
Method Article
Mapping Alzheimer's Disease Variants to Their Target Genes Using Computational Analysis of Chromatin Configuration
In This Article
Summary
We present a protocol to identify functional implications of non-coding variants identified by genome-wide association studies (GWAS) using three-dimensional chromatin interactions.
Abstract
Genome-wide association studies (GWAS) have successfully identified hundreds of genomic loci that are associated with human traits and disease. However, because the majority of the genome-wide significant (GWS) loci fall onto the non-coding genome, the functional impact of many remain unknown. Three-dimensional chromatin interactions identified by Hi-C or its derivatives can provide useful tools to annotate these loci by linking non-coding variants to their actionable genes. Here, we outline a protocol to map GWAS non-coding variants to their putative genes using Alzheimer's disease (AD) GWAS and Hi-C datasets from human adult brain tissue. Putative causal single-nucleotide polymorphisms (SNPs) are identified by application of fine-mapping algorithms. SNPs are then mapped to their putative target genes using enhancer-promoter interactions based on Hi-C. The resulting gene set represents AD risk genes, as they are potentially regulated by AD risk variants. To garner further biological insights into molecular mechanisms underlying AD, we characterize AD risk genes using developmental brain expression data and brain single-cell expression profiles. This protocol can be expanded to any GWAS and Hi-C datasets to identify putative target genes and molecular mechanisms underlying various human traits and diseases.
Introduction
Genome-wide association studies (GWAS) have played a pivotal role in revealing the genetic basis of a range of human traits and diseases. This large-scale genotyping has uncovered thousands of genomic variants associated with phenotypes ranging from height to schizophrenia risk. However, despite the enormous success of GWAS in identifying disease and trait associated loci, a mechanistic understanding of how these variants contribute to phenotype has been challenging because most phenotype associated variants reside in the non-coding fraction of the human genome. Since these variants often overlap with predicted regulatory elements, they are likely to alter transcriptional control of a nearby gene. However, non-coding loci can influence transcription of genes at linear distances exceeding one megabase, making the genes affected by each variant difficult to identify. Three-dimensional (3D) chromatin structure plays an important role in mediating connections between distant regulatory loci and gene promoters and can be used to identify genes affected by phenotype associated single-nucleotide polymorphisms (SNPs).
Gene regulation is mediated by a complex process, which involves enhancer activation and chromatin loop formation that physically connect enhancers to gene promoters to which the transcriptional machinery can be directed1,2,3. Because chromatin loops often span several hundred kilobases (kb), detailed maps of 3D chromatin architecture are required to decipher gene regulatory mechanisms. Multiple chromatin conformation capture technologies have been invented to identify the 3D chromatin architecture4. Among these technologies, Hi-C provides the most comprehensive architecture, as it captures genome-wide 3D chromatin interaction profiles. Hi-C datasets have been quickly adapted to interpret non-coding genome-wide significant (GWS) loci5,6,7,8,9,10,11,12,13, as it can link non-coding variants to their putative target genes based on chromatin interaction profiles.
In this article, we outline a protocol to computationally predict putative target genes of GWAS risk variants using chromatin interaction profiles. We apply this protocol to map AD GWS loci14 to their target genes using Hi-C datasets in the adult human brain9. The resulting AD risk genes are characterized by other functional genomic datasets that include single cell transcriptomic and developmental expression profiles.
Protocol
1. Workstation Setup
- Install R (version 3.5.0) and RStudio Desktop. Open RStudio.
- Install the following libraries in R by typing the following code into the console window in RStudio.
if (!"BiocManager" %in% rownames(installed.packages()))
install.packages("BiocManager", repos="https://cran.r-project.org")
BiocManager::install("GenomicRanges")
BiocManager::install("biomaRt")
BiocManager::install("WGCNA")
install.packages("reshape")
install.packages("ggplot2")
install.packages("corrplot")
install.packages("gProfileR")
install.packages("tidyverse")
install.packages("ggpubr") - Download files.
NOTE: In this protocol, all files are required to be downloaded to ~/work directory.- Download the following files by clicking the links provided in Table of Materials.
- Download fine-mapped credible SNPs for AD (Supplementary Table 8 from Jansen et al.14).
NOTE: Before analysis, open sheet eight in 41588_2018_311_MOESM3_ESM.xlsx, remove the first three rows and save the sheet as Supplementary_Table_8_Jansen.txt with tab separated format. - Download 10 kb resolution Hi-C interaction profiles in the adult brain from psychencode (described as Promoter-anchored_chromatin_loops.bed below).
NOTE: This file has the following format: chromosome, TSS_start, TSS_end, Enhancer_start, and Enhancer_end. In case other Hi-C datasets are used, this protocol requires Hi-C datasets processed at high resolution (5−20 kb). - Download single cell expression datasets from the PsychENCODE.
NOTE: These are from neurotypical control samples. - Download developmental expression datasets from the BrainSpan (described as devExpr.rda below).
NOTE: 267666527 is a zipped file, so unzip the 267666527 to extract "columns_metadata.csv", "expression_matrix.csv", and "rows_metadata.csv" to generate devExpr.rda (see section 3).
- Download fine-mapped credible SNPs for AD (Supplementary Table 8 from Jansen et al.14).
- Download exonic coordinates (see Supplementary Files, described as Gencode19_exon.bed and Gencode19_promoter.bed below) from Gencode version 19.
NOTE: Promoters are defined as 2 kb upstream of transcription start site (TSS). These files have the following format: chromosome, start, end, and gene. - Download gene annotation file (see Supplementary Files, described as geneAnno.rda below) from biomart.
NOTE: This file can be used to match genes based on Ensembl gene IDs and HUGO Gene Nomenclature Committee (HGNC) symbol.
- Download the following files by clicking the links provided in Table of Materials.
2. Generation of a GRanges Object for Credible SNPs
- Set up in R by typing the following code into the console window in RStudio.
library(GenomicRanges)
options(stringsAsFactors = F)
setwd("~/work") # This is the path to the working directory.
credSNP = read.delim("Supplementary_Table_8_Jansen.txt", header=T)
credSNP = credSNP[credSNP$Credible.Causal=="Yes",] - Make a GRanges object by typing the following code into the console window in RStudio.
credranges = GRanges(credSNP$Chr, IRanges(credSNP$bp, credSNP$bp), rsid=credSNP$SNP, P=credSNP$P)
save(credranges, file="AD_credibleSNP.rda")
3. Positional Mapping
NOTE: For each step, type the corresponding code into the console window in RStudio.
- Set up in R.
options(stringsAsFactors=F)
library(GenomicRanges)
load("AD_credibleSNP.rda") # (see 2) - Positional mapping of promoter/exonic SNPs to genes
- Load promoter and exonic region and generate a GRange object.
exon = read.table("Gencode19_exon.bed")
exonranges = GRanges(exon[,1],IRanges(exon[,2],exon[,3]),gene=exon[,4])
promoter = read.table("Gencode19_promoter.bed")
promoterranges = GRanges(promoter[,1], IRanges(promoter[,2], promoter[,3]), gene=promoter[,4]) - Overlap credible SNPs with exonic regions.
olap = findOverlaps(credranges, exonranges)
credexon = credranges[queryHits(olap)]
mcols(credexon) = cbind(mcols(credexon), mcols(exonranges[subjectHits(olap)])) - Overlap credible SNPs with promoter regions.
olap = findOverlaps(credranges, promoterranges)
credpromoter = credranges[queryHits(olap)]
mcols(credpromoter) = cbind(mcols(credpromoter), mcols(promoterranges[subjectHits(olap)]))
- Load promoter and exonic region and generate a GRange object.
- Link SNPs to their putative target genes using chromatin interactions.
- Load Hi-C dataset and generate a GRange object.
hic = read.table("Promoter-anchored_chromatin_loops.bed ", skip=1)
colnames(hic) = c("chr", "TSS_start", "TSS_end", "Enhancer_start", "Enhancer_end")
hicranges = GRanges(hic$chr, IRanges(hic$TSS_start, hic$TSS_end), enhancer=hic$Enhancer_start)
olap = findOverlaps(hicranges, promoterranges)
hicpromoter = hicranges[queryHits(olap)]
mcols(hicpromoter) = cbind(mcols(hicpromoter), mcols(promoterranges[subjectHits(olap)]))
hicenhancer = GRanges(seqnames(hicpromoter), IRanges(hicpromoter$enhancer, hicpromoter$enhancer+10000), gene=hicpromoter$gene) - Overlap credible SNPs with Hi-C GRange object.
olap = findOverlaps(credranges, hicenhancer)
credhic = credranges[queryHits(olap)]
mcols(credhic) = cbind(mcols(credhic), mcols(hicenhancer[subjectHits(olap)]))
- Load Hi-C dataset and generate a GRange object.
- Compile AD candidate genes defined by positional mapping and chromatin interaction profiles.
### The resulting candidate genes for AD:
ADgenes = Reduce(union, list(credhic$gene, credexon$gene, credpromoter$gene))
### to convert Ensembl Gene ID to HGNC symbol
load("geneAnno.rda")
ADhgnc = geneAnno1[match(ADgenes, geneAnno1$ensembl_gene_id), "hgnc_symbol"]
ADhgnc = ADhgnc[ADhgnc!=""]
save(ADgenes, ADhgnc, file="ADgenes.rda")
write.table(ADhgnc, file="ADgenes.txt", row.names=F, col.names=F, quote=F, sep="\t")
4. Developmental Expression Trajectories
NOTE: For each step, type the corresponding code into the console window in RStudio.
- Set up in R.
library(reshape); library(ggplot2); library(GenomicRanges); library(biomaRt)
library("WGCNA")
options(stringsAsFactors=F) - Process expression and meta data.
datExpr = read.csv("expression_matrix.csv", header = FALSE)
datExpr = datExpr[,-1]
datMeta = read.csv("columns_metadata.csv")
datProbes = read.csv("rows_metadata.csv")
datExpr = datExpr[datProbes$ensembl_gene_id!="",]
datProbes = datProbes[datProbes$ensembl_gene_id!="",]
datExpr.cr= collapseRows(datExpr, rowGroup = datProbes$ensembl_gene_id, rowID= rownames(datExpr))
datExpr = datExpr.cr$datETcollapsed
gename = data.frame(datExpr.cr$group2row)
rownames(datExpr) = gename$group- Specify developmental stages.
datMeta$Unit = "Postnatal"
idx = grep("pcw", datMeta$age)
datMeta$Unit[idx] = "Prenatal"
idx = grep("yrs", datMeta$age)
datMeta$Unit[idx] = "Postnatal"
datMeta$Unit = factor(datMeta$Unit, levels=c("Prenatal", "Postnatal")) - Select cortical regions.
datMeta$Region = "SubCTX"
r = c("A1C", "STC", "ITC", "TCx", "OFC", "DFC", "VFC", "MFC", "M1C", "S1C", "IPC", "M1C-S1C", "PCx", "V1C", "Ocx")
datMeta$Region[datMeta$structure_acronym %in% r] = "CTX"
datExpr = datExpr[,which(datMeta$Region=="CTX")]
datMeta = datMeta[which(datMeta$Region=="CTX"),]
save(datExpr, datMeta, file="devExpr.rda")
- Specify developmental stages.
- Extract developmental expression profiles of AD risk genes.
load("ADgenes.rda")
exprdat = apply(datExpr[match(ADgenes, rownames(datExpr)),],2,mean,na.rm=T)
dat = data.frame(Region=datMeta$Region, Unit=datMeta$Unit, Expr=exprdat) - Compare prenatal versus postnatal expression levels of AD risk genes.
pdf(file="developmental_expression.pdf")
ggplot(dat,aes(x=Unit, y=Expr, fill=Unit, alpha=Unit)) + ylab("Normalized expression") + geom_boxplot(outlier.size = NA) + ggtitle("Brain Expression") + xlab("") + scale_alpha_manual(values=c(0.2, 1)) + theme_classic() + theme(legend.position="na")
dev.off()
5. Cell-type Expression Profiles
NOTE: For each step, type the corresponding code into the console window in RStudio.
- Set up in R.
options(stringsAsFactors=F)
load("ADgenes.rda")
load("geneAnno.rda")
targetname = "AD"
targetgene = ADhgnc
cellexp = read.table("DER-20_Single_cell_expression_processed_TPM_backup.tsv",header=T,fill=T)
cellexp[1121,1] = cellexp[1120,1]
cellexp = cellexp[-1120,]
rownames(cellexp) = cellexp[,1]
cellexp = cellexp[,-1]
datExpr = scale(cellexp,center=T, scale=F)
datExpr = datExpr[,789:ncol(datExpr)] - Extract cellular expression profiles of AD risk genes.
exprdat = apply(datExpr[match(targetgene, rownames(datExpr)),],2,mean,na.rm=T)
dat = data.frame(Group=targetname, cell=names(exprdat), Expr=exprdat)
dat$celltype = unlist(lapply(strsplit(dat$cell, split="[.]"),'[[',1))
dat = dat[-grep("Ex|In",dat$celltype),]
dat$celltype = gsub("Dev","Fetal",dat$celltype)
dat$celltype = factor(dat$celltype, levels=c("Neurons","Astrocytes","Microglia","Endothelial",
Oligodendrocytes","OPC","Fetal"))
pdf(file="singlecell_expression_ADgenes.pdf")
ggplot(dat,aes(x=celltype, y=Expr, fill=celltype)) +
ylab("Normalized expression") + xlab("") + geom_violin() + theme(axis.text.x=element_text(angle = 90, hjust=1)) + theme(legend.position="none") +
ggtitle(paste0("Cellular expression profiles of AD risk genes"))
dev.off()
6. Gene Annotation Enrichment Analysis of AD Risk Genes
- Download and configure HOMER by typing the commands below in terminal.
mkdir homer
cd homer
wget http://homer.ucsd.edu/homer/configureHomer.pl
perl ./configureHomer.pl -install
perl ./configureHomer.pl -install human-p
perl ./configureHomer.pl -install human-o - Run HOMER by typing the commands below in terminal.
export PATH=$PATH:~/work/homer/bin
findMotifs.pl ~/work/ADgenes.txt human ~/work/ - Plot the enriched terms by typing the following code into the console window in RStudio.
library(ggpubr)
options(stringsAsFactors=F)
pdf("GO_enrichment.pdf",width=15,height=8)
plot_barplot = function(dbname,name,color){
input = read.delim(paste0(dbname,".txt"),header=T)
input = input[,c(-1,-10,-11)]
input = unique(input)
input$FDR = p.adjust(exp(input$logP))
input_sig = input[input$FDR < 0.1,]
input_sig$FDR = -log10(input_sig$FDR)
input_sig = input_sig[order(input_sig$FDR),]
p = ggbarplot(input_sig, x = "Term", y = "FDR", fill = color, color = "white", sort.val = "asc", ylab = expression(-log[10](italic(FDR))), xlab = paste0(name," Terms"), rotate = TRUE, label = paste0(input_sig$Target.Genes.in.Term,"/",input_sig$Genes.in.Term), font.label = list(color = "white", size = 9), lab.vjust = 0.5, lab.hjust = 1)
p = p+geom_hline(yintercept = -log10(0.05), linetype = 2, color = "lightgray")
return(p)
}
p1 = plot_barplot("biological_process","GO Biological Process","#00AFBB")
p2 = plot_barplot("kegg","KEGG","#E7B800")
p3 = plot_barplot("reactome","Reactome","#FC4E07")
ggarrange(p1, p2, p3, labels = c("A", "B", "C"), ncol = 2, nrow = 2)
dev.off()
Results
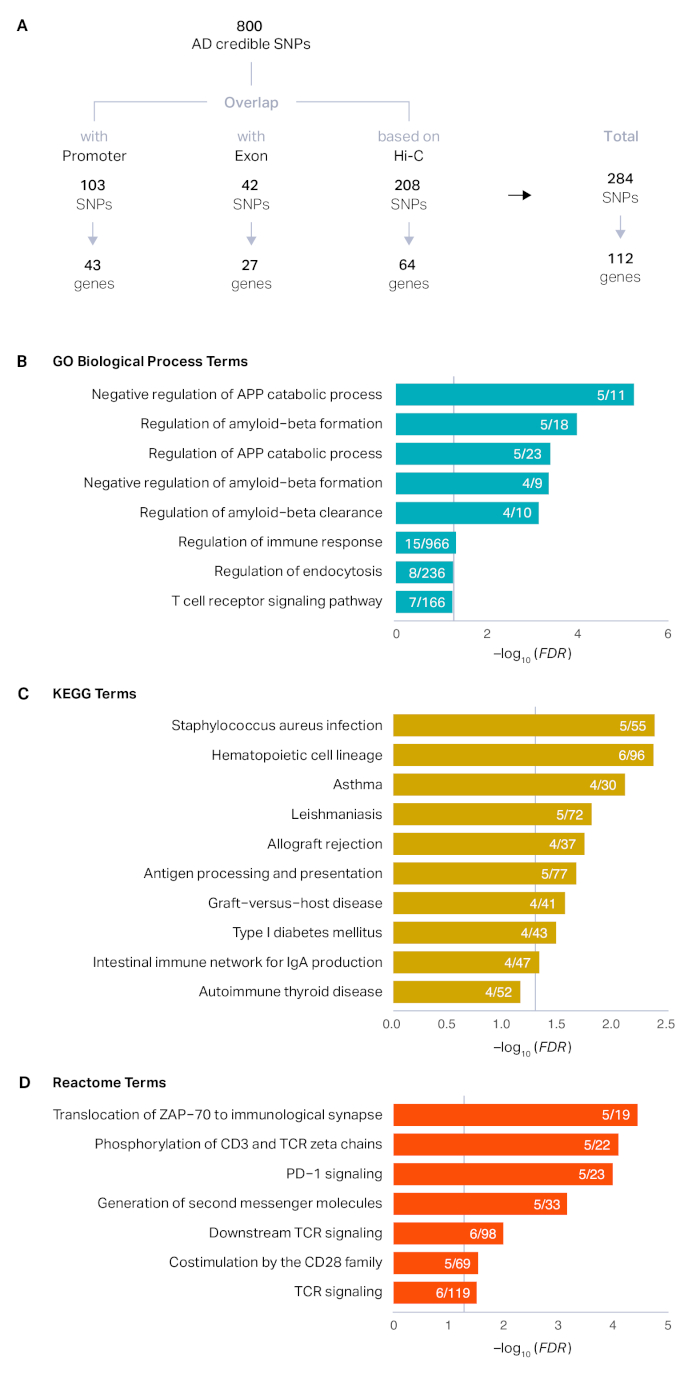
The process described here was applied to a set of 800 credible SNPs that were defined by the original study14. Positional mapping revealed that 103 SNPs overlapped with promoters (43 unique genes) and 42 SNPs overlapped with exons (27 unique genes). After positional mapping, 84% (669) SNPs remained unannotated. Using Hi-C datasets in the adult brain, we were able to link an additional 208 SNPs to 64 genes based on physical proximity. In total, we mapped 284 AD credible SNPs to 112 AD risk genes (Figure 1A). AD risk genes were associated with amyloid precursor proteins, amyloid-beta formation, and immune response, reflecting the known biology of AD15,16,17,18 (Figure 1B-D). Developmental expression profiles of AD risk genes showed marked postnatal enrichment, indicative of the age-associated elevated risk of AD (Figure 2A). Finally, AD risk genes were highly expressed in microglia, primary immune cells in the brain (Figure 2B). This is in agreement with the recurrent findings that AD has a strong immune basis and microglia are the central player in AD pathogenesis14,19,20.

Figure 1: Defining putative target genes of AD GWS loci. (A) Credible SNPs derived from the top 29 AD loci were categorized into promoter SNPs, exonic SNPs, and unannotated non-coding SNPs. Promoter and exonic SNPs were directly assigned to their target genes by positional mapping, while chromatin interaction profiles in the adult brain were additionally used to map SNPs based on physical interactions. (B-D) Enrichment of GO (B), KEGG (C), and Reactome (D) terms in AD risk genes was performed using HOMER as described in protocol section 6. The x axis represents the false discovery rate (FDR) corrected -log10 (P-value). Enriched terms with FDR < 0.1 were plotted. Grey vertical lines represent FDR = 0.05. APP amyloid precursor protein. Numerator, the number of AD risk genes represented in each term; denominator, the number of genes in each term. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Characterization of AD risk genes. (A) AD risk genes are highly expressed in the postnatal cortex compared to the prenatal cortex. (B) Violin plots depicting distributions of gene expression values (normalized expression) in different cell types from the cortex. These results show that AD risk genes are highly expressed in microglia, consistent with previous studies14. Please click here to view a larger version of this figure.
{kind=link}
Supplementary File 1. Please click here to view this file (Right click to download).
Supplementary File 2. Please click here to view this file (Right click to download).
Supplementary File 3. Please click here to view this file (Right click to download).
Discussion
Here we describe an analytic framework that can be used to functionally annotate GWS loci based on positional mapping and chromatin interactions. This process involves multiple steps (for more details see this review13). First, given that chromatin interaction profiles are highly cell-type specific, Hi-C data obtained from the appropriate cell/tissue types that best capture underlying biology of the disorder needs to be used. Given that AD is a neurodegenerative disorder, we used adult brain Hi-C data9 to annotate GWS loci. Second, each GWS locus often has up to hundreds of SNPs that are associated with the trait because of linkage disequilibrium (LD), so it is important to obtain putative causal ('credible') SNPs by computationally predicting the causality through the use of fine-mapping algorithms21,22 or experimentally testing regulatory activities using high-throughput approaches such as massively parallel reporter assays (MPRA)23 or self-transcribing active regulatory region sequencing (STARR-seq)24. For the work described here, we used credible SNPs reported in Jansen et al.14. Third, promoter and exonic SNPs are annotated based on positional mapping. We used a simple positional mapping strategy in which SNPs were mapped to the genes when they overlapped with promoters (defined as 2 kb upstream of transcription start site) or exons. However, this approach can be further elaborated by assessing the functional consequences of exonic SNPs, such as whether the SNP induces nonsense mediated decay, missense variation, or nonsense variation. Fourth, chromatin interaction profiles from the appropriate tissue/cell type can be used to assign SNPs to their putative target genes based on physical proximity. We used interaction profiles anchored to promoters, but we can further refine or expand the interaction profiles by taking enhancer activities (guided by histone H3 K27 acetylation or chromatin accessibility) or exonic interactions into account. One important consideration in this process is to use consistent human genome build. For example, if genomic positions of summary statistics are not based on hg19 (i.e., hg18 or hg38), an appropriate version of the reference genome should be obtained or the summary statistics need to be converted to hg19 using liftover25.
We applied this framework to identify putative target genes for AD GWAS, assigning 284 SNPs to 112 AD risk genes. Using developmental expression profiles26 and cell-type specific expression profiles9, we then demonstrated that this gene set was consistent with what is known about AD pathology, revealing the cell types (microglia), biological functions (immune response and amyloid beta), and elevated risk upon age.
While we presented a framework that delineates potential target genes of AD and its underlying biology, it is of note that Hi-C based annotation can be expanded to annotate any non-coding variation. As more whole-genome sequencing data becomes available and our understanding about the non-coding rare variation grows, Hi-C will provide a key resource for interpretation of disease-associated genetic variants. A compendium of Hi-C resources obtained from multiple tissue and cell types will be therefore critical to facilitating a wide application of this framework to garner biological insights into various human traits and disease.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This work was supported by the NIH grant R00MH113823 (to H.W.) and R35GM128645 (to D.H.P.), NARSAD Young Investigator Award (to H.W.), and SPARK grant from the Simons Foundation Autism Research Initiative (SFARI, to N.M. and H.W.).
Materials
| Name | Company | Catalog Number | Comments |
| 10 kb resolution Hi-C interaction profiles in the adult brain from psychencode | http://adult.psychencode.org/ | ||
| Developmental expression datasets | http://www.brainspan.org/ | ||
| Fine-mapped credible SNPs for AD (Supplementary Table 8 from Jansen et al.14) | https://static-content.springer.com/ | ||
| HOMER | http://homer.ucsd.edu/ | ||
| R (version 3.5.0) | https://www.r-project.org/ | ||
| RStudio Desktop | https://www.rstudio.com/ | ||
| Single cell expression datasets | http://adult.psychencode.org/ |
References
- Dekker, J., Misteli, T. Long-Range Chromatin Interactions. Cold Spring Harbor Perspectives in Biology. 7 (10), a019356 (2015).
- Sanyal, A., Lajoie, B. R., Jain, G., Dekker, J. The long-range interaction landscape of gene promoters. Nature. 489 (7414), 109-113 (2012).
- Plank, J. L., Dean, A. Enhancer function: mechanistic and genome-wide insights come together. Molecular Cell. 55 (1), 5-14 (2014).
- Dekker, J., Marti-Renom, M. A., Mirny, L. A. Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data. Nature Reviews Genetics. 14 (6), 390-403 (2013).
- Martin, P., et al. Capture Hi-C reveals novel candidate genes and complex long-range interactions with related autoimmune risk loci. Nature Communications. 6, 10069 (2015).
- Won, H., et al. Chromosome conformation elucidates regulatory relationships in developing human brain. Nature. 538 (7626), 523-527 (2016).
- Jäger, R., et al. Capture Hi-C identifies the chromatin interactome of colorectal cancer risk loci. Nature Communications. 6, 6178 (2015).
- Chen, J. A. A., et al. Joint genome-wide association study of progressive supranuclear palsy identifies novel susceptibility loci and genetic correlation to neurodegenerative diseases. Molecular Neurodegeneration. 13 (1), 41 (2018).
- Wang, D., et al. Comprehensive functional genomic resource and integrative model for the adult brain. Science. 362 (6420), eaat8464 (2018).
- Demontis, D., et al. Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nature Genetics. 51 (1), 63-75 (2019).
- Grove, J., et al. Identification of common genetic risk variants for autism spectrum disorder. Nature Genetics. 51 (3), 431-444 (2019).
- Lee, P. H., et al. Genome wide meta-analysis identifies genomic relationships, novel loci, and pleiotropic mechanisms across eight psychiatric disorders. bioRxiv. , 528117 (2019).
- Mah, W., Won, H. The three-dimensional landscape of the genome in human brain tissue unveils regulatory mechanisms leading to schizophrenia risk. Schizophrenia Research. , (2019).
- Jansen, I. E., et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer's disease risk. Nature Genetics. 51 (3), 404-413 (2019).
- Viola, K. L., Klein, W. L. Amyloid β oligomers in Alzheimer's disease pathogenesis, treatment, and diagnosis. Acta Neuropathologica. 129 (2), 183-206 (2015).
- Mroczko, B., Groblewska, M., Litman-Zawadzka, A., Kornhuber, J., Lewczuk, P. Amyloid β oligomers (AβOs) in Alzheimer's disease. Journal of Neural Transmission. 125 (2), 177-191 (2018).
- Heneka, M. T., et al. Neuroinflammation in Alzheimer's disease. Lancet Neurology. 14 (4), 388-405 (2015).
- Minter, M. R., Taylor, J. M., Crack, P. J. The contribution of neuroinflammation to amyloid toxicity in Alzheimer's disease. Journal of Neurochemistry. 136 (3), 457-474 (2016).
- Hansen, D. V., Hanson, J. E., Sheng, M. Microglia in Alzheimer's disease. The Journal of Cell Biology. 217 (2), 459-472 (2018).
- Gjoneska, E., et al. Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer's disease. Nature. 518 (7539), 365-369 (2015).
- Benner, C., et al. FINEMAP: efficient variable selection using summary data from genome-wide association studies. Bioinformatics. 32 (10), 1493-1501 (2016).
- Hormozdiari, F., Kostem, E., Kang, E. Y., Pasaniuc, B., Eskin, E. Identifying causal variants at loci with multiple signals of association. Genetics. 198 (2), 497-508 (2014).
- Tewhey, R., et al. Direct Identification of Hundreds of Expression-Modulating Variants using a Multiplexed Reporter Assay. Cell. 165 (6), 1519-1529 (2016).
- Arnold, C. D., et al. Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science. 339 (6123), 1074-1077 (2013).
- Kent, W. J., et al. The human genome browser at UCSC. Genome Research. 12 (6), 996-1006 (2002).
- Kang, H. J., et al. Spatio-temporal transcriptome of the human brain. Nature. 478 (7370), 483-489 (2011).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved