Method Article
Medición de la unión de nucleótidos a proteínas de membrana intactas y funcionales en tiempo real
En este artículo
Resumen
Este protocolo presenta un método para medir la unión de nucleótidos de adenina a los receptores en tiempo real en un entorno celular. La unión se mide como transferencia de energía de resonancia de Förster (FRET) entre derivados de nucleótidos de trinitrofenilo y proteínas marcadas con un aminoácido fluorescente no canónico.
Resumen
Hemos desarrollado un método para medir la unión de nucleótidos de adenina a receptores transmembrana intactos y funcionales en un entorno celular o de membrana. Este método combina la expresión de proteínas marcadas con el aminoácido fluorescente no canónico ANAP, y FRET entre ANAP y derivados de nucleótidos fluorescentes (trinitrofenil). Presentamos ejemplos de unión de nucleótidos a canales iónicos KATP marcados con ANAP medidos en membranas plasmáticas sin techo y parches de membrana extirpados de adentro hacia afuera bajo abrazadera de voltaje. Este último permite mediciones simultáneas de unión al ligando y corriente del canal, una lectura directa de la función de la proteína. El tratamiento y análisis de datos se discuten ampliamente, junto con posibles trampas y artefactos. Este método proporciona una rica visión mecanicista de la activación dependiente del ligando de los canales KATP y puede adaptarse fácilmente al estudio de otras proteínas reguladas por nucleótidos o cualquier receptor para el que se pueda identificar un ligando fluorescente adecuado.
Introducción
Varias clases importantes de proteínas están directamente reguladas por la unión de ligandos. Estos van desde enzimas solubles hasta proteínas incrustadas en la membrana, incluidas las tirosina quinasas receptoras, los receptores acoplados a proteínas G (GPCR) y los canales iónicos. Los GPCR y los canales representan ~ 34% y ~ 15% de todos los objetivos farmacológicos actuales, respectivamente 1,2. Por lo tanto, existe un considerable interés bioquímico, así como médico, en el desarrollo de métodos que proporcionen información mecanicista sobre las interacciones ligando-receptor. Los métodos tradicionales para medir la unión de ligandos, incluido el etiquetado de fotoafinidad y los estudios de unión a radioligandos, requieren grandes cantidades de proteína parcialmente purificada y generalmente se realizan en condiciones y escalas de tiempo no fisiológicas. Un método ideal requeriría solo pequeñas cantidades de proteína, podría realizarse en proteínas intactas expresadas en un entorno celular o de membrana, podría monitorearse en tiempo real y sería compatible con lecturas directas de la función de la proteína.
La transferencia de energía de resonancia de Förster (FRET) es un método que detecta la proximidad entre dos moléculas marcadas con fluorescencia3. FRET ocurre cuando un fluoróforo donante excitado transfiere energía de una manera no radiativa a una molécula aceptora (típicamente otro fluoróforo). La transferencia de energía da como resultado la extinción de la emisión de fluorescencia del donante y la sensibilización de la emisión aceptora (si el aceptor es un fluoróforo). La eficiencia de transferencia depende de la6ª potencia de la distancia entre el donante y el aceptor. Además, el donante y el aceptor deben estar cerca (generalmente menos de 10 nm) para que se produzca la FRET. Como tal, FRET puede ser explotado para medir la unión directa entre un receptor de proteína marcado fluorescentemente y un ligando fluorescente.
Varias proteínas diferentes están reguladas o activadas por la unión de nucleótidos de adenina intracelulares o extracelulares (ATP, ADP, AMP, cAMP). Muchas proteínas transportadoras requieren hidrólisis de ATP para su ciclo de reacción, incluidos los transportadores de casetes de unión a ATP y las ATPasas de tipo P como la bomba Na+/K+ 4,5. Los canales K+ sensibles al ATP (KATP), el regulador de la conductancia transmembrana de la fibrosis quística (CFTR) y los canales regulados por nucleótidos cíclicos son todos canales iónicos que están bloqueados por la unión de nucleótidos de adenina intracelular, lo que los hace exquisitamente sensibles a los cambios en el metabolismo celular y la transducción de señales 6,7,8. Los receptores purinérgicos P2X y P2Y responden a cambios en el ATP extracelular, que puede ser liberado como neurotransmisor o como resultado del daño tisular9. Hemos desarrollado un ensayo basado en FRET para la medición de la unión de nucleótidos de adenina a proteínas de membrana en tiempo real. Hemos aplicado previamente este método para estudiar la unión de nucleótidos a los canales KATP 10,11.
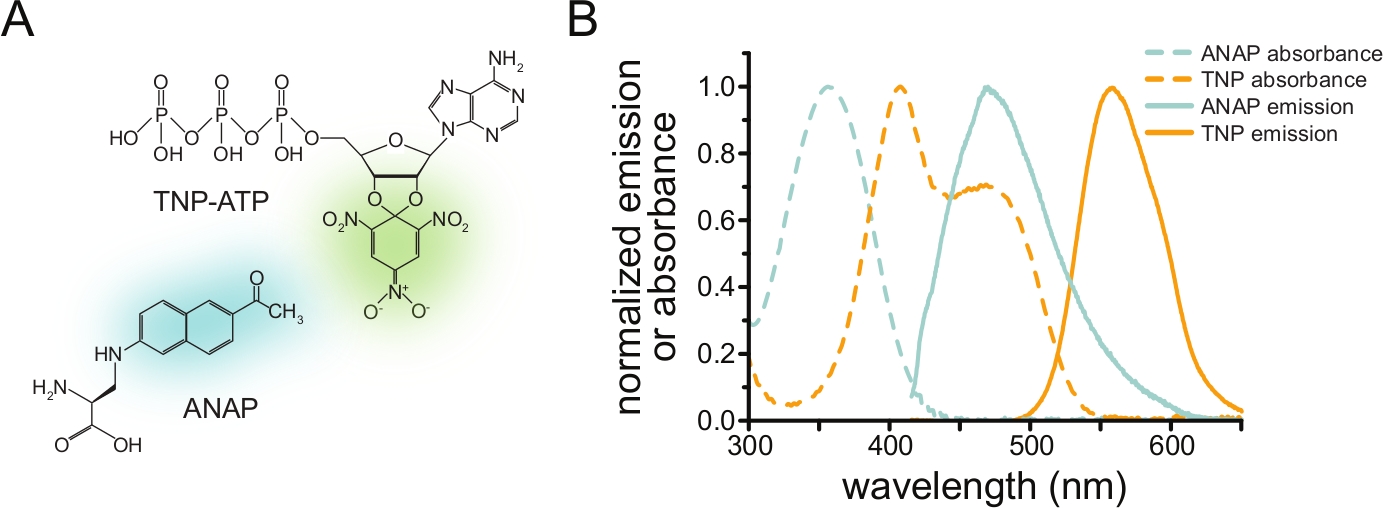
Para medir la unión de nucleótidos a través de FRET, una proteína de interés primero debe marcarse con un fluoróforo. La etiqueta fluorescente debe insertarse específicamente en la proteína de interés, de modo que esté lo suficientemente cerca del sitio de unión del ligando para que se produzca FRET, con especial cuidado para garantizar que la etiqueta no afecte la estructura y función general de la proteína. Para lograr esto, empleamos una técnica desarrollada por Chatterjee et al., utilizando supresión ámbar stop-codon para insertar un aminoácido fluorescente no canónico (l-3-(6-acetilnaftalen-2-ilamino)-2-aminopropiónico; ANAP) en el sitio deseado12. Medimos la unión a nucleótidos como FRET entre la proteína marcada con ANAP y los derivados de nucleótidos fluorescentes de trinitrofenilo (TNP) (Figura 1A). El espectro de emisión para ANAP se superpone con el espectro de absorbancia de los nucleótidos TNP, una condición necesaria para que ocurra FRET (Figura 1B). Aquí describimos dos tipos diferentes de experimento de enlace. En el primero, la unión de nucleótidos al lado intracelular de los canales KATP marcados con ANAP se mide en células que han sido descubiertas por sonicación dejando fragmentos adherentes de membrana plasmática en una cubierta de vidriodeslizamiento 10,11,13,14.
En el segundo método, la unión de nucleótidos a los canales KATP marcados con ANAP se mide en un parche de membrana bajo abrazadera de voltaje, lo que permite la medición simultánea de corrientes iónicas y fluorescencia. Al combinar estos dos enfoques experimentales, los cambios en la unión pueden correlacionarse directamente con los cambios en la función del canal11. Se discuten los resultados típicos, las posibles dificultades y el análisis de datos.
Protocolo
1. Preparación de los cuadernos de cobertura
NOTA: Estos pasos deben realizarse en una campana de cultivo de tejidos estéril. Se dan cantidades para la preparación de 10 platos.
- Coloque diez vidrios de cubierta de borosilicato de 30 mm en autoclave individualmente en diez platos estériles no tratados de 35 mm y enjuague una vez con 2 ml de agua destilada estéril.
- Diluir 1 ml de solución de poli-L-lisina al 0,1% p/v en agua destilada estéril hasta un volumen total de 10 ml (concentración final de 0,01% p/v). Mezclar bien, luego pipetear 1 ml en cada tapa e incubar a temperatura ambiente durante 20 min.
- Aspire la poli-L-lisina y lave cada cubierta dos veces con al menos 2 ml de agua destilada estéril. Dejar hasta que esté completamente seco, es decir, al menos 3 h.
2. Siembra de células HEK-293T
NOTA: Estos pasos deben realizarse en una campana de cultivo de tejidos. Las células HEK-293T fueron elegidas por su bajo fondo actual y facilidad de crecimiento en cultivo. Este protocolo puede adaptarse a otros tipos de células.
- Enjuagar un matraz T75 confluente al 80-90% de células HEK-293T una vez con solución salina tamponada con fosfato (PBS) de 12 ml antes de incubar con 2 ml de tripsina durante 2-5 min, o hasta que las células estén completamente separadas y casi completamente disociadas.
- Resuspender las células agregando 10 ml de Dulbecco's Modified Eagle Medium (DMEM) suplementado con 10% de suero bovino fetal, 100 U/ml de penicilina y 100 μg/ml de estreptomicina. Pipetear suavemente contra el fondo del matraz para romper los grupos restantes de células.
- Agregue 2 ml de DMEM suplementado al número deseado de platos de 35 mm que contengan cubreobjetos recubiertos. Agregue 100 μL de células resuspendidas a cada plato. Incubar durante la noche a 37 °C.
3. Transfección
NOTA: Estos pasos deben realizarse en una campana de cultivo de tejidos. Se dan cantidades para la transfección de 10 platos. Para la incorporación de ANAP específica del sitio, el codón de ADN en la posición prevista para el etiquetado debe reemplazarse con el codón de parada ámbar (TAG). Este constructo es co-transfectado con dos plásmidos: pANAP y peRF1-E55D12,15. pANAP codifica varias copias de un par de ARNt/ARNt sintetasa específico de ANAP. En presencia de ANAP, la transfección de este plásmido produce ARNt cargado con ANAP que reconoce el codón de parada ámbar. peRF1-E55D codifica un factor de liberación ribosómica negativo dominante que aumenta el rendimiento de la proteína marcada con ANAP de longitud completa.
- Prepare un tubo de 1,5 ml con 10 μg de pANAP, 10 μg de peRF1-E55D y ADN para la construcción destinada al etiquetado con ANAP. Llevar a un volumen final de 500 μL con DMEM no suplementado.
- En un tubo separado, prepare 3 μL de reactivo de transfección a base de lípidos (consulte la Tabla de materiales) por cada 1 μg de ADN y lleve a un volumen final de 500 μL con DMEM no suplementado.
- Combinar las mezclas de ADN y reactivos de transfección en un solo tubo e incubar durante 20 min a temperatura ambiente.
- Añadir 400 μL del stock ANAP de 1 mM (sal de trifluoroacetato en 30 mM de NaOH) a 20 ml de DMEM suplementado para una concentración final de 20 μM ANAP. Reemplace los medios viejos de las celdas plateadas con 2 ml de los medios que contienen ANAP por placa.
- Pipetear el 10% de la mezcla de transfección de ADN en cada plato. Incubar a 33 °C durante 2-4 días antes de los experimentos. La incubación a 33 °C ralentiza la división celular y aumenta el rendimiento proteico por célula16.
4. Experimentos con membranas sin techo
- Use un par de pinzas para romper un cubreobjetos con células transfectadas en fragmentos más pequeños.
- Siga uno de los procedimientos a continuación para destechar las celdas.
- Si usa cubreobjetos prerecubiertos, enjuague un fragmento con PBS y luego colóquelo en el fondo de un plato de 35 mm que contenga 2 ml de PBS. Sonicar brevemente utilizando un sonicador de sonda (50 W, 20% -40% de amplitud, sonda de 3 mm) colocado 3-5 mm por encima de la muestra para destechar las células y dejar fragmentos de membrana plasmática adherentes (Figura 2A, C).
NOTA: La potencia, la duración y la altura de la sonda del sintonizador sobre la muestra se pueden variar para obtener un alto rendimiento de membranas sin techo sin despojar completamente el cubreobjetos. - Si no utiliza hojas de cubierta precubiertas, enjuague un fragmento de cubierta con PBS, luego sumérjala en un tubo que contenga 0,1% p/v de poli-L-lisina durante ~30 s antes de sonicar brevemente (como en el paso 4.2.1) para destechar las celdas y dejar fragmentos de membrana plasmática sin techo/parcialmente sin techo (Figura 2A, C, D). Se ha demostrado que exposiciones breves a poli-L-lisina mejoran la adherencia al cubreobjetos13.
- Si usa cubreobjetos prerecubiertos, enjuague un fragmento con PBS y luego colóquelo en el fondo de un plato de 35 mm que contenga 2 ml de PBS. Sonicar brevemente utilizando un sonicador de sonda (50 W, 20% -40% de amplitud, sonda de 3 mm) colocado 3-5 mm por encima de la muestra para destechar las células y dejar fragmentos de membrana plasmática adherentes (Figura 2A, C).
- Coloque el fragmento sonicado en una cubierta con fondo de vidrio de 35 mm que contenga 2 ml de solución de baño y móntelo en un microscopio invertido equipado con un objetivo de inmersión en agua de 60x de alta NA. El puerto de la cámara del microscopio está conectado a un espectrógrafo en serie con una cámara CCD de alta sensibilidad. Perfundir la cámara de baño (0,5 – 1 ml / min) con tampón utilizando una bomba peristáltica. La composición del tampón variará dependiendo de la proteína en estudio.
NOTA: Si el usuario no tiene acceso a un objetivo con una larga distancia de trabajo, puede ser imposible enfocar los fragmentos de membrana sin techo debido a la altura adicional del deslizamiento de la cubierta. Una alternativa es sembrar células directamente en platos con fondos de vidrio de poli-L-lisina (ver Tabla de materiales para un ejemplo). Esto también reducirá las posibles aberraciones en la imagen asociadas con el enfoque a través de dos piezas de vidrio. Estas aberraciones no afectan a la forma de los espectros adquiridos. - Identifique fragmentos de membrana sin techo que expresan el canal marcado con ANAP buscando fluorescencia de canal (Figura 2C, D).
NOTA: Se recomienda utilizar una etiqueta fluorescente adicional (donde el espectro de emisión se distingue del espectro de emisión ANAP) para ayudar a identificar las membranas sin techo que contienen la proteína de interés. Los experimentos en la Figura 2C,D se realizaron en canales marcados con ANAP con etiquetas de proteínas fluorescentes C-terminales. - Acople parcialmente la máscara del espectrómetro (eleve ~ 10%) entre el puerto de la cámara en el microscopio y el espectrógrafo. La sombra de la máscara aparecerá en la imagen de la cámara. Alinee la membrana sin techo con la máscara del espectrómetro, ajustando la etapa del microscopio. Adquiera una imagen de campo brillante y fluorescencia de la membrana sin techo. Estos se utilizarán para seleccionar una región de interés para el análisis.
- Acerque la punta del sistema de perfusión de microvolúmenes a la membrana sin techo.
NOTA: Para reducir la fluorescencia de fondo, el flujo de salida del sistema de perfusión se reemplazó con una punta personalizada hecha de vidrio de borosilicato. - Para obtener imágenes de espectros de fluorescencia, excite la membrana con un LED de 385 nm a través de un filtro de excitación de paso de banda de 390/18 nm y un dicroico de borde de 416 nm. Recoger la luz emitida a través de un filtro de emisión de paso largo de 400 nm (Figura 2B).
- Acople la máscara del espectrómetro y asegúrese de que la luz emitida pase a través de ella. Acople las rejillas del espectrómetro (300 ranuras/mm). Con las rejillas en su lugar, la luz difractada por el espectrómetro se proyectará en el chip de la cámara CCD para producir imágenes espectrales (Figura 3A). Estas imágenes retienen información espacial en la dimensión y . La dimensión x se sustituye por longitud de onda.
- Opcionalmente, si la proteína de interés se marca con una proteína fluorescente, adquiera una imagen espectral de la proteína fluorescente utilizando el conjunto de filtros apropiado.
- Tome una o más exposiciones de 0.1-10 s al comienzo del experimento mientras perfunde una solución tampón libre de nucleótidos. Estos se utilizarán para corregir y normalizar los datos durante el resto del experimento (consulte la sección 5 a continuación).
NOTA: La elección del tiempo de exposición dependerá del nivel de expresión alcanzado, el brillo del fluoróforo y la óptica. El tiempo de exposición debe elegirse para maximizar la señal y minimizar la tasa de blanqueamiento observada. El intervalo de tiempo de exposición indicado en 4.10 es adecuado para mediciones de unión de equilibrio, pero puede ser útil para medir cambios cinéticos más lentos10. La capacidad de utilizar tiempos de exposición cortos para rastrear cinéticas más rápidas estará limitada por los niveles de expresión de proteínas y el fotoblanqueo, en lugar de hardware. - Aplicar un rango de concentraciones de TNP-ATP (generalmente preparado en solución de baño) para establecer una curva de concentración-respuesta. Perfundir cada solución durante al menos 1 minuto para asegurarse de que se alcanza un estado estacionario y lavar cada concentración con solución de baño durante al menos 1 minuto.
NOTA: Es importante asegurarse de que el sistema de perfusión pueda alcanzar rápidamente el equilibrio (Figura 2E) y alcanzar la concentración local correcta de TNP-ATP (Figura 2F). - Tomar una exposición (con la misma duración que se utiliza en el paso 4.10) en cada concentración y al final de cada lavado.
5. Análisis espectral
NOTA: Estas instrucciones están escritas para su uso con el código de análisis "pcf.m", que se puede encontrar en GitHub. https://github.com/mpuljung/spectra-analysis10. El código adicional y alternativo se puede encontrar en https://github.com/smusher/KATP_paper_201911. Hemos descrito las operaciones realizadas por el software aquí para que el usuario pueda crear su propio código o elegir analizar los datos manualmente.
- Inicie el programa de análisis escribiendo el nombre del programa ("pcf") en la línea de comandos.
- Cuando se abra un cuadro de diálogo de archivo/carpeta abierto con el mensaje: "Seleccionar archivos para ROI", seleccione los nombres de archivo asociados con las imágenes de campo claro y fluorescencia de la membrana sin techo. Aparecerá un mensaje en la línea de comandos para escribir el nombre del archivo de salida.
- Escriba el nombre del archivo y presione enter.
- Cuando el software muestre las imágenes de campo claro y fluorescencia, seleccione una región de interés (ROI) en la imagen espectral correspondiente a la ubicación del fragmento de membrana sin techo o del parche extirpado (consulte la sección 6) siguiendo las indicaciones del software. Seleccione una región de fondo en la misma imagen espectral (que representa el mismo rango de longitud de onda que en el ROI) correspondiente a una sección de cubierta o plato sin membrana unida (Figura 3A). El software le pedirá que haga clic en la parte superior del ROI y presione Entrar, haga clic en la parte inferior del ROI y presione Enter y luego repita este proceso para la región en segundo plano.
- Cuando se abra un cuadro de diálogo de abrir archivo/carpeta con el mensaje: "Seleccionar archivo para espectro FP", seleccione el nombre de archivo asociado con el espectro de proteínas fluorescentes (FP) (paso opcional 4.9). Si no se adquirió espectro FP, seleccione un archivo de espectro diferente. El espectro FP sirve como control de calidad para distinguir entre la proteína marcada y la fluorescencia de fondo.
- Cuando se abra un cuadro de diálogo de archivo/carpeta abierta con el mensaje: "Seleccionar archivos para Analyisis", seleccione todos los archivos correspondientes a los espectros ANAP (de los pasos 4.10 a 4.12), incluidos los archivos necesarios para la corrección de lejía.
- Cuando se abra un cuadro de diálogo de archivo/carpeta abierto con el mensaje: "Seleccionar archivos para la recolección de blanqueo", seleccione el subconjunto de archivos del paso 5.6 correspondiente a los espectros iniciales adquiridos en solución libre de nucleótidos al comienzo del experimento o espectros adquiridos durante los lavados en solución libre de nucleótidos que se utilizarán para la corrección (de los pasos 4.10 a 4.12).
- Promedie la línea de cada imagen para producir espectros, es decir, promedie la intensidad de todos los píxeles en la dimensión y de un ROI o región de fondo en cada longitud de onda. (Figura 3B). Reste el espectro de fondo promediado resultante del espectro promediado adquirido del ROI para eliminar la fluorescencia de fondo y la fluorescencia del TNP-ATP no unido (Figura 3C). Estos pasos son realizados automáticamente por el software.
- Determine la intensidad del ANAP para cada exposición promediando la intensidad de una ventana de 5 nm centrada alrededor del pico ANAP de los espectros sustraídos (típicamente ~ 470 nm, pero puede variar dependiendo del microambiente local del residuo ANAP).
NOTA: La Figura 3D muestra 6 espectros obtenidos de exposiciones consecutivas de 10 s de un fragmento de membrana sin techo que expresa canales marcados con ANAP. El recuadro muestra la intensidad promediada del pico de cada espectro. El software encontrará automáticamente la longitud de onda máxima en el primer espectro adquirido y utilizará este valor en todo momento. La intensidad será calculada automáticamente por el software. - Normalice las intensidades ANAP para cada experimento dividiendo la intensidad ANAP de una exposición dada (F) por la intensidad ANAP de la primera exposición en la serie temporal, que se tomó en el paso 4.10 (Fmax). Una vez más, el software realiza estos cálculos automáticamente.
- Realice los pasos siguientes para obtener datos.
- Para corregir el fotoblanqueo ANAP, primero ajuste una sola disminución exponencial, (F/Fmax) = A*exp(-t/τ)+(1-A), donde t es el tiempo de exposición acumulado, τ es la constante de tiempo y A es la amplitud) a los pasos de lavado intermedios entre aplicaciones TNP-ATP o a múltiples exposiciones iniciales tomadas antes del lavado en TNP-ATP (Figura 3D, recuadro).
NOTA: El software mostrará este ajuste y se le pedirá que lo acepte o lo rechace. Si se rechaza el ajuste, se proporcionará otra oportunidad para seleccionar archivos para la corrección del blanqueamiento. - Divida los espectros ANAP normalizados (en el paso 5.10) por el valor previsto del ajuste exponencial del paso 5.11.1 en cada punto de tiempo (Figura 3E).
NOTA: Para el ejemplo mostrado, la fluorescencia máxima normalizada observada a 50 s es 0.65 y la fluorescencia predicha del ajuste exponencial es 0.64. Para corregir el blanqueo, divida el valor observado (0.65, recuadro de la Figura 3E, círculo vacío) por el valor predicho (0.64, recuadro de la Figura 3E, línea discontinua) para producir el valor corregido (~1, recuadro de la Figura 3E, círculo coloreado). Si la corrección del blanqueamiento es adecuada, la intensidad del ANAP de todas las exposiciones adquiridas en ausencia de nucleótidos debe ser aproximadamente igual (Figura 3E). Estos cálculos son realizados automáticamente por el software. - Obtenga la salida como una imagen que traza los datos y una hoja de cálculo con pestañas que contiene los espectros sin procesar, los espectros restados, los espectros corregidos para el fotoblanqueo y los datos máximos para cada archivo para que se pueda realizar un análisis adicional.
- Para corregir el fotoblanqueo ANAP, primero ajuste una sola disminución exponencial, (F/Fmax) = A*exp(-t/τ)+(1-A), donde t es el tiempo de exposición acumulado, τ es la constante de tiempo y A es la amplitud) a los pasos de lavado intermedios entre aplicaciones TNP-ATP o a múltiples exposiciones iniciales tomadas antes del lavado en TNP-ATP (Figura 3D, recuadro).
6. Experimentos de fluorometría de patch-clamp
- Extraiga las pipetas del parche de los capilares de vidrio de borosilicato de paredes gruesas a una resistencia de 1,5 MΩ a 2,5 MΩ cuando se llenen con solución de pipeta. La composición de la solución de pipeta variará dependiendo de la proteína en estudio.
- Transfiera un cubreobjetos con células transfectadas a un plato de 35 mm con fondo de vidrio que contenga 2 ml de solución de baño y móntelo en un microscopio invertido equipado con un objetivo de inmersión en agua de 60x y alta NA. Perfundir la cámara de baño (0,5 – 1 ml / min) con solución de baño utilizando una bomba peristáltica. En cuanto a la solución de pipeta, la solución de baño variará dependiendo de la proteína en estudio.
- Identificar una célula que expresa canales marcados con ANAP buscando fluorescencia en la membrana celular.
- Llene una pipeta de parche con solución de pipeta. Aplique una presión positiva suave a la pipeta y colóquela en la cámara de baño. Presione la pipeta contra la membrana de la célula y aplique una succión suave para lograr un sello GΩ (Figura 4A).
- Extirpe el parche alejando rápidamente el soporte de la pipeta de la celda (figura 4A).
NOTA: La extirpación del parche de esta manera debe formar un parche de adentro hacia afuera, con los dominios citosólicos de la proteína expuestos al sistema de perfusión. Si la ubicación del sitio de unión al nucleótido en estudio no es citosólica, será necesario utilizar parches de afuera hacia afuera o registros de células enteras para realizar experimentos de PCF. - Acerque la punta de la pipeta del parche a la punta del sistema de perfusión y compruebe que el parche está dentro de la hendidura de la máscara del espectrómetro (figura 4A).
- Aplique TNP-ATP y espectros de imagen como en los pasos 4.10-4.12, mientras registra simultáneamente la respuesta de la corriente iónica a la aplicación de nucleótidos.
NOTA: El vidrio pipeta puede introducir aberraciones espaciales y reflejos en las imágenes adquiridas. Sin embargo, estas aberraciones no afectarán la forma de los espectros adquiridos y la luz de excitación reflejada se separa fácilmente de la fluorescencia utilizando el espectrógrafo o un filtro de emisión de paso largo. - Analiza los espectros. Los espectros obtenidos de parches extirpados pueden exhibir una sustracción excesiva de fluorescencia de TNP-ATP no unida debido a la exclusión de TNP-ATP del vidrio de la pipeta del parche (Figura 4C-E). Esta resta excesiva no afecta el espectro de emisión ANAP y, por lo tanto, puede ignorarse.
NOTA: Como la señal de fluorescencia en los parches extirpados será menor que en las membranas sin techo, es importante utilizar un tiempo de exposición que proporcione una relación señal-ruido lo suficientemente alta sin blanquear ANAP demasiado rápido.
Resultados
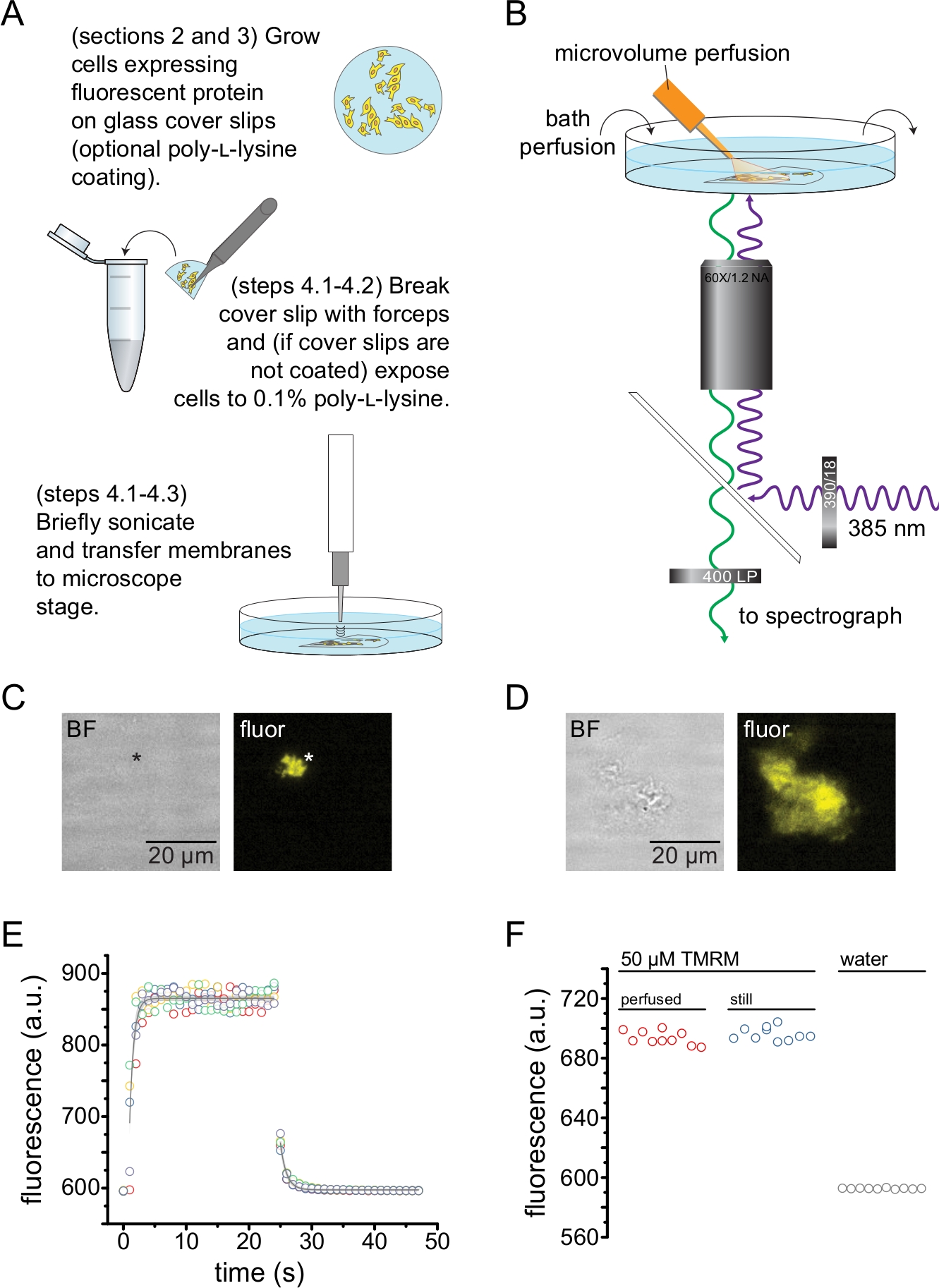
La Figura 2 muestra la configuración experimental básica para medir la unión de nucleótidos a proteínas fluorescentes en fragmentos de membrana sin techo obtenidos por sonicación (Figura 2A, B). Se utilizaron dos enfoques diferentes para obtener membranas sin techo, cultivando directamente células en cubrevestimientos recubiertos de poli-L-lisina o cultivando células en vidrio no tratado y exponiéndolas brevemente a poli-L-lisina (0,1% en agua) antes de destecharlas. La Figura 2C muestra un fragmento típico de membrana sin techo de una célula HEK-293T que expresa canales KATP marcados con proteína fluorescente naranja (OFP). Las membranas sin techo eran prácticamente invisibles en las imágenes de campo claro y se identificaron por la fluorescencia de proteínas de membrana marcadas o por contratinción con un colorante de membrana como octadecil rodamina B13. Además de las membranas sin techo, la sonicación de las células HEK-293T también produjo fragmentos celulares parcialmente sin techo (Figura 2D)10,17. Estos fragmentos eran visibles en campo brillante. Esto podría ser el resultado de membranas plasmáticas erizadas que solo se adhieren mal al vidrio de la cubierta. Alternativamente, estos fragmentos pueden contener vesículas y membranas de orgánulos intracelulares. Como tal, es preferible adquirir imágenes solo de membranas "verdaderas" sin techo, ya que la proteína diana marcada asociada con las membranas intracelulares puede reflejar etapas intermedias de procesamiento y ensamblaje postraduccional. Se recomienda cultivar células en vidrio recubierto de poli-L-lisina, ya que esto resultó en un mayor rendimiento de membranas "verdaderas" sin techo tras la sonicación.
Se aplicó un sistema de perfusión de microvolúmenes a nucleótidos fluorescentes para minimizar las cantidades necesarias en un experimento típico (Figura 2B). La punta de vidrio recubierta de poliimida proporcionada se reemplazó con una punta de vidrio de borosilicato tirada a mano en nuestra configuración de perfusión, lo que redujo el fondo de fluorescencia. Para minimizar la acumulación de nucleótidos alrededor de las membranas sin techo que se estaban fotografiando, toda la cámara de baño se perfundió lentamente con tampón. Como tal, queríamos medir la tasa de cambio de solución de nuestro sistema de perfusión de microvolumen y verificar que pudimos lograr la concentración de ligando prevista en nuestra región de interés, es decir, que el ligando de nuestro sistema de perfusión no se diluyó directamente en el medio de baño antes de alcanzar la membrana sin techo. Para controlar estas posibilidades, se midió el lavado y lavado de una solución de 50 μM de tetrametilrodamina-5-maleimida (TMRM) de nuestro sistema de perfusión de microvolumen dirigido a la superficie de un plato cubierto con fondo de vidrio perfundido con agua (Figura 2E). La cinética de intercambio de soluciones fue reproducible y bien descrita por una sola disminución exponencial con constantes de tiempo inferiores a 1 s tanto para el lavado como para el lavado. Tales tiempos de intercambio de soluciones limitan nuestra capacidad para medir la cinética de unión y desunión de ligandos en nuestra configuración actual. Para verificar que pudimos alcanzar la concentración de ligando deseada en la superficie del cubreobjetos, comparamos la intensidad de fluorescencia de 50 μM TMRM entregada al cubreobjetos por nuestro sistema de perfusión de microvolumen con 50 μM TMRM en un baño inmóvil (Figura 2F). No se observó diferencia de intensidad, verificando que se pueden lograr concentraciones de ligando adecuadas en la superficie del cubreobjetos con nuestro sistema de perfusión de microvolumen, incluso cuando el baño está perfundido.
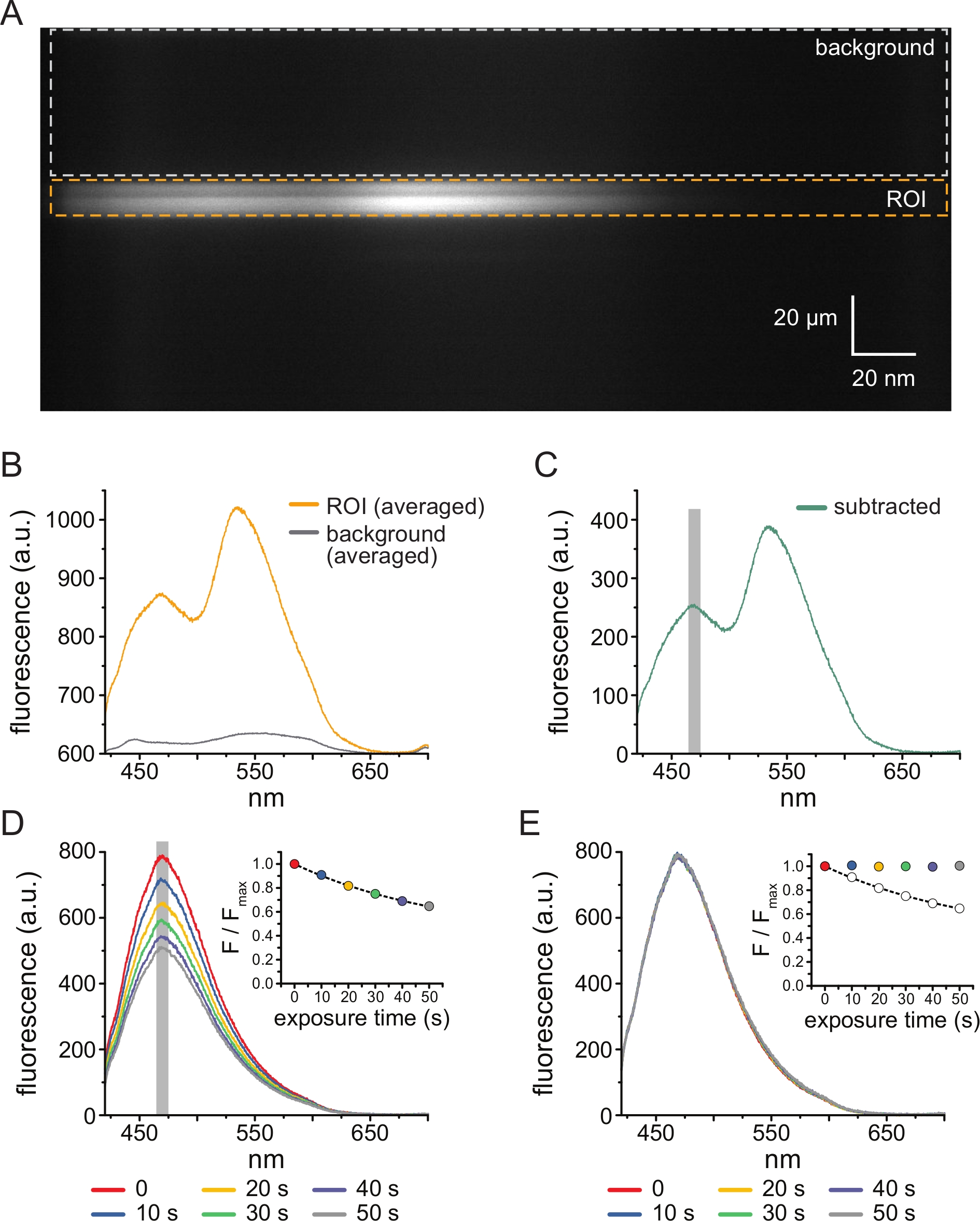
La Figura 3A muestra una imagen espectral obtenida de canales KATP marcados con ANAP en una membrana sin techo de una célula HEK-239T expuesta a 5 μM TNP-ATP. Para obtener este tipo de imágenes, la luz emitida desde la membrana sin techo se dirigía a través de un espectrómetro en serie con una cámara CCD. La fluorescencia emitida se difractó de las rejillas y se proyectó en el chip de la cámara, produciendo espectros. Las imágenes resultantes retienen información espacial en la dimensión y , pero la dimensión x fue reemplazada por longitud de onda. La región de interés (ROI), correspondiente a la membrana sin techo, está delineada en naranja. Dos regiones de alta intensidad son evidentes en la imagen, correspondientes a la emisión máxima de ANAP y TNP-ATP. Esto se apreció mejor en el espectro promediado longitud de onda por longitud de onda (sobre todo el ROI) que se muestra en la Figura 3B. El pico ~470 nm corresponde a ANAP incorporado en KATP; el pico ~535 nm corresponde a TNP-ATP. Para corregir la fluorescencia de fondo y la excitación directa de TNP-ATP en solución, se seleccionó una región de fondo (Figura 3A, gris) de cada imagen. El espectro de fondo promediado se muestra en la Figura 3B. El espectro final se obtuvo restando el espectro de fondo promediado del espectro ROI promediado (Figura 3C).
ANAP es propenso a los artefactos de fotoblanqueo. La Figura 3D muestra la reducción en la fluorescencia máxima de ANAP después de múltiples exposiciones. La fluorescencia máxima de varias exposiciones en ausencia de TNP-ATP (o de lavados entre concentraciones de TNP-ATP) se ajustó a una desintegración exponencial única y esto se utilizó para corregir los artefactos de fotoblanqueo (Figura 3E). Se recomienda realizar experimentos de concentración-respuesta de concentraciones de nucleótidos de baja a alta y de alta a baja. Si la corrección del blanqueamiento no introduce ningún artefacto adicional, los resultados deben ser comparables11.
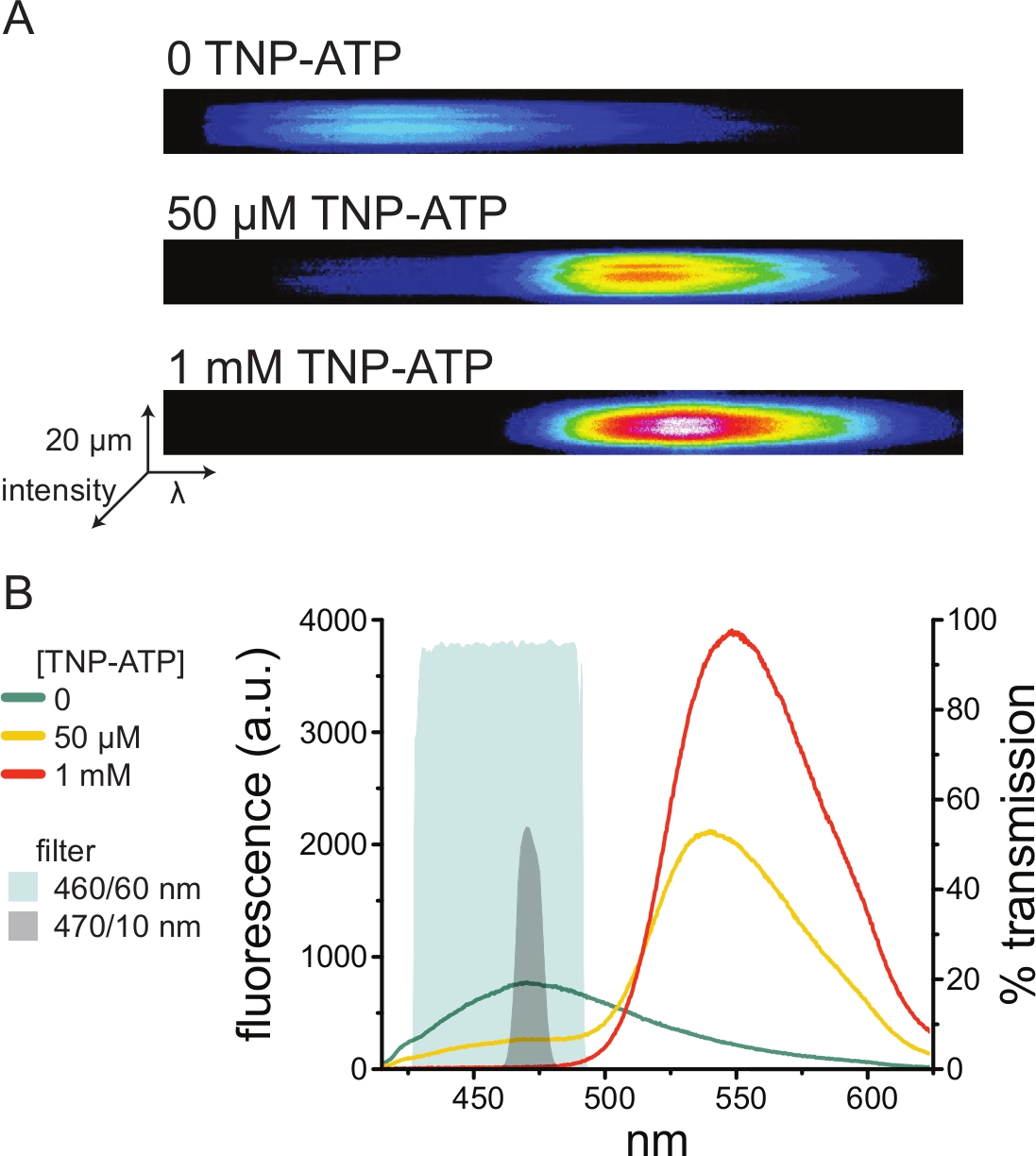
La Figura 5A muestra imágenes espectrales representativas de una membrana sin techo obtenidas de una célula que expresa canales KATP marcados con ANAP en ausencia y presencia de TNP-ATP. Los espectros corregidos se muestran en la Figura 5B. Al observar los espectros de emisión, hubo una clara separación entre la emisión de fluorescencia donante y aceptora. Como se observó cierta unión no específica de TNP-ATP a membranas plasmáticas naïve de células HEK-293T no transfectadas, se recomienda cuantificar FRET como una reducción en la fluorescencia del donante (ANAP)10,11. Este pico fue específico para el receptor marcado.
Para los ligandos que inducen un cambio conformacional en su receptor, los estudios de unión aislados no proporcionan información directa y mecánicamente significativa sobre el proceso de unión del ligando18. La relación concentración-respuesta para la unión al ligando depende no sólo de la afinidad de unión intrínseca, sino también del cambio conformacional inducido por la unión del ligando, y la propensión inherente del receptor a cambiar la conformación en ausencia de ligando. Para comprender mejor los procesos que subrayan las interacciones ligando-receptor, las mediciones de unión se pueden combinar con experimentos que proporcionen una lectura de la función de la proteína. Con este fin, los canales iónicos son un sistema modelo ideal, ya que sus corrientes se pueden medir con una resolución de tiempo inferior a ms hasta el nivel de una sola molécula utilizando una abrazadera de voltaje. Históricamente, las mediciones de corriente y fluorescencia pareadas han proporcionado información significativa sobre la apertura y cierre (gating) de los canales iónicos dependientes de voltaje y ligando 19,20,21. Se han realizado experimentos para medir simultáneamente las corrientes iónicas y la unión de nucleótidos cíclicos fluorescentes a varios canales regulados por nucleótidos cíclicos22,23,24. Estos estudios emplearon un ligando que aumentó su rendimiento cuántico al unirse. La fluorescencia del ligando no unido en el volumen de solución cerca del parche se puede restar mediante imágenes de los parches utilizando microscopía confocal22,23. En nuestros estudios, la unión se midió utilizando la reducción de la fluorescencia ANAP. Como esta señal es específica del canal y FRET entre ANAP y TNP-ATP depende fuertemente de la distancia (la mitad máxima a ~ 43 Å), se evitó la contaminación de nuestra señal por nucleótidos no unidos y no unidos específicamente.
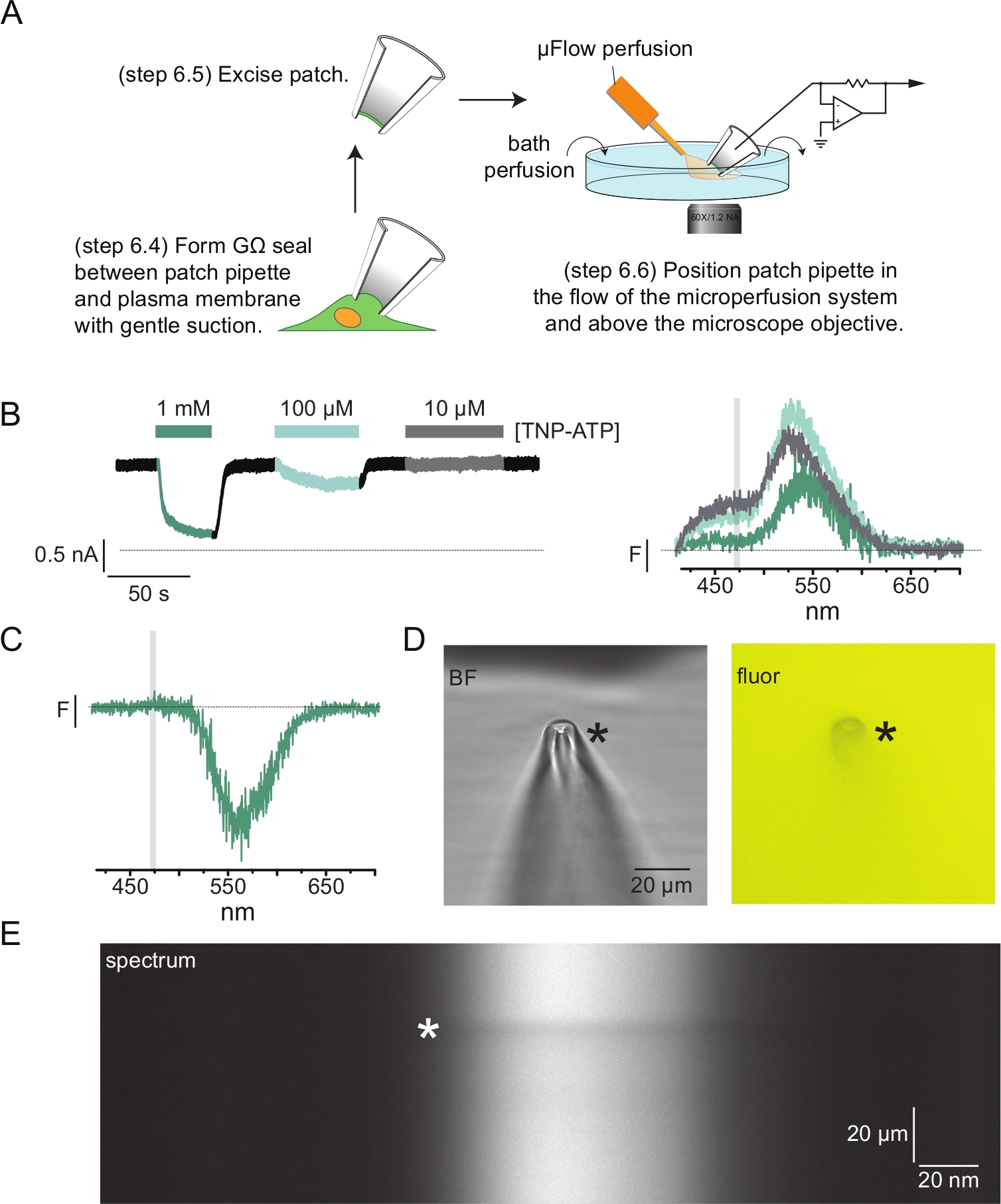
La figura 4A muestra un experimento típico de fluorometría de pinza de parche (PCF). Se formó un sello de alta resistencia (GΩ) entre una pipeta de vidrio de borosilicato rellena de solución salina (conectada a un amplificador de pinza de voltaje) y una celda que expresaK ATP marcado con ANAP. Después de la formación del sello, la pipeta se separó de la célula, permitiendo el acceso a los sitios de unión de nucleótidos intracelulares. A continuación, la pipeta se colocó sobre el objetivo del microscopio, centrado en la hendidura de la máscara del espectrómetro y el flujo de salida del sistema de perfusión de microvolumen (modificado con una punta de vidrio de borosilicato) se acercó a la pipeta (Figura 4D). Se controló el voltaje y se midieron las corrientes de los canales en el parche. Las corrientes y espectros representativos de los canales KATP marcados con ANAP se muestran en la Figura 4B, codificados por colores para que coincidan con los espectros con las corrientes. Los espectros de emisión se corrigieron para el fondo y el blanqueamiento como para las membranas sin techo.

Figura 1: ANAP y TNP-ATP forman un par FRET adecuado. a) Estructuras de ANAP y TNP-ATP. Se resaltan las fracciones fluorescentes. (B) Espectros de absorbancia y emisión de fluorescencia de ANAP y TNP-ATP. Se requiere una superposición entre la emisión ANAP y la absorbancia TNP-ATP para FRET. Adaptado de Puljung et al. (publicado bajo la Creative Commons Attribution License, https://creativecommons.org/licenses/by/4.0/)10. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2: Medición de la unión de nucleótidos en membranas plasmáticas sin techo. (A) Esquema para la preparación de membranas plasmáticas sin techo a partir de células adherentes que expresan una proteína de membrana fluorescente. Se proporcionan instrucciones para las células cultivadas en hojas de cobertura recubiertas de poli-L-lisina o no tratadas. (B) Configuración experimental para medir la unión de nucleótidos en membranas sin techo. (C) Imágenes fluorescentes y de campo brillante de una membrana plasmática completamente descubierta derivada de una célula que expresa la proteína fluorescente naranja (OFP) marcada con canales KATP. El asterisco marca la posición de la membrana, que es casi invisible en la imagen de campo claro. OFP se excitó con un amplio LED de 565 nm a través de un filtro de paso de banda de 531/40 nm y una luz dicroica de borde de 562 nm y se recogió a través de un filtro de paso de banda de 593/40 nm. (D) Imágenes fluorescentes y de campo brillante de un fragmento de membrana parcialmente sin techo derivado de una célula que expresa la proteína fluorescente naranja (OFP) marcada con canales KATP. (E) Curso de tiempo de intercambio de soluciones adquirido utilizando la configuración descrita en B. Se muestran cinco réplicas técnicas. El sistema de perfusión de microvolumen se cargó con tetrametilrodamina-5-maleimida (TMRM) de 50 μM. El baño se perfundió con agua a una velocidad de ~ 0.5 ml / min. Los datos de los cursos de tiempo de lavado (fluorescencia creciente) y lavado (fluorescencia decreciente) se ajustaron con una sola disminución exponencial de la forma F = A * exp (-x / τ) + y0. La constante de tiempo (τ) para el lavado fue ~0.6 s. La constante de tiempo para el lavado fue ~ 1.0 s. TMRM se excitó con un amplio LED de 565 nm a través de un filtro de paso de banda de 540/25 nm y un borde dicroico de 565 nm y la luz emitida se recogió a través de un filtro de paso de banda de 605/55 nm. (F) Comparación de la intensidad de fluorescencia de una solución de 50 μM de TMRM aplicada utilizando el sistema de perfusión de microvolumen como en B y un baño inmóvil que contiene 50 μM TMRM. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3: Resta de fondo y corrección de blanqueo. (A) Imagen espectral (información espacial en la dimensión y, longitud de onda en la dimensión x) de una membrana plasmática sin techo de una célula que expresa canales KATP marcados con ANAP. Se aplicó tnp-ATP de 5 μM utilizando la configuración descrita en la Figura 2B. La caja naranja denota la región de interés (ROI), correspondiente a la membrana sin techo. El cuadro gris denota la región de fondo utilizada para corregir el espectro. (B) Espectros de emisión derivados de promedios longitud de onda por longitud de onda de las regiones de ROI y fondo en A. (C) Espectro obtenido restando el espectro de fondo promediado del espectro de ROI promediado en B. La ventana de 5 nm alrededor del pico ANAP utilizada para determinar la intensidad promedio se muestra como un área sombreada gris. (D) Espectros adquiridos a partir de seis exposiciones consecutivas 10-s de una membrana plasmática sin techo de una célula que expresa canales KATP marcados con ANAP. Tenga en cuenta la disminución de la fluorescencia resultante del fotoblanqueo. El recuadro muestra el ajuste de fluorescencia máxima normalizada con un solo decaimiento exponencial de la forma F/Fmax = A*exp(-t/τ) + (1-A). Los símbolos en el recuadro están codificados por colores para que coincidan con los espectros. (E) Los mismos espectros que en D corregidos para el fotoblanqueo. El recuadro muestra la fluorescencia máxima normalizada de D como círculos abiertos, con la fluorescencia máxima corregida mostrada usando círculos rellenos. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 4: Mediciones simultáneas de unión de nucleótidos y corrientes de canal utilizando fluorometría de patch-clamp (PCF). (A) Esquema que muestra la configuración experimental para medir la unión de nucleótidos y las corrientes iónicas. (B) Ejemplo de corrientes (izquierda) y espectros (derecha) adquiridos de un parche de membrana extirpado de una célula que expresa canales KATP marcados con ANAP. Las corrientes se registraron a un potencial de retención de -60 mV, se digitalizaron a 20 kHz y se filtraron a 5 kHz. El área sombreada en gris corresponde al rango de longitud de onda a partir del cual se cuantificó la intensidad del ANAP. Adaptado de Usher et al. (publicado bajo la Creative Commons Attribution License, https://creativecommons.org/licenses/by/4.0/)11. (C) Espectro adquirido de un parche de membrana extirpado de una célula que expresa canales KATP marcados con ANAP expuestos a 1 mM TNP-ATP. Tenga en cuenta el pico negativo correspondiente al rango de longitud de onda sobre el cual se observa la fluorescencia TNP-ATP. El área sombreada gris denota el rango de longitud de onda utilizado para cuantificar la fluorescencia ANAP como en B. Adaptado de Usher et al. (publicado bajo la Creative Commons Attribution License, https://creativecommons.org/licenses/by/4.0/)11. (D) Imágenes fluorescentes y de campo claro de una pipeta de parche expuesta a 1 mM TNP-ATP. El asterisco marca la punta de la pipeta. (E) Imagen espectral de la misma pipeta de parche en 1 mM TNP-ATP. El asterisco marca la posición de la pipeta. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 5: Unión TNP-ATP a canales KATP marcados con ANAP. (A) Imágenes espectrales de una membrana plasmática sin techo de una célula que expresa canales K ATP marcados con ANAP en ausencia de TNP-ATP o en presencia de 50 μM o 1 mM TNP-ATP. Las intensidades se muestran como un mapa de calor. (B) Espectros promediados longitud de onda por longitud de onda de las imágenes en A que muestran la extinción de la fluorescencia ANAP por TNP-ATP. Las áreas sombreadas representan dos filtros de paso de banda diferentes que se pueden usar para medir el enfriamiento ANAP si no se dispone de un espectrómetro. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

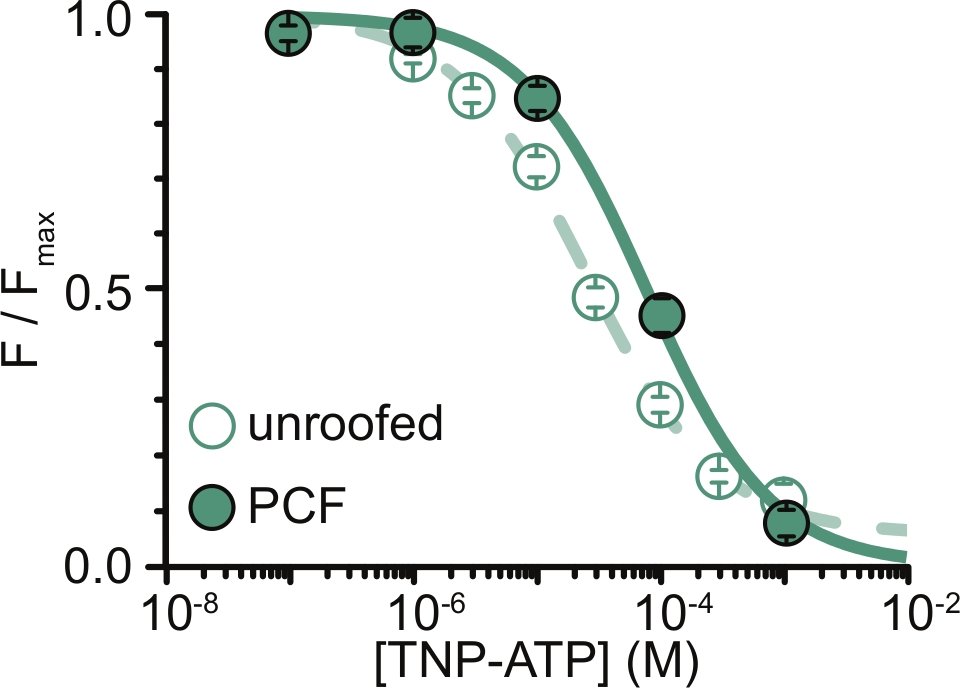
Figura 6: Enfriamiento de canales KATP marcados con ANAP por TNP-ATP en membranas sin techo y PCF. Superposición de datos de Usher et al. (publicados bajo la Creative Commons Attribution License, https://creativecommons.org/licenses/by/4.0/)11. Los datos se ajustaron a la ecuación de Hill: F / F max = E max + (1 – E max) / (1+10(EC50 –[TNP-ATP])*h). F es la fluorescencia medida, F max es la fluorescencia máxima en ausencia de nucleótido, Emax es el enfriamiento máximo a concentraciones de nucleótidos saturantes y h es la pendiente de Hill. EC50, (la concentración de nucleótidos a la que el enfriamiento es la mitad máxima) y [TNP-ATP] son valores logarítmicos. Membranas sin techo: EC50 = -4.59 (25.7 μM), h = 0.82, Emax = 0.93. PCF: EC50 = -4.11 (77.6 μM), h = 0.87, Emax = 1.00. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Discusión
Hemos desarrollado un método para medir la unión de nucleótidos de adenina en tiempo real a proteínas de membrana intactas. Nuestro método se basa en varias otras técnicas establecidas, incluido el etiquetado de proteínas con ANAP utilizando supresión de codón de parada ámbar 12, destechamiento celular 14 y fluorometría de pinza de voltaje / PCF 19,20,21,22,23,24,25 . La síntesis de estos enfoques permite la medición de la unión de nucleótidos con alta resolución espacial y temporal. De hecho, en nuestro trabajo anterior, pudimos distinguir entre diferentes sitios de unión en el mismo complejo proteico utilizando este enfoque10,11. Es importante destacar que esta técnica se puede aplicar directamente a pequeñas cantidades de proteína en un entorno celular en condiciones que preservan la función de la proteína. El uso de nuestro método de unión junto con la lectura electrofisiológica directa de las corrientes del canal iónico nos permite obtener información valiosa sobre los fundamentos moleculares de la activación del canal11.
Como los espectrómetros son una pieza no estándar del equipo de laboratorio, la intensidad de ANAP también se puede monitorear en aislamiento relativo utilizando filtros de paso de banda. La figura 5B muestra las propiedades espectrales de dos de estos filtros. El filtro de paso de banda de 470/10 nm filtra eficazmente la señal de fluorescencia de TNP-ATP y se superpone bien con la fluorescencia máxima ANAP. Sin embargo, la transmitancia máxima de este filtro es solo de alrededor del 50%, lo que puede dificultar la obtención de buenas señales de membranas tenues (o en parches de membrana extirpados bajo abrazadera de voltaje). Otra opción es un filtro de paso de banda de 460/60 nm. Hay un poco más de superposición entre el filtro de 460/60 nm y el pie del pico de emisión TNP-ATP en comparación con el filtro de 470/10 nm. Sin embargo, el paso de banda de 460/60 nm tiene una transmitancia del 90-95% en un amplio rango del pico ANAP, lo que se espera que aumente la señal de emisión de fluorescencia.
ANAP es un fluoróforo ambientalmente sensible 12,26,27. La emisión máxima y el rendimiento cuántico varían dependiendo del sitio de incorporación en la proteína de interés y pueden cambiar a medida que la proteína cambia de conformación. Tales cambios serían inmediatamente evidentes a partir de los espectros de emisión, pero no serían tan obvios cuando la intensidad del ANAP se mide utilizando filtros. En cualquier caso, se requieren controles apropiados para demostrar que la señal de fluorescencia no varía debido a cambios en el entorno local alrededor de ANAP posteriores a la unión de nucleótidos. Los experimentos de control con nucleótidos no marcados pueden ayudar a verificar que cualquier cambio en la intensidad de ANAP es el resultado de FRET entre ANAP y nucleótidos TNP. Los nucleótidos TNP pueden unirse no específicamente a las membranas derivadas de células no transfectadas (ya sea a la membrana plasmática o a proteínas de membrana nativas)10. Cuantificamos la unión como una disminución en la fluorescencia del donante, ya que esta señal es específica del canal marcado. Sin embargo, recomendamos realizar experimentos de control adicionales para cada par agonista/receptor, por ejemplo, mutar el sitio de unión de nucleótidos si se conoce, para verificar que el cambio en la fluorescencia del donante es realmente el resultado de la unión directa al receptor marcado11. Finalmente, se recomienda trabajar con construcciones que contengan una etiqueta de proteína fluorescente además de la etiqueta ANAP. Esto ayuda a diferenciar la fluorescencia del receptor marcado del fondo/autofluorescencia. La fluorescencia de fondo se puede distinguir de ANAP por el pico y la forma de los espectros de emisión10, pero tales determinaciones pueden ser muy difíciles cuando solo se utilizan conjuntos de filtros. Además, las células y las membranas sin techo que expresan receptores fluorescentes se pueden identificar utilizando la etiqueta de proteína fluorescente sin tener que excitar ANAP y arriesgarse a un fotoblanqueo excesivo.
En muchos de nuestros registros de PCF, observamos un fuerte pico negativo en nuestros espectros a altas concentraciones de TNP-ATP (Figura 4C). Este pico negativo es un artefacto de nuestro protocolo de resta de fondo. La figura 4D muestra imágenes fluorescentes y de campo claro de una pipeta de parche expuesta a 1 mM TNP-ATP. Una sombra en la punta de la pipeta es evidente, como resultado de la exclusión de TNP-ATP del volumen de las paredes de la pipeta, que es más evidente dentro del plano de enfoque. La imagen espectral de la Figura 4E muestra una banda oscura, correspondiente a esta sombra. Cuando una región por encima o por debajo de esta banda oscura se utiliza para la resta de fondo, produce un pico negativo. Es importante destacar que este pico ocurrió en un rango de longitud de onda correspondiente a la emisión de TNP-ATP y no afectó nuestras mediciones de enfriamiento ANAP.
La principal limitación de nuestros experimentos fue obtener una expresión adecuada de la membrana plasmática de construcciones marcadas con ANAP para medir la fluorescencia. En general, fue más fácil adquirir espectros de alta calidad de membranas sin techo que en PCF, debido a su mayor tamaño y nuestra capacidad para escanear rápidamente un plato completo de membranas sin techo, a diferencia de PCF, donde los parches solo se pueden obtener uno a la vez. En nuestros experimentos, los datos de membranas sin techo y experimentos de PCF fueron similares pero no equivalentes (Figura 6)11. Sin embargo, no hay ninguna razón a priori por la que esto deba ser una observación universal, ya que las proteínas en una pipeta de parche pueden estar en un estado funcional diferente al de las membranas sin techo.
Aquí, se han hecho intentos para maximizar la expresión de nuestras construcciones marcadas con ANAP, en particular reduciendo la temperatura de cultivo celular a 33 ° C10,11,16. En nuestra experiencia, intentar identificar sitios en la proteína en los que ANAP sería una sustitución conservadora no resultó consistentemente en construcciones que se expresaran bien. Tuvimos más éxito escaneando sistemáticamente regiones proteicas enteras en busca de sitios de incorporación ANAP y seleccionando candidatos para la expresión superficial10. El sistema de etiquetado ANAP también funciona en ovocitos de Xenopus laevis, lo que permite extirpar parches de membrana mucho más grandes, aumentando así la señal a ruido26,27,28.
Mientras que se espera que los niveles más grandes de expresión den como resultado señales más brillantes, el número mínimo de canales requeridos para medir la fluorescencia depende de varios factores, incluido el brillo del fluoróforo, el grado de fotoblanqueo, la intensidad de la luz de excitación y el plano de enfoque. En teoría, las estimaciones podrían hacerse correlacionando la intensidad de fluorescencia y la corriente del canal como se ha demostrado anteriormente28,29. Sin embargo, la fiabilidad de tales estimaciones requiere cierto conocimiento de la conductancia de un solo canal y la probabilidad de apertura del canal. Además de los factores enumerados anteriormente, la señal de fluorescencia también se verá afectada por canales asociados con vesículas o secciones de la membrana plasmática adheridas al vidrio de pipeta que no están bajo pinza de voltaje.
Este método se adapta fácilmente al estudio de otros canales iónicos sensibles a nucleótidos. CFTR es estructuralmente similar a la subunidad accesoria del receptor de sulfonilurea de KATP30,31. Al igual que KATP, CFTR la activación está controlada por la unión de nucleótidos, lo que la convierte en un objetivo futuro obvio de nuestro método7. Los receptores purinérgicos P2X son canales iónicos cerrados por ATP9 extracelular. El TNP-ATP actúa como antagonista de los receptores P2X32,33. Por lo tanto, no será útil para estudiar la activación P2X, aunque puede ser utilizado en ensayos de competición con agonistas P2X. Alternativamente, otros derivados fluorescentes de ATP con suficiente superposición espectral con la emisión ANAP pueden usarse para estudiar la activación. Alexa-647-ATP es un agonista P2X fluorescente34. El R0 calculado entre Alexa-647 y ANAP es ~ 85 Å, lo que significa que la unión directa a P2X debería resultar en un enfriamiento sustancial de ANAP incorporado en el canal. Sin embargo, un R0 tan largo también resultará en la extinción de Alexa-647-ATP unido a subunidades vecinas y aumenta la probabilidad de que la unión de nucleótidos no específicos resulte en FRET. Como el sitio de unión del ligando en los receptores P2X es extracelular, las mediciones de unión se realizarían en células intactas, en pinzas de voltaje de células completas o en parches de membrana de afuera hacia afuera. Nuestro método también se puede ampliar para estudiar la unión y activación de transportadores y bombas electrogénicas y no electrogénicas que dependen del ATP para su ciclo de reacción, así como de los receptores P2Y acoplados a proteínas G. Finalmente, aunque hemos desarrollado este método para medir la unión a nucleótidos de adenina (TNP-ATP, TNP-ADP, TNP-AMP), el mismo enfoque se puede utilizar para estudiar la unión a prácticamente cualquier receptor para el que se haya identificado un ligando fluorescente adecuado.
Divulgaciones
Los autores declaran no tener conflictos de intereses.
Agradecimientos
Deseamos agradecer a Raúl Terrón Exposito por su excelente asistencia técnica. Este trabajo fue financiado por el Consejo de Investigación en Biotecnología y Ciencias Biológicas (BB/R002517/1; MCP y FMA) y el Wellcome Trust (203731/Z/16/A; SGU)
Materiales
| Name | Company | Catalog Number | Comments |
| T75 tissue-culture treated flask | StarLab | CC7682-4875 | |
| 0.1% w/v poly-L-lysine | Sigma-Aldrich | P8920 | |
| 30 mm borosilicate cover glass slips | VWR | 631-0174 | |
| 35 mm non-treated sterile dishes | CytoOne | CC7672-3340 | |
| 35 mm cover glass bottom dish | WPI | FD35-PDL-100 | |
| Dulbecco’s Modified Eagle Medium (DMEM) | Gibco | 31966021 | |
| Foetal bovine serum (FBS) | Gibco | 10500-064 | |
| Penicillin/Streptomycin | Gibco | 15140-122 | |
| TrypLE select (tryosin) | Gibco | 12563-011 | Trypsin/EDTA reagent |
| Phosphate buffered saline (PBS) | Gibco | 14040-091 | |
| UltraPure distilled water | Invitrogen | 10977-035 | |
| HEK293T cells | ATTC | CRL-3216 | Used between passages 5-30 |
| ANAP-TFA | AsisChem | ASIS-0014 | Reconstituted in 30 mM NaOH to a final concentration of 1 mM |
| pANAP expression plasmid | Addgene | Plasmid #48696 | Encodes tRNA/tRNA synthetase pair for expression of ANAP-tagged protein |
| peRF1-E55D | Chin Lab (MRC Laboratory of Molecular Biology, Cambridge, UK) | Jason Chin: DOI: 10.1021/ja5069728 | Encodes dominant-negative eukaryotic ribosomal release factor |
| TransIT-LT1 | Mirus Bio | MIR 2300 | Lipopolyplex transfection reagent |
| Thick-walled borosilicate glass capillaries | Harvard Apparatus | GC150F-15 | |
| Tetramethylrhodamine-5-maleimide | Sigma-Aldrich | 94506 | |
| TNP-ATP | Jena Bioscience | NU-221L | Delivered at 10 mM in water |
| Nikon Eclipse TE2000-U inverted microscope microscope | Nikon | ||
| 60x water immersion objective (1.4 NA) | Nikon | MRD07602 | |
| 4-Wavelength High-Power LED Head | ThorLabs | LED4D245 | 385/490/565/625 nm LEDs |
| Four-Channel LED Driver | ThorLabs | DC4100 | |
| 390/18 nm band-pass excitation filter | ThorLabs | MF390-18 | For ANAP excitation |
| 400 nm long-pass emission filter | ThorLabs | FEL0400 | For imaging ANAP spectra |
| 416 nm edge dichroic | ThorLabs | MD416 | For imaging ANAP spectra |
| 460/60 nm band-pass emission filter | ThorLabs | MF460-60 | Suggested wide band-pass filter for imaging ANAP fluorescence (Figure 4B) |
| 470/10 nm band-pass emission filter | ThorLabs | FB470-10 | Suggested narrow band-pass filter for imaging ANAP fluorescence (Figure 4B) |
| 531/40 band-pass excitation filter | Brightline | FF01-531/40-25 | For orange fluorescent protein (OFP) excitation |
| 540/25 nm band-pass excitation filter | Chroma | D540/25X | For tetramethylrhodamine-5-maleimide (TMRM) excitation |
| 562 nm edge dichroic | Semrock | FF562-Di03 | For imaging OFP fluorescence |
| 565 nm edge dichroic | Chroma | 565DC | For imaging TMRM fluorescence |
| 593/40 nm band-pass excitation filter | Brightline | FF01-387/11-25 | For imaging OFP fluorescence |
| 605/55 nm band-pass emission filter | Chroma | D605/55M | For imaging TMRM fluorescence |
| IsoPlane-160 Imaging Spectrometer | Princeton Instruments | IsoPlane-160 | |
| PIXIS 400BR_eXcelon Camera | Princeton Instruments | PIXIS: 400BR_eXcelon | |
| Axopatch 200B amplifier | Molecular Devices | Axopatch 200B-2 | |
| Digidata 1440A digitizer | Molecular Devices | Digidata 1440A | |
| Probe sonicator | Sonics & Materials | VC-50 | For unroofing |
| REGLO digital peristaltic pump | Ismatec | ISM 832 | For bath perfusion |
| Microvolume perfusion system | ALA Scientific Instruments | ALA μFlow-8 | For TNP-ATP perfusion |
| pClamp 10.6.2 | Molecular Devices | Recording and analysing currents | |
| Lightfield 5.20.1507 | Princeton Instruments | Acquisition software for images and spectra | |
| Matlab | Mathworks | For data analysis | |
| Python 3.8.1 | Python Software Foundation | For data analysis |
Referencias
- Garcia, M. L., Kaczorowski, G. J. Ion channels find a pathway for therapeutic success. Proceedings of the National Academy of Sciences of the United States of America. 113 (20), 5472-5474 (2016).
- Hauser, A. S., Attwood, M. M., Rask-Andersen, M., Schioth, H. B., Gloriam, D. E. Trends in GPCR drug discovery: new agents, targets and indications. Nature Reviews Drug Discovery. 16 (12), 829-842 (2017).
- Lakowicz, J. R. . Principles of fluorescence spectroscopy. 3rd edn. , (2006).
- Higgins, C. F., Linton, K. J. The ATP switch model for ABC transporters. Nature Structural & Molecular Biology. 11 (10), 918-926 (2004).
- Toyoshima, C., Cornelius, F. New crystal structures of PII-type ATPases: excitement continues. Current Opinion in Structural Biology. 23 (4), 507-514 (2013).
- Craven, K. B., Zagotta, W. N. CNG and HCN channels: two peas, one pod. Annual Review of Physiology. 68, 375-401 (2006).
- Csanady, L., Vergani, P., Gadsby, D. C. Strict coupling between CFTR's catalytic cycle and gating of its Cl- ion pore revealed by distributions of open channel burst durations. Proceedings of the National Academy of Sciences of the United States of America. 107 (3), 1241-1246 (2010).
- Vedovato, N., Ashcroft, F. M., Puljung, M. C. The Nucleotide-Binding Sites of SUR1: A Mechanistic Model. Biophysical Journal. 109 (12), 2452-2460 (2015).
- Burnstock, G. Introduction to the Special Issue on Purinergic Receptors. Advances in Experimental Medicine and Biology. 1051, 1-6 (2017).
- Puljung, M., Vedovato, N., Usher, S., Ashcroft, F. Activation mechanism of ATP-sensitive K(+) channels explored with real-time nucleotide binding. Elife. 8, 41103 (2019).
- Usher, S. G., Ashcroft, F. M., Puljung, M. C. Nucleotide inhibition of the pancreatic ATP-sensitive K+ channel explored with patch-clamp fluorometry. Elife. 9, 52775 (2020).
- Chatterjee, A., Guo, J., Lee, H. S., Schultz, P. G. A genetically encoded fluorescent probe in mammalian cells. Journal of the American Chemical Society. 135 (34), 12540-12543 (2013).
- Gordon, S. E., Senning, E. N., Aman, T. K., Zagotta, W. N. Transition metal ion FRET to measure short-range distances at the intracellular surface of the plasma membrane. Journal of General Physiology. 147 (2), 189-200 (2016).
- Heuser, J. The production of 'cell cortices' for light and electron microscopy. Traffic. 1 (7), 545-552 (2000).
- Schmied, W. H., Elsasser, S. J., Uttamapinant, C., Chin, J. W. Efficient multisite unnatural amino acid incorporation in mammalian cells via optimized pyrrolysyl tRNA synthetase/tRNA expression and engineered eRF1. Journal of the American Chemical Society. 136 (44), 15577-15583 (2014).
- Lin, C. Y., et al. Enhancing Protein Expression in HEK-293 Cells by Lowering Culture Temperature. PloS One. 10 (4), 0123562 (2015).
- Usukura, J., et al. Use of the unroofing technique for atomic force microscopic imaging of the intra-cellular cytoskeleton under aqueous conditions. Journal of Electron Microscopy. 61 (5), 321-326 (2012).
- Colquhoun, D. Binding, gating, affinity and efficacy: the interpretation of structure-activity relationships for agonists and of the effects of mutating receptors. British Journal of Pharmacology. 125 (5), 924-947 (1998).
- Mannuzzu, L. M., Moronne, M. M., Isacoff, E. Y. Direct physical measure of conformational rearrangement underlying potassium channel gating. Science. 271 (5246), 213-216 (1996).
- Zheng, J., Zagotta, W. N. Gating rearrangements in cyclic nucleotide-gated channels revealed by patch-clamp fluorometry. Neuron. 28 (2), 369-374 (2000).
- Zheng, J., Zagotta, W. N. Patch-clamp fluorometry recording of conformational rearrangements of ion channels. Science's STKE. 2003 (176), 7 (2003).
- Biskup, C., et al. Relating ligand binding to activation gating in CNGA2 channels. Nature. 446 (7134), 440-443 (2007).
- Kusch, J., et al. Interdependence of receptor activation and ligand binding in HCN2 pacemaker channels. Neuron. 67 (1), 75-85 (2010).
- Wu, S., et al. State-dependent cAMP binding to functioning HCN channels studied by patch-clamp fluorometry. Biophysical Journal. 100 (5), 1226-1232 (2011).
- Cha, A., Bezanilla, F. Characterizing voltage-dependent conformational changes in the Shaker K+ channel with fluorescence. Neuron. 19 (5), 1127-1140 (1997).
- Kalstrup, T., Blunck, R. Dynamics of internal pore opening in K(V) channels probed by a fluorescent unnatural amino acid. Proceedings of the National Academy of Sciences of the United States of America. 110 (20), 8272-8277 (2013).
- Kalstrup, T., Blunck, R. S4-S5 linker movement during activation and inactivation in voltage-gated K(+) channels. Proceedings of the National Academy of Sciences of the United States of America. 115 (29), 6751-6759 (2018).
- Dai, G., Aman, T. K., DiMaio, F., Zagotta, W. N. The HCN channel voltage sensor undergoes a large downward motion during hyperpolarization. Nature Structural & Molecular Biology. 26 (8), 686-694 (2019).
- Liu, C., et al. Patch-clamp fluorometry-based channel counting to determine HCN channel conductance. Journal of General Physiology. 148 (1), 65-76 (2016).
- Hwang, T. C., et al. Structural mechanisms of CFTR function and dysfunction. Journal of General Physiology. 150 (4), 539-570 (2018).
- Puljung, M. C. Cryo-electron microscopy structures and progress toward a dynamic understanding of KATP channels. Journal of General Physiology. 150 (5), 653-669 (2018).
- Kasuya, G., et al. Structural insights into the competitive inhibition of the ATP-gated P2X receptor channel. Nature Communications. 8 (1), 876 (2017).
- Virginio, C., Robertson, G., Surprenant, A., North, R. A. Trinitrophenyl-substituted nucleotides are potent antagonists selective for P2X1, P2X3, and heteromeric P2X2/3 receptors. Molecular Pharmacology. 53 (6), 969-973 (1998).
- Bhargava, Y., Nicke, A., Rettinger, J. Validation of Alexa-647-ATP as a powerful tool to study P2X receptor ligand binding and desensitization. Biochemical and Biophysical Research Communications. 438 (2), 295-300 (2013).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados