
Method Article
Cartographier les variantes de la maladie d'Alzheimer à leurs gènes cibles à l'aide de l'analyse computationnelle de la configuration de la chromatine
Dans cet article
Résumé
Nous présentons un protocole pour identifier les implications fonctionnelles des variantes non codantes identifiées par des études d'association à l'échelle du génome (GWAS) utilisant des interactions tridimensionnelles de chromatine.
Résumé
Des études d'association à l'échelle du génome (GWAS) ont permis d'identifier avec succès des centaines de loci génomiques qui sont associés à des traits humains et à des maladies. Cependant, parce que la majorité des loci significatifs de génome (GWS) tombent sur le génome non-codant, l'impact fonctionnel de beaucoup restent inconnus. Les interactions tridimensionnelles de chromatine identifiées par Hi-C ou ses dérivés peuvent fournir des outils utiles pour annoter ces loci en reliant les variantes non codantes à leurs gènes exploitables. Ici, nous énoncions un protocole pour cartographier les variantes non codantes gWAS à leurs gènes putatifs en utilisant la maladie d'Alzheimer (MA) GWAS et Hi-C ensembles de données à partir de tissu cérébral adulte humain. Les polymorphismes causals putatifs à un seul nucléotide (SNP) sont identifiés par l'application d'algorithmes de cartographie fine. Les SNP sont ensuite cartographiés selon leurs gènes cibles putatifs à l'aide d'interactions enhancer-promoteur basées sur Hi-C. L'ensemble de gènes qui en résulte représente les gènes de risque de la MA, car ils sont potentiellement réglementés par des variantes de risque aD. Afin d'obtenir d'autres connaissances biologiques sur les mécanismes moléculaires sous-jacents à la MA, nous caractérisons les gènes à risque de la MA à l'aide de données sur l'expression cérébrale développementale et de profils d'expression monocellulaire du cerveau. Ce protocole peut être étendu à n'importe quel ensemble de données GWAS et Hi-C afin d'identifier les gènes cibles putatifs et les mécanismes moléculaires sous-jacents à divers traits et maladies humains.
Introduction
Les études d'association à l'échelle du génome (GWAS) ont joué un rôle essentiel en révélant la base génétique d'une gamme de traits et de maladies humains. Ce génotypage à grande échelle a mis au jour des milliers de variantes génomiques associées à des phénotypes allant de la hauteur au risque de schizophrénie. Cependant, en dépit de l'énorme succès de GWAS dans l'identification des loci associés à la maladie et au trait, une compréhension mécaniste de la façon dont ces variantes contribuent au phénotype a été difficile parce que la plupart des variantes associées au phénotype résident dans le non-codage fraction du génome humain. Étant donné que ces variantes se chevauchent souvent avec les éléments réglementaires prévus, elles sont susceptibles de modifier le contrôle transcriptionnel d'un gène voisin. Cependant, les loci non codants peuvent influencer la transcription des gènes à des distances linéaires dépassant une mégabase, ce qui rend les gènes affectés par chaque variante difficiles à identifier. La structure tridimensionnelle (3D) de chromatine joue un rôle important dans la médiation des connexions entre les loci régulateurs éloignés et les promoteurs de gènes et peut être utilisée pour identifier les gènes affectés par les polymorphismes mononucléotides associés au phénotype (SNP).
La régulation génique est médiée par un processus complexe, qui implique l'activation de l'améliorateur et la formation de boucles de chromatine qui relient physiquement les exhausteurs aux promoteurs de gènes auxquels la machinerie transcriptionnelle peut être dirigée1,2,3. Étant donné que les boucles de chromatine couvrent souvent plusieurs centaines de kilobases (kb), des cartes détaillées de l'architecture de chromatine 3D sont nécessaires pour déchiffrer les mécanismes de régulation des gènes. Plusieurs technologies de capture de conformation de chromatine ont été inventées pour identifier l'architecture de chromatine 3D4. Parmi ces technologies, Hi-C fournit l'architecture la plus complète, car elle capture des profils d'interaction de chromatine 3D à l'échelle du génome. Les ensembles de données Hi-C ont été rapidement adaptés pour interpréter les variantes non codantes significatives (GWS) loci5,7,8,9,10,11,12,13, car il peut lier les variantes non codantes à leurs gènes cibles putatives basées sur des profils d'interaction de chromatine.
Dans cet article, nous énoncions un protocole pour prévoir computationnellement les gènes cibles putatifs des variantes de risque de GWAS utilisant des profils d'interaction de chromatine. Nous appliquons ce protocole pour cartographier AD GWS loci14 à leurs gènes cibles en utilisant des ensembles de données Hi-C dans le cerveau humain adulte9. Les gènes de risque de la MA qui en résultent sont caractérisés par d'autres ensembles de données génomiques fonctionnelles qui comprennent des profils d'expression transcriptomique et développementale à cellule unique.
Protocole
1. Configuration de poste de travail
- Installez R (version 3.5.0) et RStudio Desktop. Ouvrez RStudio.
- Installez les bibliothèques suivantes en R en tapant le code suivant dans la fenêtre de la console dans RStudio.
si (!" BiocManager" %in% rownames (installed.packages()))
install.packages("BiocManager", repos'"https://cran.r-project.org")
BiocManager::install("GenomicRanges")
BiocManager::install("biomaRt")
BiocManager::install("WGCNA")
install.packages ("reshape")
install.packages ("ggplot2")
install.packages ("corrplot")
install.packages ("gProfileR")
install.packages ("tidyverse")
install.packages ("ggpubr") - Téléchargez des fichiers.
REMARQUE : Dans ce protocole, tous les fichiers doivent être téléchargés vers l'annuaire de travail.- Téléchargez les fichiers suivants en cliquant sur les liens fournis dans tableau des matériaux.
- Télécharger des SNP crédibles pour AD (tableau complémentaire 8 de Jansen etal.14).
REMARQUE : Avant l'analyse, ouvrez la feuille huit dans 41588_2018_311_MOESM3_ESM.xlsx, retirez les trois premières rangées et enregistrez la feuille comme Supplementary_Table_8_Jansen.txt avec le format séparé d'onglet. - Téléchargez des profils d'interaction Hi-C de résolution de 10 kb dans le cerveau adulte à partir de psychencode (décrit comme Promoteur-anchored_chromatin_loops.ld ci-dessous).
REMARQUE : Ce fichier a le format suivant : chromosome, TSS_start, TSS_end, Enhancer_start et Enhancer_end. Dans le cas où d'autres jeux de données Hi-C sont utilisés, ce protocole nécessite des jeux de données Hi-C traités à haute résolution (5 à 20 kb). - Téléchargez des jeux de données d'expression cellulaire unique à partir du PsychENCODE.
REMARQUE : Il s'agit d'échantillons témoins neurotypiques. - Téléchargez les ensembles de données d'expression développementale du BrainSpan (décrit comme devExpr.rda ci-dessous).
REMARQUE: 267666527 est un fichier zippé, donc décompresser le 267666527 pour extraire "columns_metadata.csv", "expression_matrix.csv", et "rows_metadata.csv" pour générer devExpr.rda (voir section 3).
- Télécharger des SNP crédibles pour AD (tableau complémentaire 8 de Jansen etal.14).
- Téléchargez les coordonnées exoniques (voir Fichiers supplémentaires, décrits comme Gencode19_exon.ld et Gencode19_promoter.ld ci-dessous) à partir de Gencode version 19.
REMARQUE : Les promoteurs sont définis comme étant de 2 kb en amont du site de démarrage de la transcription (TSS). Ces fichiers ont le format suivant : chromosome, début, fin et gène. - Télécharger le fichier d'annotation génétique (voir Fichiers supplémentaires, décrit comme geneAnno.rda ci-dessous) à partir de biomart.
REMARQUE : Ce fichier peut être utilisé pour faire correspondre les gènes basés sur les ID génétiques Ensembl et le symbole DU Comité de Nomenclature génique HUGO (HGNC).
- Téléchargez les fichiers suivants en cliquant sur les liens fournis dans tableau des matériaux.
2. Génération d'un objet GRanges pour les SNP crédibles
- Configurez en R en tapant le code suivant dans la fenêtre de la console dans RStudio.
bibliothèque (GenomicRanges)
options (stringsAsFactors ' F)
setwd (" / travail ") - C'est le chemin vers le répertoire de travail.
credSNP - read.delim ("Supplementary_Table_8_Jansen.txt", header-T)
credSNP - credSNP[credSNP$Credible.Causal"Oui",] - Faites un objet GRanges en tapant le code suivant dans la fenêtre de la console dans RStudio.
credranges - GRanges (credSNP$Chr, IRanges(credSNP$bp, credSNP$bp), rsid-credSNP$SNP, P-credSNP$P)
enregistrer (credranges, fichier "AD_credibleSNP.rda")
3. Cartographie positionnelle
REMARQUE : Pour chaque étape, tapez le code correspondant dans la fenêtre de la console dans RStudio.
- Installé en R.
options (stringsAsFactors-F)
bibliothèque (GenomicRanges)
charge ("AD_credibleSNP.rda") (voir 2) - Cartographie positionnelle des SNP promoteurs/exoniques aux gènes
- Chargez le promoteur et la région exonique et génèrez un objet GRange.
exon -read.table ("Gencode19_exon.bed")
exonranges - GRanges (exon[,1],IRanges(exon[,2],exon[,3]),gene-exon[,4])
promoteur - read.table ("Gencode19_promoter.bed")
promoterranges - GRanges (promoteur[,1], IRanges (promoteur[,2], promoteur[,3]), gene-promoteur[,4]) - Overlap SNPs crédibles avec des régions exoniques.
olap - findOverlaps (credranges, exonranges)
credexon - credranges[queryHits(olap)]
mcols (credexon) ' cbind(mcols(credexon), mcols(exonranges[subjectHits(olap)))) - Chevauchez les SNP crédibles avec les régions promoteurs.
olap - findOverlaps (credranges, promoterranges)
credpromoter - credranges[queryHits(olap)]
mcols (credpromoter) ' cbind(mcols(credpromoter), mcols(promoterranges[subjectHits(olap)))) )
- Chargez le promoteur et la région exonique et génèrez un objet GRange.
- Liez les SNP à leurs gènes cibles putatifs à l'aide d'interactions de chromatine.
- Chargez le jeu de données Hi-C et générez un objet GRange.
hic - read.table("Promoteur-anchored_chromatin_loops.ld ", skip 1)
colnames (hic) 'c("chr", "TSS_start", "TSS_end", "Enhancer_start", "Enhancer_end")
hicranges - GRanges (hic$chr, IRanges (hic$TSS_start, hic$TSS_end), enhancer'hic$Enhancer_start)
olap - findOverlaps (hicranges, promoterranges)
hicpromoter - hicranges[queryHits(olap)]
mcols (hicpromoter) ' cbind(mcols(hicpromoter), mcols(promoterranges[subjectHits(olap))))
hicenhancer - GRanges (seqnames(hicpromoter), IRanges (hicpromoter$enhancer, hicpromoter$enhancer-10000), gene-hicpromoter$gene) - Overlap SNPs crédibles avec l'objet Hi-C GRange.
olap - findOverlaps (credranges, hicenhancer)
credhic - credranges[queryHits(olap)]
mcols (credhic) - cbind (mcols(credhic), mcols(hicenhancer[subjectHits(olap)))) )
- Chargez le jeu de données Hi-C et générez un objet GRange.
- Compiler les gènes candidats AD définis par la cartographie positionnelle et les profils d'interaction de chromatine.
Les gènes candidats résultants pour l'AD :
ADgenes - Réduire (union, liste(gène credhic$, credexon$gene, credpromoter$gene))
Pour convertir l'ID Gène Ensembl en symbole HGNC
charge ("geneAnno.rda")
ADhgnc - geneAnno1[match(ADgenes, geneAnno$ensembl_gene_id), "hgnc_symbol"]
ADhgnc - ADhgnc[ADhgnc!
enregistrer (ADgenes, ADhgnc, fichier "ADgenes.rda")
write.table (ADhgnc, fichier "ADgenes.txt", row.names-F, col.names-F, devisF, sep'"t")
4. Trajectoires d'expression développementale
REMARQUE : Pour chaque étape, tapez le code correspondant dans la fenêtre de la console dans RStudio.
- Installé en R.
bibliothèque (remodelage); bibliothèque(ggplot2); bibliothèque (GenomicRanges); bibliothèque (biomaRt)
bibliothèque ("WGCNA")
options (stringsAsFactors-F) - Expression du processus et méta données.
datExpr - read.csv ("expression_matrix.csv", en-tête - FALSE)
datExpr et datExpr[,-1]
datMeta - read.csv ("columns_metadata.csv")
datProbes - read.csv ("rows_metadata.csv")
datExpr - datExpr[datProbes$ensembl_gene_id!
datProbes - datProbes[datProbes$ensembl_gene_id!
datExpr.crSD collapseRows(datExpr, rowGroup - datProbes$ensembl_gene_id, rowIDMD rownames (datExpr))
datExpr - datExpr.cr$datETcollapsed
gename - data.frame (datExpr.cr$group2row)
rownames (datExpr) - gename$group- Spécifier les stades de développement.
datMeta$Unité - "Postnatal"
idx et grep ("pcw", datMeta$age)
datMeta$Unit[idx] - "Prénatal"
idx et grep ("yrs", datMeta$age)
datMeta$Unit[idx] - "Postnatal"
datMeta$Unit -facteur (datMeta$Unit, levels'c("Prenatal", "Postnatal")) - Sélectionnez les régions corticales.
datMeta$Region - "SubCTX"
c ("A1C", "STC", "ITC", "TCx", "OFC", "DFC", "VFC", "MFC", "M1C", "S1C", "IPC", "M1C-S1C", "PCx", "V1C", "Ocx")
datMeta$Region[datMeta$structure_acronym %in% r] - "CTX"
datExpr - datExpr[,qui(datMeta$Region" "CTX")]
datMeta et datMeta[qui(datMeta$Region" "CTX"),]
enregistrer (datExpr, datMeta, fichier "devExpr.rda")
- Spécifier les stades de développement.
- Extraire les profils d'expression développementale des gènes de risque d'AD.
charge ("ADgenes.rda")
exprdat - appliquer(datExpr[match(ADgenes, rownames(datExpr)),2,mean,na.rm-T)
dat -data.frame (Region-datMeta$Region, Unit-datMeta$Unit, Expr-exprdat) - Comparez les niveaux d'expression prénatale par rapport aux niveaux postnatals des gènes à risque de MA.
pdf (fichier"developmental_expression.pdf")
ggplot (dat,aes(x-Unit, y-Expr, fill-Unit, alpha-Unit)) - ylab ("Normalized expression") - geom_boxplot (outlier.size - NA) - ggtitle ("Brain Expression") - xlab ("") - scale_alpha_manual (values-c(0.2, 1)theme_classic) )
dev.off()
5. Profils d'expression de type cellulaire
REMARQUE : Pour chaque étape, tapez le code correspondant dans la fenêtre de la console dans RStudio.
- Installé en R.
options (stringsAsFactors-F)
charge ("ADgenes.rda")
charge ("geneAnno.rda")
nom de cible 'AD'
targetgene - ADhgnc
cellexp - read.table ("DER-20_Single_cell_expression_processed_TPM_backup.tsv", header-T,fill-T)
cellexp[1121,1] - cellexp[1120,1]
cellexp - cellexp[-1120,]
rownames (cellexp) - cellexp[,1]
cellexp et cellexp[,-1]
datExpr -échelle (cellexp,centre-T, échelle F)
datExpr - datExpr[,789:ncol(datExpr)] - Extraire les profils d'expression cellulaire des gènes de risque d'ANNONCE.
exprdat - appliquer(datExpr[match(targetgene, rownames(datExpr)),2,mean,na.rm-T)
dat 'data.frame(Group'targetname, cell-names(exprdat), Expr'exprdat)
dat$celltype -unlist(lapply(strsplit(dat$cell, split"[.]"), '[[',1)))
dat et dat[-grep) («Ex» In"dat$celltype),]
dat$celltype 'gsub',"Dev","Fetal,"dat$celltype)
dat$celltype - facteur (dat$celltype, levels'c("Neurons","Astrocytes","Microglia","Endothelial",
Oligodendrocytes","OPC","Fetal"))
pdf (fichier"singlecell_expression_ADgenes.pdf")
ggplot (dat,aes(x-celltype, y-Expr, fill-celltype))
ylab (« Expression normalisée ») ' xlab(« » - geom_violin () - thème (axis.text.x -element_text (angle 90, hjust-1)) - thème (legend.position ' "no")
ggtitle (paste0("Profils d'expression cellulaire des gènes de risque de la MA"))
dev.off()
6. Analyse de l'enrichissement de l'annotation génétique des gènes de risque de la MA
- Téléchargez et configurez HOMER en tapant les commandes ci-dessous en terminal.
homer mkdir
cd homer
wget http://homer.ucsd.edu/homer/configureHomer.pl
perl ./configureHomer.pl -installer
perl ./configureHomer.pl -installer humain-p
perl ./configureHomer.pl -installer humain-o - Exécutez HOMER en tapant les commandes ci-dessous en terminal.
exportation DE PATH-$PATH : '/travail/homer/bin'
findMotifs.pl '/travail/ADgenes.txt humain'/travail/ - Tracez les termes enrichis en tapant le code suivant dans la fenêtre de la console dans RStudio.
bibliothèque (ggpubr)
options (stringsAsFactors-F)
pdf ("GO_enrichment.pdf", largeur 15,hauteur 8)
plot_barplot fonction (dbname,name,color)
entrée - read.delim (paste0(dbname,".txt"),header-T)
entrée [,c(-1,-10,-11)]
entrée unique (entrée)
entrée$FDR ' p.adjust(exp(input$logP))
input_sig ' input[input$FDR 'lt; 0.1,]
input_sig$FDR -log10(input_sig$FDR)
input_sig et input_sig[commande(input_sig $FDR),]
p ggbarplot (input_sig, x ' "Term", y ' 'FDR', 'remplir', couleur - "blanc", sort.val -"asc", ylab - expression (-log[10](italique (FDR))), xlab -paste0 (nom," Conditions"), rotation - TRUE, étiquette 'paste0(input_sig$Target.Genes.in.Term,"/"input_sig$Genes.in.Term), police.label 'list(color '"white", taille '9), lab.vjust '0.5, lab.hjust
p ' p 'geom_hline(yintercept ' -log10(0.05), linetype '2, couleur 'lightgray')
retour(p)
}
p1 - plot_barplot ("biological_process","GO Biological Process","#00AFBB")
p2 - plot_barplot ("kegg","KEGG","#E7B800")
p3 - plot_barplot ("reactome","Reactome","#FC4E07")
ggarrange(p1, p2, p3, étiquettes 'c("A", "B", "C"), ncol '2, nrow '2)
dev.off()
Résultats
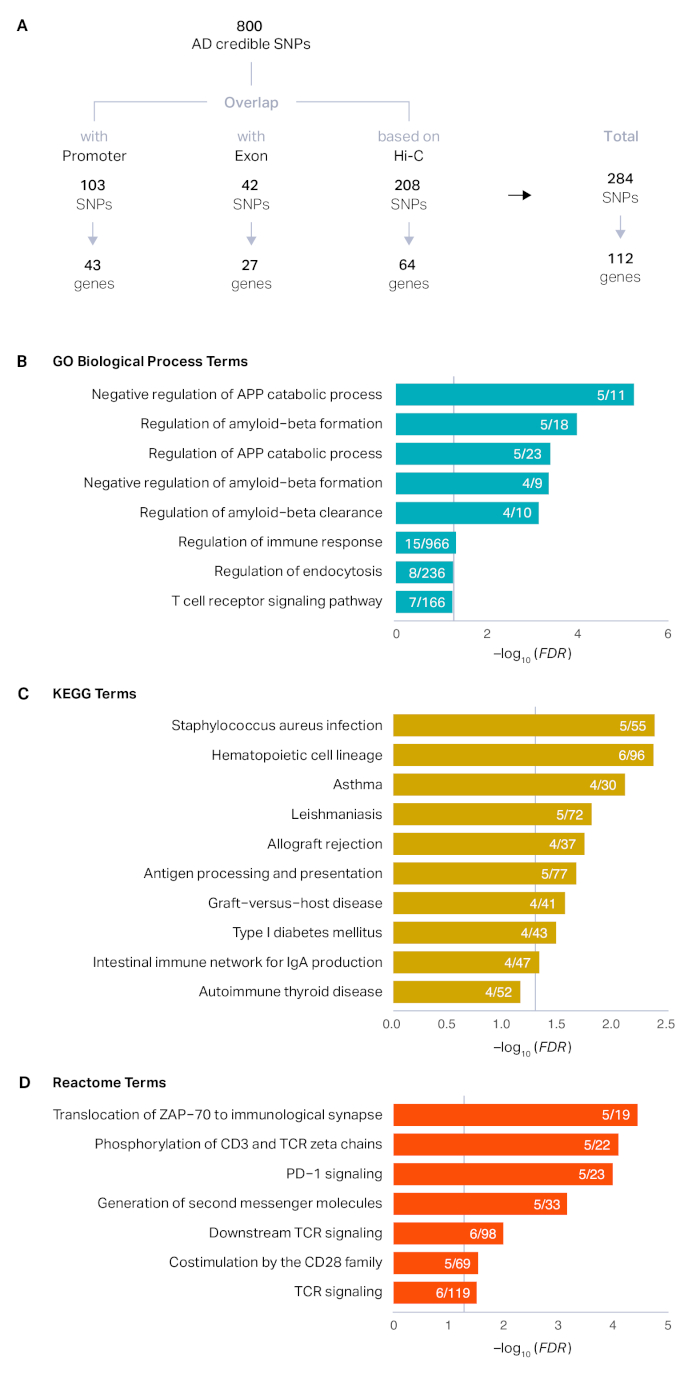
Le processus décrit ici a été appliqué à un ensemble de 800 SNP crédibles qui ont été définis par l'étude originale14. La cartographie de position a révélé que 103 SNP chevauchaient les promoteurs (43 gènes uniques) et 42 SNP chevauchés avec des exons (27 gènes uniques). Après cartographie positionnelle, 84 % (669) SNP sont restés sans annotated. En utilisant des ensembles de données Hi-C dans le cerveau adulte, nous avons pu relier 208 SNP supplémentaires à 64 gènes basés sur la proximité physique. Au total, nous avons cartographié 284 SNP crédibles adm. 100 à 112 gènes à risque AD(figure 1A). Les gènes de risque d'AD ont été associés aux protéines de précurseur amyloïde, à la formation amyloïde-bêta, et à la réponse immunitaire, reflétant la biologie connue de l'AD15,16,17,18 ( figure1B-D). Les profils d'expression développementale des gènes de risque de MA ont montré l'enrichissement postnatal marqué, indicatif du risque élevé associé à l'âge de la MA (figure 2A). Enfin, les gènes du risque de MA ont été fortement exprimés dans les microglies, les cellules immunitaires primaires du cerveau (Figure 2B). Ceci est en accord avec les résultats récurrents que la MA a une base immunitaire forte et les microglies sont l'acteur central dans la pathogénie AD14,19,20.

Figure 1 : Définir les gènes cibles putatifs des loci AD GWS. (A) Les SNP crédibles dérivés des 29 principaux loci ads ont été classés dans les SNP promoteurs, les SNP exoniques et les SNP non codants non annotés. Le promoteur et les SNP exoniques ont été directement affectés à leurs gènes cibles par cartographie positionnelle, tandis que les profils d'interaction de chromatine dans le cerveau adulte ont été en outre utilisés pour cartographier les SNP en fonction des interactions physiques. (B-D) L'enrichissement de GO (B), KEGG (C), et Reactome (D) termes dans les gènes de risque de LA D a été effectué en utilisant HOMER comme décrit dans la section 6 du protocole. L'axe x représente le faux taux de découverte (FDR) corrigé -log10 (valeur P). Des termes enrichis avec FDR et 0,1 ont été tracés. Les lignes verticales grises représentent le FDR 0,05. Protéine précurseur amyloïde d'APP. Numérateur, le nombre de gènes de risque de MA représentés dans chaque terme ; dénominateur, le nombre de gènes dans chaque terme. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 2 : Caractérisation des gènes à risque de la MA. (A) les gènes de risque d'AD sont fortement exprimés dans le cortex postnatal comparé au cortex prénatal. (B) Des parcelles de violon représentant des distributions de valeurs d'expression génique (expression normalisée) dans différents types de cellules du cortex. Ces résultats montrent que les gènes de risque de LAD sont fortement exprimés dans les microglies, conformément aux études précédentes14. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}
Dossier supplémentaire 1. S'il vous plaît cliquez ici pour voir ce fichier (Clic droit pour télécharger).
Dossier supplémentaire 2. S'il vous plaît cliquez ici pour voir ce fichier (Clic droit pour télécharger).
Dossier supplémentaire 3. S'il vous plaît cliquez ici pour voir ce fichier (Clic droit pour télécharger).
Discussion
Ici, nous décrivons un cadre analytique qui peut être utilisé pour annoter fonctionnellement GWS loci basé sur la cartographie positionnelle et les interactions de chromatine. Ce processus comporte plusieurs étapes (pour plus de détails voir cet examen13). Tout d'abord, étant donné que les profils d'interaction de chromatine sont fortement de type cellulaire spécifique, les données Hi-C obtenues à partir des types appropriés de cellules/tissus qui capturent le mieux la biologie sous-jacente du trouble doivent être utilisées. Étant donné que la MA est un trouble neurodégénératif, nous avons utilisé le cerveau adulte Hi-C données9 pour annoter GWS loci. Deuxièmement, chaque locus GWS a souvent jusqu'à des centaines de SNP qui sont associés au trait en raison du déséquilibre de liaison (LD), il est donc important d'obtenir des SNP causals putatifs (« crédibles ») en prédisant causalité par l'utilisation d'algorithmes de cartographie fine21,22 ou de tester expérimentalement des activités de réglementation en utilisant des approches à haut débit telles que des essais de reporter massivement parallèles (MPRA)23 ou l'auto-transcrire le séquençage actif de la région réglementaire ( STARR-seq)24. Pour le travail décrit ici, nous avons utilisé des SNP crédibles rapportés dans Jansen et al.14. Troisièmement, les SNP promoteurs et exoniques sont annotés en fonction de la cartographie de position. Nous avons utilisé une stratégie de cartographie positionnelle simple dans laquelle les SNP ont été cartographiés aux gènes lorsqu'ils se chevauchaient avec des promoteurs (définis comme 2 kb en amont du site de démarrage de transcription) ou des exons. Cependant, cette approche peut être plus approfondie en évaluant les conséquences fonctionnelles des SNP exoniques, telles que si le SNP induit la décomposition médiatisée de non-sens, la variation de mauvais sens, ou la variation absurde. Quatrièmement, les profils d'interaction de chromatine du type approprié de tissu/cellule peuvent être employés pour assigner des SNP à leurs gènes cibles putatifs basés sur la proximité physique. Nous avons utilisé des profils d'interaction ancrés aux promoteurs, mais nous pouvons affiner ou élargir davantage les profils d'interaction en tenant compte des activités d'amélioration (guidées par l'acétylation ou l'accessibilité de la chromatine h3 K27) ou des interactions exoniques. Une considération importante dans ce processus est d'utiliser la construction cohérente du génome humain. Par exemple, si les positions génomiques des statistiques sommaires ne sont pas fondées sur hg19 (c.-à-d. hg18 ou hg38), une version appropriée du génome de référence devrait être obtenue ou les statistiques sommaires doivent être converties en hg19 à l'aide de l'élévateur25.
Nous avons appliqué ce cadre pour identifier les gènes cibles putatifs pour AD GWAS, en attribuant 284 SNP à 112 gènes de risque ad. En utilisant les profils d'expression développementale26 et les profils d'expression spécifiques de type cellulaire9, nous avons alors démontré que cet ensemble de gènes était compatible avec ce que l'on sait sur la pathologie de la MA, révélant les types cellulaires (microglies), les fonctions biologiques (réponse immunitaire et bêta amyloïde), et le risque élevé à l'âge.
Bien que nous ayons présenté un cadre qui délimite les gènes cibles potentiels de la MA et de sa biologie sous-jacente, il est à noter que l'annotation basée sur le Hi-C peut être élargie pour annoter toute variation non codante. Au fur et à mesure que de plus en plus de données sur le séquençage du génome entier seront disponibles et que notre compréhension de la variation rare non codante s'accroît, Le Hi-C constituera une ressource clé pour l'interprétation des variantes génétiques associées à la maladie. Un recueil de ressources Hi-C obtenues à partir de plusieurs types de tissus et de cellules sera donc essentiel pour faciliter une large application de ce cadre afin d'obtenir des informations biologiques sur divers traits humains et maladies.
Déclarations de divulgation
Les auteurs n'ont rien à révéler.
Remerciements
Ce travail a été soutenu par la subvention des NIH R00MH13823 (à H.W.) et R35GM128645 (à D.H.P.), le prix NARSAD jeune chercheur (à H.W.), et la subvention SPARK de la Simons Foundation Autism Research Initiative (SFARI, à N.M. et H.W.).
matériels
| Name | Company | Catalog Number | Comments |
| 10 kb resolution Hi-C interaction profiles in the adult brain from psychencode | http://adult.psychencode.org/ | ||
| Developmental expression datasets | http://www.brainspan.org/ | ||
| Fine-mapped credible SNPs for AD (Supplementary Table 8 from Jansen et al.14) | https://static-content.springer.com/ | ||
| HOMER | http://homer.ucsd.edu/ | ||
| R (version 3.5.0) | https://www.r-project.org/ | ||
| RStudio Desktop | https://www.rstudio.com/ | ||
| Single cell expression datasets | http://adult.psychencode.org/ |
Références
- Dekker, J., Misteli, T. Long-Range Chromatin Interactions. Cold Spring Harbor Perspectives in Biology. 7 (10), a019356(2015).
- Sanyal, A., Lajoie, B. R., Jain, G., Dekker, J. The long-range interaction landscape of gene promoters. Nature. 489 (7414), 109-113 (2012).
- Plank, J. L., Dean, A. Enhancer function: mechanistic and genome-wide insights come together. Molecular Cell. 55 (1), 5-14 (2014).
- Dekker, J., Marti-Renom, M. A., Mirny, L. A. Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data. Nature Reviews Genetics. 14 (6), 390-403 (2013).
- Martin, P., et al. Capture Hi-C reveals novel candidate genes and complex long-range interactions with related autoimmune risk loci. Nature Communications. 6, 10069(2015).
- Won, H., et al. Chromosome conformation elucidates regulatory relationships in developing human brain. Nature. 538 (7626), 523-527 (2016).
- Jäger, R., et al. Capture Hi-C identifies the chromatin interactome of colorectal cancer risk loci. Nature Communications. 6, 6178(2015).
- Chen, J. A. A., et al. Joint genome-wide association study of progressive supranuclear palsy identifies novel susceptibility loci and genetic correlation to neurodegenerative diseases. Molecular Neurodegeneration. 13 (1), 41(2018).
- Wang, D., et al. Comprehensive functional genomic resource and integrative model for the adult brain. Science. 362 (6420), eaat8464(2018).
- Demontis, D., et al. Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nature Genetics. 51 (1), 63-75 (2019).
- Grove, J., et al. Identification of common genetic risk variants for autism spectrum disorder. Nature Genetics. 51 (3), 431-444 (2019).
- Lee, P. H., et al. Genome wide meta-analysis identifies genomic relationships, novel loci, and pleiotropic mechanisms across eight psychiatric disorders. bioRxiv. , 528117(2019).
- Mah, W., Won, H. The three-dimensional landscape of the genome in human brain tissue unveils regulatory mechanisms leading to schizophrenia risk. Schizophrenia Research. , In press (2019).
- Jansen, I. E., et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer's disease risk. Nature Genetics. 51 (3), 404-413 (2019).
- Viola, K. L., Klein, W. L. Amyloid β oligomers in Alzheimer's disease pathogenesis, treatment, and diagnosis. Acta Neuropathologica. 129 (2), 183-206 (2015).
- Mroczko, B., Groblewska, M., Litman-Zawadzka, A., Kornhuber, J., Lewczuk, P. Amyloid β oligomers (AβOs) in Alzheimer's disease. Journal of Neural Transmission. 125 (2), 177-191 (2018).
- Heneka, M. T., et al. Neuroinflammation in Alzheimer's disease. Lancet Neurology. 14 (4), 388-405 (2015).
- Minter, M. R., Taylor, J. M., Crack, P. J. The contribution of neuroinflammation to amyloid toxicity in Alzheimer's disease. Journal of Neurochemistry. 136 (3), 457-474 (2016).
- Hansen, D. V., Hanson, J. E., Sheng, M. Microglia in Alzheimer's disease. The Journal of Cell Biology. 217 (2), 459-472 (2018).
- Gjoneska, E., et al. Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer's disease. Nature. 518 (7539), 365-369 (2015).
- Benner, C., et al. FINEMAP: efficient variable selection using summary data from genome-wide association studies. Bioinformatics. 32 (10), 1493-1501 (2016).
- Hormozdiari, F., Kostem, E., Kang, E. Y., Pasaniuc, B., Eskin, E. Identifying causal variants at loci with multiple signals of association. Genetics. 198 (2), 497-508 (2014).
- Tewhey, R., et al. Direct Identification of Hundreds of Expression-Modulating Variants using a Multiplexed Reporter Assay. Cell. 165 (6), 1519-1529 (2016).
- Arnold, C. D., et al. Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science. 339 (6123), 1074-1077 (2013).
- Kent, W. J., et al. The human genome browser at UCSC. Genome Research. 12 (6), 996-1006 (2002).
- Kang, H. J., et al. Spatio-temporal transcriptome of the human brain. Nature. 478 (7370), 483-489 (2011).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.