
Method Article
Mapeo de variantes de la enfermedad de Alzheimer a sus genes objetivo mediante el análisis computacional de la configuración de la cromatina
En este artículo
Resumen
Presentamos un protocolo para identificar las implicaciones funcionales de las variantes no codificacionales identificadas por los estudios de asociación del genoma (GWAS) utilizando interacciones tridimensionales de cromatina.
Resumen
Los estudios de asociación en todo el genoma (GWAS) han identificado con éxito cientos de loci genómicos que están asociados con rasgos humanos y enfermedades. Sin embargo, debido a que la mayoría de los loci significativos en todo el genoma (GWS) caen sobre el genoma no codificante, el impacto funcional de muchos sigue siendo desconocido. Las interacciones tridimensionales de cromatina identificadas por Hi-C o sus derivados pueden proporcionar herramientas útiles para anotar estos loci vinculando variantes no codificantes a sus genes accionables. Aquí, delineamos un protocolo para mapear variantes no codificantes de GWAS a sus genes putativos usando conjuntos de datos GWAS y Hi-C de la enfermedad de Alzheimer (AD) del tejido cerebral adulto humano. Los polimorfismos de un solo nucleótido (SNP) de causalidad putativa se identifican mediante la aplicación de algoritmos de asignación fina. A continuación, los SNP se asignan a sus genes de destino putativos mediante interacciones potenciadoro-promotor basadas en Hi-C. El conjunto de genes resultante representa genes de riesgo de AD, ya que están potencialmente regulados por variantes de riesgo de AD. Para obtener más información biológica sobre los mecanismos moleculares subyacentes a la AD, caracterizamos los genes de riesgo de AD utilizando datos de expresión cerebral del desarrollo y perfiles de expresión de una sola célula cerebral. Este protocolo se puede ampliar a cualquier conjunto de datos GWAS e Hi-C para identificar genes de destino putativos y mecanismos moleculares subyacentes a diversos rasgos y enfermedades humanas.
Introducción
Los estudios de asociación en todo el genoma (GWAS) han desempeñado un papel fundamental en la revelación de la base genética de una serie de rasgos y enfermedades humanas. Este genotipado a gran escala ha descubierto miles de variantes genómicas asociadas con fenotipos que van desde la altura hasta el riesgo de esquizofrenia. Sin embargo, a pesar del enorme éxito de gWAS en la identificación de loci asociados a enfermedades y rasgos, una comprensión mecanicista de cómo estas variantes contribuyen al fenotipo ha sido un reto porque la mayoría de las variantes asociadas al fenotipo residen en la no codificación fracción del genoma humano. Dado que estas variantes a menudo se superponen con los elementos reglamentarios previstos, es probable que alteren el control transcripcional de un gen cercano. Sin embargo, los loci no codificantes pueden influir en la transcripción de genes a distancias lineales superiores a una megabase, lo que hace que los genes afectados por cada variante sean difíciles de identificar. La estructura tridimensional de la cromatina (3D) desempeña un papel importante en la mediación de las conexiones entre los loci reguladores distantes y los promotores de genes y se puede utilizar para identificar genes afectados por polimorfismos de un solo nucleótido (SNP) asociados al fenotipo.
La regulación genética está mediada por un proceso complejo, que implica la activación del potenciador y la formación del bucle de cromatina que conecta físicamente potenciadores a los promotores genéticos a los que se puede dirigir la maquinaria transcripcional1,2,3. Debido a que los bucles de cromatina a menudo abarcan varios cientos de kilobases (kb), se requieren mapas detallados de la arquitectura de cromatina 3D para descifrar los mecanismos reguladores de genes. Se han inventado múltiples tecnologías de captura de conformación de cromatina para identificar la arquitectura de cromatina 3D4. Entre estas tecnologías, Hi-C proporciona la arquitectura más completa, ya que captura perfiles de interacción de cromatina 3D en todo el genoma. Los conjuntos de datos Hi-C se han adaptado rápidamente parainterpretar loci5,6,7,8,9,10,11,12,13, ya que puede vincular variantes no codificantes a sus genes de destino putativos basados en perfiles de interacción con cromatina.
En este artículo, delineamos un protocolo para predecir computacionalmente los genes de destino putativos de las variantes de riesgo de GWAS utilizando perfiles de interacción con cromatina. Aplicamos este protocolo para asignar AD GWS loci14 a sus genes de destino utilizando conjuntos de datos Hi-C en el cerebro humano adulto9. Los genes de riesgo de AD resultantes se caracterizan por otros conjuntos de datos genómicos funcionales que incluyen perfiles de expresión transcriptomica y de desarrollo de una sola célula.
Protocolo
1. Configuración de la estación de trabajo
- Instale R (versión 3.5.0) y RStudio Desktop. Abra RStudio.
- Instale las siguientes bibliotecas en R escribiendo el siguiente código en la ventana de la consola en RStudio.
si (!" BiocManager" %in% rownames(installed.packages()))
install.packages("BiocManager", repos-"https://cran.r-project.org")
BiocManager::install("GenomicRanges")
BiocManager::install("biomaRt")
BiocManager::install("WGCNA")
install.packages("reshape")
install.packages("ggplot2")
install.packages("corrplot")
install.packages("gProfileR")
install.packages("tidyverse")
install.packages("ggpubr") - Descargar archivos.
NOTA: En este protocolo, es necesario descargar todos los archivos en el directorio de trabajo.- Descargue los siguientes archivos haciendo clic en los enlaces proporcionados en Tabla de materiales.
- Descargue los SNP creíbles de mapa fino para AD (Tabla complementaria 8 de Jansen et al.14).
NOTA: Antes del análisis, abra la hoja ocho en 41588_2018_311_MOESM3_ESM.xlsx, elimine las tres primeras filas y guarde la hoja como Supplementary_Table_8_Jansen.txt con formato separado por tabulaciones. - Descargar perfiles de interacción Hi-C resolución de 10 kb en el cerebro adulto de psychencode (descrito como Promoter-anchored_chromatin_loops.bed a continuación).
NOTA: Este archivo tiene el siguiente formato: cromosoma, TSS_start, TSS_end, Enhancer_start y Enhancer_end. En caso de que se utilicen otros conjuntos de datos Hi-C, este protocolo requiere conjuntos de datos Hi-C procesados a alta resolución (5 a 20 kb). - Descargue datasets de expresión de celda única desde PsychENCODE.
NOTA: Se trata de muestras de control neurotípicas. - Descargue conjuntos de datos de expresiones de desarrollo desde BrainSpan (descrito como devExpr.rda a continuación).
NOTA: 267666527 es un archivo comprimido, por lo que descomprima el 267666527 para extraer "columns_metadata.csv", "expression_matrix.csv" y "rows_metadata.csv" para generar devExpr.rda (ver sección 3).
- Descargue los SNP creíbles de mapa fino para AD (Tabla complementaria 8 de Jansen et al.14).
- Descargue las coordenadas exonicas (consulte Archivos complementarios, descritos como Gencode19_exon.bed y Gencode19_promoter.bed a continuación) de Gencode versión 19.
NOTA: Los promotores se definen como 2 kb aguas arriba del sitio de inicio de la transcripción (TSS). Estos archivos tienen el siguiente formato: cromosoma, inicio, fin y gen. - Descargue el archivo de anotación de genes (consulte Archivos complementarios, descrito como geneAnno.rda a continuación) de biomart.
NOTA: Este archivo se puede utilizar para que coincida con los genes basados en los datos de genes de Ensembl y el símbolo del Comité de Nomenclatura Genética (HGNC) de HUGO.
- Descargue los siguientes archivos haciendo clic en los enlaces proporcionados en Tabla de materiales.
2. Generación de un objeto GRanges para SNP creíbles
- Configure en R escribiendo el siguiente código en la ventana de la consola en RStudio.
library(GenomicRanges)
options(stringsAsFactors - F)
setwd("/work") - Esta es la ruta de acceso al directorio de trabajo.
credSNP á read.delim("Supplementary_Table_8_Jansen.txt", header-T)
credSNP á credSNP[credSNP$Credible.Causal-"Sí",] - Cree un objeto GRanges escribiendo el código siguiente en la ventana de la consola de RStudio.
credranges á GRanges(credSNP$Chr, IRanges(credSNP$bp, credSNP$bp), rsid-credSNP$SNP, P-credSNP$P)
save(credranges, file-"AD_credibleSNP.rda")
3. Mapeo posicional
NOTA: Para cada paso, escriba el código correspondiente en la ventana de la consola en RStudio.
- Configurar en R.
options(stringsAsFactors-F)
library(GenomicRanges)
load("AD_credibleSNP.rda") ( ver 2) - Mapeo posicional de SNP promotores/exonicos a genes
- Cargue el promotor y la región exónica y genere un objeto GRange.
exon á read.table("Gencode19_exon.bed")
exonranges - GRanges(exon[,1],IRanges(exon[,2],exon[,3]),gene-exon[,4])
promotor: read.table("Gencode19_promoter.bed")
promoters á GRanges(promoter[,1], IRanges(promoter[,2], promoter[,3]), gene-promoter[,4]) - Superponer sNP creíbles con regiones exónicas.
olap: findOverlaps(credranges, exonranges)
credexon - credranges[queryHits(olap)]
mcols(credexon) á cbind(mcols(credexon), mcols(exonranges[subjectHits(olap)])) - Superponer SNP creíbles con regiones promotoras.
olap- findOverlaps(credranges, promoterranges)
credpromoter á credranges[queryHits(olap)]
mcols(credpromoter) á cbind(mcols(credpromoter), mcols(promoterranges[subjectHits(olap)]))
- Cargue el promotor y la región exónica y genere un objeto GRange.
- Vincula los SNP a sus genes de destino putativos mediante interacciones de cromatina.
- Cargue el conjunto de datos Hi-C y genere un objeto GRange.
hic á read.table("Promoter-anchored_chromatin_loops.bed ", skip-1)
colnames(hic) á c("chr", "TSS_start", "TSS_end", "Enhancer_start", "Enhancer_end")
hicranges - GRanges(hic$chr, IRanges(hic$TSS_start, hic$TSS_end), enhancer-hic$Enhancer_start)
olap - findOverlaps(hicranges, promoterranges)
hicpromoter á hicranges[queryHits(olap)]
mcols(hicpromoter) á cbind(mcols(hicpromoter), mcols(promoterranges[subjectHits(olap)]))
hicenhancer - GRanges(seqnames(hicpromoter), IRanges(hicpromoter$enhancer, hicpromoter$enhancer+10000), gene-hicpromoter$gene) - Superponer SNP creíbles con Hi-C GRange objeto.
olap: findOverlaps(credranges, hicenhancer)
credhic - credranges[queryHits(olap)]
mcols(credhic) - cbind(mcols(credhic), mcols(hicenhancer[subjectHits(olap)]))
- Cargue el conjunto de datos Hi-C y genere un objeto GRange.
- Compile genes candidatos a AD definidos por cartografía posicional y perfiles de interacción de cromatina.
Los genes candidatos resultantes para AD:
ADgenes - Reducir(union, list(credhic$gene, credexon$gene, credpromoter$gene))
• para convertir El ID de gen de Ensembl en símbolo HGNC
load("geneAnno.rda")
ADhgnc á geneAnno1[match(ADgenes, geneAnno1$ensembl_gene_id), "hgnc_symbol"]
ADhgnc - ADhgnc[ADhgnc!-""]
save(ADgenes, ADhgnc, file-"ADgenes.rda")
write.table(ADhgnc, file-"ADgenes.txt", row.names-F, col.names-F, quote-F, sep-"-t")
4. Trayectorias de expresión del desarrollo
NOTA: Para cada paso, escriba el código correspondiente en la ventana de la consola en RStudio.
- Configurar en R.
biblioteca (remodelar); library(ggplot2); library(GenomicRanges); biblioteca (biomaRt)
biblioteca("WGCNA")
options(stringsAsFactors-F) - Procesar la expresión y los metadatos.
datExpr á read.csv("expression_matrix.csv", encabezado ? FALSE)
datExpr á datExpr[,-1]
datMeta á read.csv("columns_metadata.csv")
datProbes á read.csv("rows_metadata.csv")
datExpr á datExpr[datProbes$ensembl_gene_id!",]
datProbes á datProbes[datProbes$ensembl_gene_id!",]
datExpr.cr- collapseRows(datExpr, rowGroup ? datProbes$ensembl_gene_id, rowID - rownames(datExpr))
datExpr á datExpr.cr$datETcollapsed
gename á data.frame(datExpr.cr$group2row)
rownames(datExpr) - gename$group- Especifique las etapas de desarrollo.
datMeta$Unit á "Postnatal"
idx á grep("pcw", datMeta$age)
datMeta$Unit[idx] á "Prenatal"
idx á grep("yrs", datMeta$age)
datMeta$Unit[idx] á "Postnatal"
datMeta$Unit á factor(datMeta$Unit, levels-c("Prenatal", "Postnatal")) - Seleccione regiones corticales.
datMeta$Region á "SubCTX"
r á c("A1C", "STC", "ITC", "TCx", "OFC", "DFC", "VFC", "MFC", "M1C", "S1C", "IPC", "M1C-S1C", "PCx", "V1C", "Ocx")
datMeta$Region[datMeta$structure_acronym %in% r] á "CTX"
datExpr á datExpr[,which(datMeta$Region-"CTX")]
datMeta á datMeta[which(datMeta$Region-"CTX"),]
save(datExpr, datMeta, file-"devExpr.rda")
- Especifique las etapas de desarrollo.
- Extraiga perfiles de expresión de desarrollo de genes de riesgo de AD.
load("ADgenes.rda")
exprdat á apply(datExpr[match(ADgenes, rownames(datExpr)),],2,mean,na.rm-T)
dat á data.frame(Region-datMeta$Region, Unit-datMeta$Unit, Expr-exprdat) - Compare los niveles de expresión prenatal frente a postnatal de los genes de riesgo de AD.
pdf(archivo"developmental_expression.pdf")
ggplot(dat,aes(x-Unit, y-Expr, fill-Unit, alpha-Unit)) + yla("Expresión normalizada") + geom_boxplot(outlier.size ? NA) + ggtitle("Expresión cerebral") + xlab("") + scale_alpha_manual(values-c(0.2, 1)) + theme_classic() + theme(legend.position-"na" )
dev.off()
5. Perfiles de expresión de tipo celular
NOTA: Para cada paso, escriba el código correspondiente en la ventana de la consola en RStudio.
- Configurar en R.
options(stringsAsFactors-F)
load("ADgenes.rda")
load("geneAnno.rda")
targetname - "AD"
targetgene - ADhgnc
cellexp á read.table("DER-20_Single_cell_expression_processed_TPM_backup.tsv",header-T,fill-T)
cellexp[1121,1] á cellexp[1120,1]
cellexp á cellexp[-1120,]
rownames(cellexp) - cellexp[,1]
cellexp á cellexp[,-1]
datExpr á scale(cellexp,center-T, scale-F)
datExpr á datExpr[,789:ncol(datExpr)] - Extraer perfiles de expresión celular de genes de riesgo de AD.
exprdat á apply(datExpr[match(targetgene, rownames(datExpr)),],2,mean,na.rm-T)
dat á data.frame(Group-targetname, cell-names(exprdat), Expr-exprdat)
dat$celltype á unlist(lapply(strsplit(dat$cell, split-"[.]),'[[',1))
dat á dat[-grep("Ex- In",dat$celltype),]
dat$celltype á gsub("Dev","Fetal",dat$celltype)
dat$celltype á factor(dat$celltype, levels-c("Neurons","Astrocytes","Microglia","Endotelial",
Oligodendrocitos","OPC","Fetal"))
pdf(archivo"singlecell_expression_ADgenes.pdf")
ggplot(dat,aes(x-celltype, y-Expr, fill-celltype)) +
ylab("Expresión normalizada") + xlab("") + geom_violin() + theme(axis.text.x-element_text(angle ? 90, hjust-1)) + theme(legend.position-"none") +
ggtitle(paste0("Perfiles de expresión celular de genes de riesgo AD"))
dev.off()
6. Análisis de enriquecimiento de anotación genética de genes de riesgo AD
- Descargue y configure HOMER escribiendo los siguientes comandos en el terminal.
mkdir homer
cd homer
http://homer.ucsd.edu/homer/configureHomer.pl
perl ./configureHomer.pl -install
perl ./configureHomer.pl -instalar humano-p
perl ./configureHomer.pl -instalar humano-o - Ejecute HOMER escribiendo los siguientes comandos en el terminal.
ruta de exportación $PATH: /trabajo/homer/bin
findMotifs.pl de trabajo/ADgenes.txt humanos/trabajo/ - Trace los términos enriquecidos escribiendo el siguiente código en la ventana de la consola en RStudio.
library(ggpubr)
options(stringsAsFactors-F)
pdf("GO_enrichment.pdf",ancho-15,altura-8)
plot_barplot de la función (nombre de la base de datos,nombre,color)
input á read.delim(paste0(dbname,".txt"),header-T)
entrada-input[,c(-1,-10,-11)]
entrada: único (entrada)
input$FDR á p.adjust(exp(input$logP))
input_sig de entrada[input$FDR < 0.1,]
input_sig$FDR -log10(input_sig$FDR)
input_sig input_sig[orden(input_sig$FDR),]
p á ggbarplot(input_sig, x á "Term", y á "FDR", relleno , color, color á "blanco", sort.val á "asc", ylab á expression(-log[10](italic(FDR))), xlab á paste0(name," Terms"), rotar , TRUE, label , paste0(input_sig$Target.Genes.in.Term,"/",input_sig$Genes.in.Term), font.label á list(color ? "white", size ?9), lab.vjust á 0.5, lab.hjust á 1)
p á p+geom_hline(yintercept - -log10(0.05), tipo de línea 2, color á "lightgray")
return(p)
}
p1 - plot_barplot("biological_process","Proceso biológico GO","#00AFBB")
p2 á plot_barplot("kegg","KEGG","#E7B800")
p3 - plot_barplot("reactome","Reactome","#FC4E07")
ggarrange(p1, p2, p3, etiquetas á c("A", "B", "C"), ncol ncol 2, nrow nrow 2)
dev.off()
Resultados
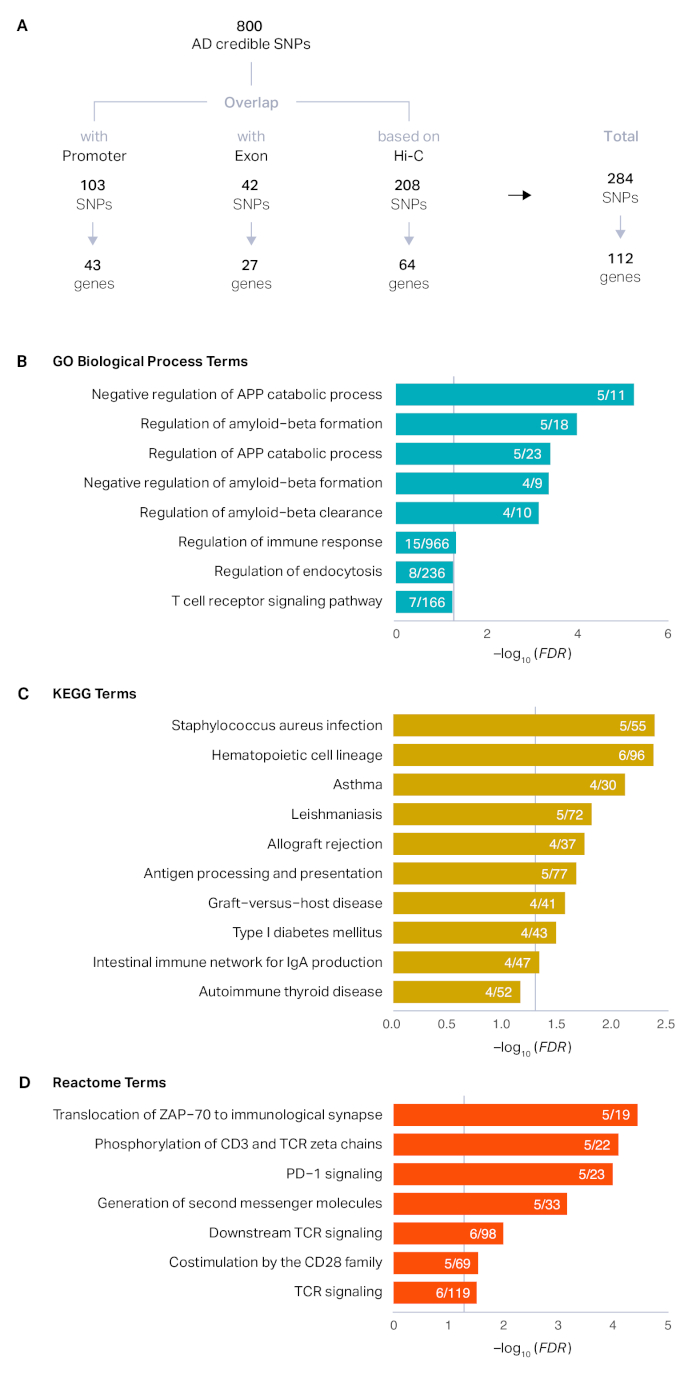
El proceso descrito aquí se aplicó a un conjunto de 800 SNP creíbles que fueron definidos por el estudio original14. El mapeo posicional reveló que 103 SNP se superponían con promotores (43 genes únicos) y 42 SNP se superponían con exones (27 genes únicos). Después de la asignación posicional, el 84% (669) SNp permaneció sin anotar. Utilizando conjuntos de datos Hi-C en el cerebro adulto, pudimos vincular 208 SNP adicionales a 64 genes basados en la proximidad física. En total, mapeamos 284 SNP creíbles de AD a 112 genes de riesgo AD(Figura 1A). Los genes de riesgo de AD se asociaron con proteínas precursoras de amiloide, formación de amiloide-beta y respuesta inmune, reflejando la biología conocida de AD15,16,17,18 (Figura 1B-D). Los perfiles de expresión de desarrollo de los genes de riesgo de AD mostraron un marcado enriquecimiento postnatal, indicativo del riesgo elevado asociado a la edad de AD(Figura 2A). Finalmente, los genes de riesgo de AD se expresaron altamente en microglia, células inmunitarias primarias en el cerebro(Figura 2B). Esto está de acuerdo con los hallazgos recurrentes de que AD tiene una fuerte base inmune y la microglia es el actor central en la patogénesis AD14,19,20.

Figura 1: Definición de genes de destino putativo s de loci AD GWS. (A) Los SNP creíbles derivados de los 29 loci de AD superiores se clasificaron en SNP promotores, SNP exónicos y SNP no codificantes sin anotar. Los Promotores y SNP exónicos se asignaron directamente a sus genes objetivo mediante mapeo posicional, mientras que los perfiles de interacción de cromatina en el cerebro adulto se utilizaron adicionalmente para mapear SNP basados en interacciones físicas. (B-D) El enriquecimiento de términos GO (B), KEGG (C) y Reactome (D) en genes de riesgo AD se realizó utilizando HOMER como se describe en la sección 6 del protocolo. El eje x representa la tasa de detección falsa (FDR) corregida -log10 (valor P). Se trazaron términos enriquecidos con FDR < 0.1. Las líneas verticales grises representan FDR a 0,05. Proteína precursora de amiloide APP. Numerador, el número de genes de riesgo de AD representados en cada término; denominador, el número de genes en cada término. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2: Caracterización de genes de riesgo de AD. (A) Los genes de riesgo de AD están muy expresados en la corteza postnatal en comparación con la corteza prenatal. (B) Gráficas de violín que representan distribuciones de valores de expresión génica (expresión normalizada) en diferentes tipos de células de la corteza. Estos resultados muestran que los genes de riesgo de AD están muy expresados en microglia, de acuerdo con estudios anteriores14. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Archivo Suplementario 1. Haga clic aquí para ver este archivo (haga clic con el botón derecho para descargar).
Archivo Suplementario 2. Haga clic aquí para ver este archivo (haga clic con el botón derecho para descargar).
Archivo Suplementario 3. Haga clic aquí para ver este archivo (haga clic con el botón derecho para descargar).
Discusión
Aquí describimos un marco analítico que se puede utilizar para anotar funcionalmente los loci de GWS basados en mapeo posicional e interacciones de cromatina. Este proceso implica varios pasos (para más detalles vea esta revisión13). En primer lugar, dado que los perfiles de interacción con cromatina son altamente específicos del tipo celular, los datos de Hi-C obtenidos de los tipos de células/tejidos apropiados que mejor capturan la biología subyacente del trastorno deben utilizarse. Dado que la AD es un trastorno neurodegenerativo, usamos datos de Hi-C del cerebro adulto9 para anotar loci GWS. En segundo lugar, cada locus de GWS a menudo tiene hasta cientos de SNP que están asociados con el rasgo debido al desequilibrio de la vinculación (LD), por lo que es importante obtener SNP putativos causales ('creíbles') prediciendo computacionalmente la causalidad mediante el uso de algoritmos de mapeo fino21,22 o pruebas experimentales de las actividades regulatorias utilizando enfoques de alto rendimiento, como ensayos de reporteros paralelos masivos (MPRA)23 o secuenciación de región reguladora activa auto-transcripción ( STARR-seq)24. Para el trabajo descrito aquí, utilizamos SNP creíbles reportados en Jansen et al.14. En tercer lugar, los SNP de promotor y exónico se anotan en función del mapeo posicional. Utilizamos una estrategia de mapeo posicional simple en la que los SNP se mapearon a los genes cuando se superponían con promotores (definidos como 2 kb aguas arriba del sitio de inicio de transcripción) o exones. Sin embargo, este enfoque puede elaborarse más detalladamente evaluando las consecuencias funcionales de los SNP exónicos, como si el SNP induce una caries mediada sin sentido, una variación de sentido erróneo o una variación sin sentido. En cuarto lugar, los perfiles de interacción con cromatina del tipo de tejido/célula apropiado se pueden utilizar para asignar SNP a sus genes de destino putativos en función de la proximidad física. Utilizamos perfiles de interacción anclados a los promotores, pero podemos refinar o ampliar aún más los perfiles de interacción teniendo en cuenta las actividades potenciadoras (guiadas por la acetilación o accesibilidad a la cromatina de histona H3 K27) o las interacciones exónicas. Una consideración importante en este proceso es utilizar la construcción consistente del genoma humano. Por ejemplo, si las posiciones genómicas de las estadísticas resumidas no se basan en hg19 (es decir, hg18 o hg38), se debe obtener una versión adecuada del genoma de referencia o las estadísticas resumidas deben convertirse a hg19 utilizando liftover25.
Aplicamos este marco para identificar genes de destino putativos para AD GWAS, asignando 284 SNP a 112 genes de riesgo AD. Utilizando perfiles de expresión de desarrollo 26 yperfiles de expresión específicos de tipo celular9, luego demostramos que este conjunto de genes era consistente con lo que se conoce sobre la patología AD, revelando los tipos de células (microglia), las funciones biológicas (respuesta inmune y beta amiloide), y el riesgo elevado a la edad.
Si bien presentamos un marco que delinea los genes objetivo potenciales de AD y su biología subyacente, es de destacar que la anotación basada en Hi-C se puede expandir para anotar cualquier variación que no sea codificante. A medida que se disponga de más datos de secuenciación del genoma completo y aumente nuestra comprensión sobre la variación rara no codificante, Hi-C proporcionará un recurso clave para la interpretación de variantes genéticas asociadas a la enfermedad. Por lo tanto, un compendio de recursos Hi-C obtenidos de múltiples tipos de tejidos y células será fundamental para facilitar una amplia aplicación de este marco para obtener información biológica sobre diversos rasgos humanos y enfermedades.
Divulgaciones
Los autores no tienen nada que revelar.
Agradecimientos
Este trabajo fue apoyado por la subvención NIH R00MH113823 (a H.W.) y R35GM128645 (a D.H.P.), el Premio NarSAD Young Investigator (a H.W.), y la subvención SPARK de la Simons Foundation Autism Research Initiative (SFARI, a N.M. y H.W.).
Materiales
| Name | Company | Catalog Number | Comments |
| 10 kb resolution Hi-C interaction profiles in the adult brain from psychencode | http://adult.psychencode.org/ | ||
| Developmental expression datasets | http://www.brainspan.org/ | ||
| Fine-mapped credible SNPs for AD (Supplementary Table 8 from Jansen et al.14) | https://static-content.springer.com/ | ||
| HOMER | http://homer.ucsd.edu/ | ||
| R (version 3.5.0) | https://www.r-project.org/ | ||
| RStudio Desktop | https://www.rstudio.com/ | ||
| Single cell expression datasets | http://adult.psychencode.org/ |
Referencias
- Dekker, J., Misteli, T. Long-Range Chromatin Interactions. Cold Spring Harbor Perspectives in Biology. 7 (10), a019356(2015).
- Sanyal, A., Lajoie, B. R., Jain, G., Dekker, J. The long-range interaction landscape of gene promoters. Nature. 489 (7414), 109-113 (2012).
- Plank, J. L., Dean, A. Enhancer function: mechanistic and genome-wide insights come together. Molecular Cell. 55 (1), 5-14 (2014).
- Dekker, J., Marti-Renom, M. A., Mirny, L. A. Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data. Nature Reviews Genetics. 14 (6), 390-403 (2013).
- Martin, P., et al. Capture Hi-C reveals novel candidate genes and complex long-range interactions with related autoimmune risk loci. Nature Communications. 6, 10069(2015).
- Won, H., et al. Chromosome conformation elucidates regulatory relationships in developing human brain. Nature. 538 (7626), 523-527 (2016).
- Jäger, R., et al. Capture Hi-C identifies the chromatin interactome of colorectal cancer risk loci. Nature Communications. 6, 6178(2015).
- Chen, J. A. A., et al. Joint genome-wide association study of progressive supranuclear palsy identifies novel susceptibility loci and genetic correlation to neurodegenerative diseases. Molecular Neurodegeneration. 13 (1), 41(2018).
- Wang, D., et al. Comprehensive functional genomic resource and integrative model for the adult brain. Science. 362 (6420), eaat8464(2018).
- Demontis, D., et al. Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nature Genetics. 51 (1), 63-75 (2019).
- Grove, J., et al. Identification of common genetic risk variants for autism spectrum disorder. Nature Genetics. 51 (3), 431-444 (2019).
- Lee, P. H., et al. Genome wide meta-analysis identifies genomic relationships, novel loci, and pleiotropic mechanisms across eight psychiatric disorders. bioRxiv. , 528117(2019).
- Mah, W., Won, H. The three-dimensional landscape of the genome in human brain tissue unveils regulatory mechanisms leading to schizophrenia risk. Schizophrenia Research. , In press (2019).
- Jansen, I. E., et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer's disease risk. Nature Genetics. 51 (3), 404-413 (2019).
- Viola, K. L., Klein, W. L. Amyloid β oligomers in Alzheimer's disease pathogenesis, treatment, and diagnosis. Acta Neuropathologica. 129 (2), 183-206 (2015).
- Mroczko, B., Groblewska, M., Litman-Zawadzka, A., Kornhuber, J., Lewczuk, P. Amyloid β oligomers (AβOs) in Alzheimer's disease. Journal of Neural Transmission. 125 (2), 177-191 (2018).
- Heneka, M. T., et al. Neuroinflammation in Alzheimer's disease. Lancet Neurology. 14 (4), 388-405 (2015).
- Minter, M. R., Taylor, J. M., Crack, P. J. The contribution of neuroinflammation to amyloid toxicity in Alzheimer's disease. Journal of Neurochemistry. 136 (3), 457-474 (2016).
- Hansen, D. V., Hanson, J. E., Sheng, M. Microglia in Alzheimer's disease. The Journal of Cell Biology. 217 (2), 459-472 (2018).
- Gjoneska, E., et al. Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer's disease. Nature. 518 (7539), 365-369 (2015).
- Benner, C., et al. FINEMAP: efficient variable selection using summary data from genome-wide association studies. Bioinformatics. 32 (10), 1493-1501 (2016).
- Hormozdiari, F., Kostem, E., Kang, E. Y., Pasaniuc, B., Eskin, E. Identifying causal variants at loci with multiple signals of association. Genetics. 198 (2), 497-508 (2014).
- Tewhey, R., et al. Direct Identification of Hundreds of Expression-Modulating Variants using a Multiplexed Reporter Assay. Cell. 165 (6), 1519-1529 (2016).
- Arnold, C. D., et al. Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science. 339 (6123), 1074-1077 (2013).
- Kent, W. J., et al. The human genome browser at UCSC. Genome Research. 12 (6), 996-1006 (2002).
- Kang, H. J., et al. Spatio-temporal transcriptome of the human brain. Nature. 478 (7370), 483-489 (2011).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados