, J. J. Regulatory regions in DNA: promoters, enhancers, silencers, and insulators. Methods in Molecular Biology. 674, 33-42 (2010).
- Thurman, R. E., et al. The accessible chromatin landscape of the human genome. Nature. 489 (7414), 75-82 (2012).
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 489 (7414), 57-74 (2012).
- Roadmap Epigenomics Consortium. Integrative analysis of 111 reference human epigenomes. Nature. 518 (7539), 317-330 (2015).
- Andersson, R., et al. An atlas of active enhancers across human cell types and tissues. Nature. 507 (7493), 455-461 (2014).
- Carninci, P., et al. The transcriptional landscape of the mammalian genome. Science. 309 (5740), 1559-1563 (2005).
- Bernstein, B. E., et al. The NIH roadmap epigenomics mapping consortium. Nature Biotechnology. 28 (10), 1045-1048 (2010).
- Zhou, S., Treloar, A. E., Lupien, M. Emergence of the Noncoding Cancer Genome: A Target of Genetic and Epigenetic Alterations. Cancer Discovery. 6 (11), 1215-1229 (2016).
- Khurana, E., et al. Role of non-coding sequence variants in cancer. Nature Reviews Genetics. 17 (2), 93-108 (2016).
- Herz, H. M., Hu, D., Shilatifard, A. Enhancer malfunction in cancer. Molecular Cell. 53 (6), 859-866 (2014).
- Willis, T. G., Dyer, M. J. The role of immunoglobulin translocations in the pathogenesis of B-cell malignancies. Blood. 96 (3), 808-822 (2000).
- Gröschel, S., et al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell. 157 (2), 369-381 (2014).
- Cheng, C. K., et al. RUNX1 upregulation via disruption of long-range transcriptional control by a novel t(5;21)(q13;q22) translocation in acute myeloid leukemia. Molecular Cancer. 17 (1), 133 (2018).
- Wiedenheft, B., Sternberg, S. H., Doudna, J. A. RNA-guided genetic silencing systems in bacteria and archaea. Nature. 482 (7385), 331-338 (2012).
- Bhaya, D., Davison, M., Barrangou, R. CRISPR-Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation. Annual Review of Genetics. 45, 273-297 (2011).
- Mali, P., et al. RNA-guided human genome engineering via Cas9. Science. 339 (6121), 823-826 (2013).
- Jinek, M., et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337 (6096), 816-821 (2012).
- Ran, F. A., et al. Genome engineering using the CRISPR-Cas9 system. Nature Protocols. 8 (11), 2281-2308 (2013).
- Cho, S. W., Lee, J., Carroll, D., Kim, J. S., Lee, J. Heritable gene knockout in Caenorhabditis elegans by direct injection of Cas9-sgRNA ribonucleoproteins. Genetics. 195 (3), 1177-1180 (2013).
- Kim, S., Kim, D., Cho, S. W., Kim, J., Kim, J. S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Research. 24 (6), 1012-1019 (2014).
- Lin, S., Staahl, B. T., Alla, R. K., Doudna, J. A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife. 3, 04766 (2014).
- Quentmeier, H., et al. Cell line OCI/AML3 bears exon-12 NPM gene mutation-A and cytoplasmic expression of nucleophosmin. Leukemia. 19 (10), 1760-1767 (2005).
- Heigwer, F., Kerr, G., Boutros, M. E-CRISP: fast CRISPR target site identification. Nature Methods. 11 (2), 122-123 (2014).
- Montague, T. G., Cruz, J. M., Gagnon, J. A., Church, G. M., Valen, E. CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Research. 42, 401-407 (2014).
- Doench, J. G., et al. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nature Biotechnology. 32 (12), 1262-1267 (2014).
- Haeussler, M., et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biology. 17 (1), 148 (2016).
- Ramlee, M. K., Wang, J., Cheung, A. M., Li, S. Using a Fluorescent PCR-capillary Gel Electrophoresis Technique to Genotype CRISPR/Cas9-mediated Knockout Mutants in a High-throughput Format. Journal of Visualized Experiments. (122), e55586 (2017).
- Canver, M. C., et al. Characterization of genomic deletion efficiency mediated by clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 nuclease system in mammalian cells. Journal of Biological Chemistry. 289 (31), 21312-21324 (2014).
- Sood, R., Kamikubo, Y., Liu, P. Role of RUNX1 in hematological malignancies. Blood. 129 (15), 2070-2082 (2017).
- Wang, H., et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 153 (4), 910-918 (2013).
- Yang, H., et al. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell. 154 (6), 1370-1379 (2013).
- Wright, J. B., Sanjana, N. E. CRISPR Screens to Discover Functional Noncoding Elements. Trends in Genetics. 32 (9), 526-529 (2016).
- Korkmaz, G., et al. Functional genetic screens for enhancer elements in the human genome using CRISPR-Cas9. Nature Biotechnology. 34 (2), 192-198 (2016).
- Cong, L., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Hwang, W. Y., et al. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nature Biotechnology. 31 (3), 227-229 (2013).
- Gratz, S. J., et al. Genome engineering of Drosophila with the CRISPR RNA-guided Cas9 nuclease. Genetics. 194 (4), 1029-1035 (2013).
- Chen, C., Fenk, L. A., de Bono, M. Efficient genome editing in Caenorhabditis elegans by CRISPR-targeted homologous recombination. Nucleic Acids Research. 41 (20), 193 (2013).
- Jacobs, T. B., LaFayette, P. R., Schmitz, R. J., Parrott, W. A. Targeted genome modifications in soybean with CRISPR/Cas9. BMC Biotechnology. 15, 16 (2015).
- Svitashev, S., et al. Targeted Mutagenesis, Precise Gene Editing, and Site-Specific Gene Insertion in Maize Using Cas9 and Guide RNA. Plant Physiology. 169 (2), 931-945 (2015).
- Schwank, G., et al. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell. 13 (6), 653-658 (2013).
- Ye, L., et al. Genome editing using CRISPR-Cas9 to create the HPFH genotype in HSPCs: An approach for treating sickle cell disease and β-thalassemia. Proceedings of the National Academy of Sciences of the United States of America. 113 (38), 10661-10665 (2016).
- Gabriel, R. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nature Biotechnology. 29 (9), 816-823 (2011).
- Gaj, T., Guo, J., Kato, Y., Sirk, S. J., Barbas, C. F. Targeted gene knockout by direct delivery of zinc-finger nuclease proteins. Nature Methods. 9 (8), 805-807 (2012).
- Zuris, J. A., et al. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nature Biotechnology. 33 (1), 73-80 (2015).
- Wang, M., et al. Efficient delivery of genome-editing proteins using bioreducible lipid nanoparticles. Proceedings of the National Academy of Sciences of the United States of America. 113 (11), 2868-2873 (2016).
- Gundry, M. C., et al. Highly Efficient Genome Editing of Murine and Human Hematopoietic Progenitor Cells by CRISPR/Cas9. Cell Reports. 17 (5), 1453-1461 (2016).
- Mout, R., et al. Direct Cytosolic Delivery of CRISPR/Cas9-Ribonucleoprotein for Efficient Gene Editing. ACS Nano. 11 (3), 2452-2458 (2017).
- Ramakrishna, S., et al. Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Research. 24 (6), 1020-1027 (2014).
- D'Astolfo, D. S., et al. Efficient intracellular delivery of native proteins. Cell. 161 (3), 674-690 (2015).
- Yen, J., et al. TRIAMF: A New Method for Delivery of Cas9 Ribonucleoprotein Complex to Human Hematopoietic Stem Cells. Scientific Reports. 8 (1), 16304 (2018).
- Sung, Y. H., et al. Highly efficient gene knockout in mice and zebrafish with RNA-guided endonucleases. Genome Research. 24 (1), 125-131 (2014).
- Martin, A., et al. CRISPR/Cas9 mutagenesis reveals versatile roles of Hox genes in crustacean limb specification and evolution. Current Biology. 26 (1), 14-26 (2016).
- Menoret, S., et al. Homology-directed repair in rodent zygotes using Cas9 and TALEN engineered proteins. Scientific Reports. 5, 14410 (2015).
- Woo, J. W., et al. DNA-free genome editing in plants with preassembled CRISPR-Cas9 ribonucleoproteins. Nature Biotechnology. 33 (11), 1162-1164 (2015).
- Malnoy, M., et al. DNA-free genetically edited grapevine and apple protoplast using CRISPR/Cas9 ribonucleoproteins. Frontiers in Plant Science. 7, 1904 (2016).
- Liang, Z., et al. Efficient DNA-free genome editing of bread wheat using CRISPR/Cas9 ribonucleoprotein complexes. Nature Communications. 8, 14261 (2017).
- Svitashev, S., Schwartz, C., Lenderts, B., Young, J. K., Mark Cigan, A. Genome editing in maize directed by CRISPR-Cas9 ribonucleoprotein complexes. Nature Communications. 7, 13274 (2016).
- Shin, S. E., et al. CRISPR/Cas9-induced knockout and knock-in mutations in Chlamydomonas reinhardtii. Scientific Reports. 6, 27810 (2016).
- 1000 Genomes Project Consortium et al. A global reference for human genetic variation. Nature. 526 (7571), 68-74 (2015).
- Klampfl, T., et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. The New England Journal of Medicine. 369 (25), 2379-2390 (2013).
- Döhner, H., et al. Diagnosis and management of AML in adults: 2017 ELN recommendations fro an international expert panel. Blood. 129 (4), 424-447 (2017).
- Barretina, J., et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 483 (7391), 603-607 (2012).
- Aguirre, A. J., et al. Genomic Copy Number Dictates a Gene-Independent Cell Response to CRISPR/Cas9 Targeting. Cancer Discovery. 6 (8), 914-929 (2016).
- Munoz, D. M., et al. CRISPR Screens Provide a Comprehensive Assessment of Cancer Vulnerabilities but Generate False-Positive Hits for Highly Amplified Genomic Regions. Cancer Discovery. 6 (8), 900-913 (2016).
- Gonçalves, E., et al. Structural rearrangements generate cell-specific, gene-independent CRISPR-Cas9 loss of fitness effects. Genome Biology. 20 (1), 27 (2019).
- Abudayyeh, O. O., et al. RNA targeting with CRISPR-Cas13. Nature. 550 (7675), 280-284 (2017).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados