
Method Article
Optimización del crecimiento de cristales de endotiapepsina para experimentos de cristalografía en serie
En este artículo
Resumen
El objetivo de este artículo es dar al espectador una sólida comprensión de cómo transformar su protocolo de difusión de vapor de pequeño volumen, para cultivar cristales grandes de una sola proteína, en un método de microcristalización por lotes de gran volumen para cristalografía en serie.
Resumen
Aquí, se presenta un protocolo para facilitar la creación de grandes volúmenes (> 100 μL) de lodos microcristalinos adecuados para experimentos de cristalografía en serie tanto en sincrotrones como en XFEL. El método se basa en una comprensión del diagrama de fase del cristal de proteína y cómo se puede utilizar ese conocimiento. El método se divide en tres etapas: (1) optimización de la morfología del cristal, (2) transición a lote y (3) escalado. La etapa 1 implica encontrar cristales individuales bien difractantes, con suerte pero no necesariamente, que se presenten en una morfología similar a un cubo. En la Etapa 2, la condición de la Etapa 1 se optimiza por el tiempo de crecimiento del cristal. Esta estrategia puede transformar cristales cultivados por difusión de vapor en lotes. Una vez que el crecimiento del cristal puede ocurrir dentro de aproximadamente 24 h, se puede trazar un morfograma de la mezcla de proteínas y precipitantes y usarlo como base para una estrategia de escalamiento (Etapa 3). Cuando los cristales se pueden cultivar en lote, se puede intentar escalar y optimizar el tamaño y la concentración del cristal a medida que aumenta el volumen. La endotiapepsina se ha utilizado como proteína de demostración para este protocolo. Algunas de las decisiones presentadas son específicas para la endotiapepsina. Sin embargo, se espera que la forma en que se han aplicado inspire una forma de pensar sobre este procedimiento que otros puedan adaptar a sus propios proyectos.
Introducción
La cristalografía macromolecular a temperatura ambiente (RT) es popular nuevamente dentro de la comunidad de biología estructural. El desarrollo de fuentes de luz láser de electrones libres de rayos X (XFEL) ha estimulado el desarrollo de enfoques de entrega de muestras RT 1,2,3,4, y estos métodos ahora se han aplicado a sincrotrones 5,6,7,8. Los métodos RT no solo abren la posibilidad de estrategias experimentales bomba-sonda 9,10,11,12, sino que también hay una creciente evidencia de que promueven estados conformacionales alternativos dentro de las proteínas 13,14,15,16,17.
Sin embargo, la razón principal por la que los criométodos ganaron tracción sobre los enfoques RT a fines de la década de 1990 fue la desaceleración del daño por radiación por temperaturas de cristales bajo cero18. Los criométodos19 comenzaron a permitir la recopilación de un conjunto de datos completo a partir de un solo cristal de proteína. Los métodos modernos de RT en XFEL y sincrotrones resolvieron el problema del daño por radiación de un solo cristal mediante el desarrollo de estrategias rápidas de administración de cristales (> 100 Hz) 1,2,3,4. Estos métodos permiten la recopilación de un conjunto de datos completo de miles de cristales expuestos individualmente. Por lo tanto, estos enfoques de administración de RT requieren la producción de grandes cantidades de soluciones que contengan microcristales homogéneos (> 100 μL de < cristales de 50 μm). Sin embargo, dado que los criométodos tienden a requerir solo cristales individuales, los métodos para crear tales lodos microcristalinos actualmente no son omnipresentes en los laboratorios de cristalografía de proteínas.
Hay ejemplos en la literatura de cómo hacer partes del procedimiento de optimización de microcristalización para muestras de cristalografía en serie. Aquí, se debe hacer una distinción entre proteínas de membrana y solubles. Los protocolos para optimizar el crecimiento de cristales de proteínas de micromembrana cultivadas en monooleína (o algún otro lípido), para fase cúbica lipídica (LCP), han sido bien descritos20,21,22. Sin embargo, generalmente faltan métodos para la microcristalización de proteínas solubles, incluidas las proteínas de membrana cultivadas en condiciones sin LCP. Estudios previos se han centrado en partes específicas del proceso, como el cribado de microcristales 23,24, la mejora de la nucleación24 y el escalamiento mediante difusión de interfaz libre 25, pero no es un método completo.
Sin embargo, recientemente se describió un método26 que intenta ofrecer un protocolo completo. Como muchos aspectos de la cristalografía de proteínas, no es nueva. Muchas de las ideas propuestas ya fueron descritas por Rayment (2002)27. El método tiene como objetivo mostrar a los cristalógrafos cómo realizar la conversión de un solo cristal cultivado mediante difusión de vapor, a una metodología por lotes para cultivar miles de microcristales. El método se centra en la difusión de vapor como punto de partida común, ya que el 95% de todas las deposiciones del Banco de Datos de Proteínas (PDB) provienen de cristales cultivados en placas de difusión de vapor26. Sin embargo, la difusión de vapor no es el método ideal para la microcristalización26, por lo que se describe una metodología para convertir la difusión de vapor en cristalización por lotes. Una vez que los cristales se pueden cultivar en lotes, las rutas de escala a volúmenes más grandes se vuelven más prácticas. Dados los caprichos de la cristalización de proteínas, los autores enfatizarían que este método no es a prueba de fallos. Sin embargo, el protocolo debería, al menos, proporcionar una idea del "espacio de cristalización" de una proteína.
Este método se basa en el diagrama de fase de cristalización de proteínas y en cómo la comprensión de ese diagrama puede actuar como guía durante la optimización de la microcristalización. Un diagrama de fase de proteína se representa comúnmente como un gráfico x/y con concentraciones de precipitante y proteína en los ejes x e y, respectivamente (Figura 1A). Desde el punto de agua pura (esquina inferior izquierda - Figura 1A), la concentración de proteína y precipitante aumenta hasta que se alcanza la línea de solubilidad. La línea de solubilidad marca el punto de sobresaturación (línea púrpura - Figura 1A). Cuando una proteína está sobresaturada, la solución se vuelve termodinámicamente inestable y comenzará a separarse en dos fases: "rica en proteínas" y una solución saturada estable. Esta separación puede ocurrir en cualquier lugar más allá de la línea de solubilidad y su cinética depende de las propiedades de la proteína y los componentes de la solución.
Cuando las concentraciones de proteína y precipitante son demasiado grandes, la proteína se descompondrá de manera inestable fuera de la solución y dará como resultado un precipitado amorfo (región rosada - Figura 1A). Sin embargo, la separación ordenada de fases puede ocurrir en la región de nucleación [ver García-Ruiz (2003)28 para una descripción detallada] y los nucleantes cristalinos tienen la propensión a formarse (región verde - Figura 1A). La nucleación y el crecimiento eliminan la proteína de la solución y mueven la gota a la región metaestable donde el crecimiento puede continuar hasta que se alcance la línea de solubilidad [ver McPherson y Kuznetsov (2014) 29 para una discusión detallada]. El diagrama es, para la gran mayoría de las condiciones de cristalización, una simplificación excesiva30. Sin embargo, independientemente de esto, el diagrama sigue siendo de gran utilidad para los microcristalógrafos, ya que el mapeo del diagrama permite determinar la línea de solubilidad y la cinética de nucleación.
En términos de creación de microcristales, los dos factores durante la cristalización que deben optimizarse son el número de cristales (Xn) y su dimensión media más larga (Xs). X n será proporcional al número de eventos de nucleación (n ) (Ec. 1).
 Ec. 1
Ec. 1
X s es proporcional a la concentración de proteína libre por encima de la línea de solubilidad (Ps) dividida por Xn (Eq. 2).
 Ec. 2
Ec. 2
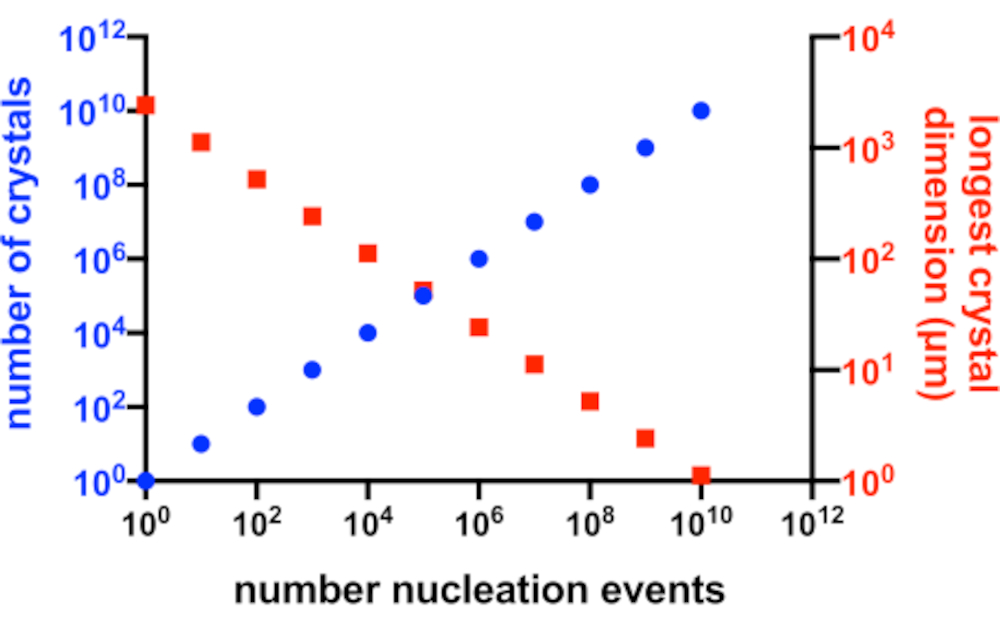
En una situación perfecta, cada evento de nucleación produciría un posible cristal y cada uno de estos cristales tendría igual acceso a la proteína disponible en solución. La figura 2 es una representación gráfica de un escenario ideal de la relación entre Xn y Xs. Prácticamente, el control principal que un cristalógrafo tiene sobre Xn y X s es influyendo en la cantidad de nucleación o mediante la adición decristales de semillas. El microcristalógrafo debe juzgar cómo aumentar Xn de tal manera que se pueda crear una concentración de cristal y un tamaño de cristal adecuados.
La mayoría de las técnicas de cristalización requieren un "período de transición" (Figura 1B). Por ejemplo, en un experimento de difusión de vapor, al mezclar la proteína y las soluciones precipitantes, las concentraciones de cada una cambiarán a medida que la gota se equilibre con la solución del pozo. Uno espera que estos cambios hagan gradualmente la transición de la gota a la zona de nucleación donde aumentará la propensión a la cristalización. A medida que los cristales comienzan a nuclearse y crecer, la cantidad de proteína en solución comenzará a disminuir, disminuyendo la probabilidad de una mayor nucleación. La cantidad final de nucleación será específica de la proteína y la condición, y también dependerá de la profundidad de penetración en la zona de nucleación. Dada la limitada penetración en la zona de nucleación de los métodos que requieren un paso de transición, el nivel de nucleación se limitará en última instancia a la velocidad de nucleación en el límite de la región de nucleación metaestable.
Debido a la importancia de poder mejorar el nivel de nucleación para un microcristalógrafo, es importante pasar a una metodología de cristalización por lotes. El lote puede aprovechar mejor toda la región de nucleación (Figura 1C). En los métodos por lotes, la idea es mezclar la proteína y el precipitante de tal manera que se cree una solución sobresaturada sin la necesidad de ningún cambio en las concentraciones de componentes. La nucleación debe ser posible inmediatamente después de la mezcla. Por lo tanto, los métodos por lotes permiten alcanzar teóricamente toda la zona de nucleación. Cualquier aumento en la cinética de nucleación más allá del límite de nucleación metaestable puede ser utilizado.
Si el nivel basal de nucleación cristalina no es suficiente para generar ungran X n, se pueden usar métodos de microsiembra. En la microsiembra, los cristales precultivados se rompen para crear una suspensión de fragmentos cristalinos que pueden actuar como un andamio para el crecimiento de cristales frescos31,32. La microsiembra se ha utilizado ampliamente en la preparación de muestras cristalográficas en serie como una forma de aumentar Xn sin la necesidad de aumentar la nucleación cristalina (Figura 1C).
La transición de la difusión de vapor a la dispersión por lotes se puede visualizar en un diagrama de fase como mover el punto de partida experimental de las regiones no sobresaturadas o metaestables a la zona de nucleación. Esto se puede hacer aumentando las concentraciones de proteínas y/o precipitantes, y/o la proporción de las dos dentro de la gota (Figura 1D), y observando en qué condiciones producen cristales que aparecen rápidamente (< 24 h)26. El equilibrio completo de la gota de difusión de vapor puede tardar días o semanas33. Por lo tanto, al buscar condiciones que muestren cristales que aparecen rápidamente, se pueden encontrar condiciones de lote sin tener que pasar a formatos alternativos de cribado de cristalización como el microlote34,35,36,37.
Una vez que se ha encontrado la zona de nucleación, se ha encontrado una condición de lote y se puede crear un morfograma, aquí, un diagrama de fase aproximado. El morfograma es de gran utilidad cuando se contempla si se debe usar un protocolo de lote sembrado o de lote recto. Al trazar elX n en función de la proteína y la concentración de precipitante, se puede hacer una evaluación de la cinética de nucleación26. Si X n permanece bajo en toda la región de nucleación, se puede requerir un lote sembrado para hacer Xn lo suficientemente grande como para limitar el crecimiento de cristales. Esta evaluación es el primer paso en el proceso de escalado a volúmenes mayores (> 100 μL).
Este método fue diseñado de tal manera que podría llevarse a cabo en la mayoría de los laboratorios de cristalización mediante el uso de equipos estándar de cristalización por difusión de vapor. También se han realizado muchos estudios que describen técnicas para facilitar muchas partes de este proceso, en caso de que el equipo esté disponible. Estos incluyen, pero no se limitan a, dispersión dinámica de luz (DLS)25,27, imágenes no lineales 20,24,25, difracción de polvo 20,24,27 y microscopía electrónica 26 [ver Cheng et al. (2020) 40 para una buena revisión].
El objetivo de este trabajo es proporcionar una demostración visual del método para la transición de la cristalización por difusión de vapor de pequeño volumen (< 500 nL) a la cristalización por lotes de gran volumen (> 100 μL). La endotiapepsina de Cryphonectria parasitica se ha utilizado como un sistema de ejemplo para demostrar esta traducción. El tipo de experimento y el método de entrega de muestras para el que se requieren los microcristales influirán en la salida idealde X s 26. Para experimentos de mezcla que requieren una resolución de milisegundos41 o boquillas virtuales con dinámica de gas42, puede ser deseable una Xs final de < 5 μm. En este caso, el objetivo era producir cristales de proteínas que difractaran a aproximadamente 1,5 Å, para un experimento de bomba-sonda activada por fotones, y utilizando un enfoque de entrega de objetivo fijo.
Para dar una ilustración de los requisitos de muestra de tal experimento de cristalografía en serie utilizando endotiapepsina, la Tabla 1 muestra los parámetros experimentales de un experimento hipotético. La información de la muestra se basó en el protocolo que se describe a continuación. Dadas algunas estimaciones conservadoras sobre las tasas de aciertos y los requisitos de recopilación de datos, 50 mg es la estimación del consumo total de la muestra para todo el experimento.
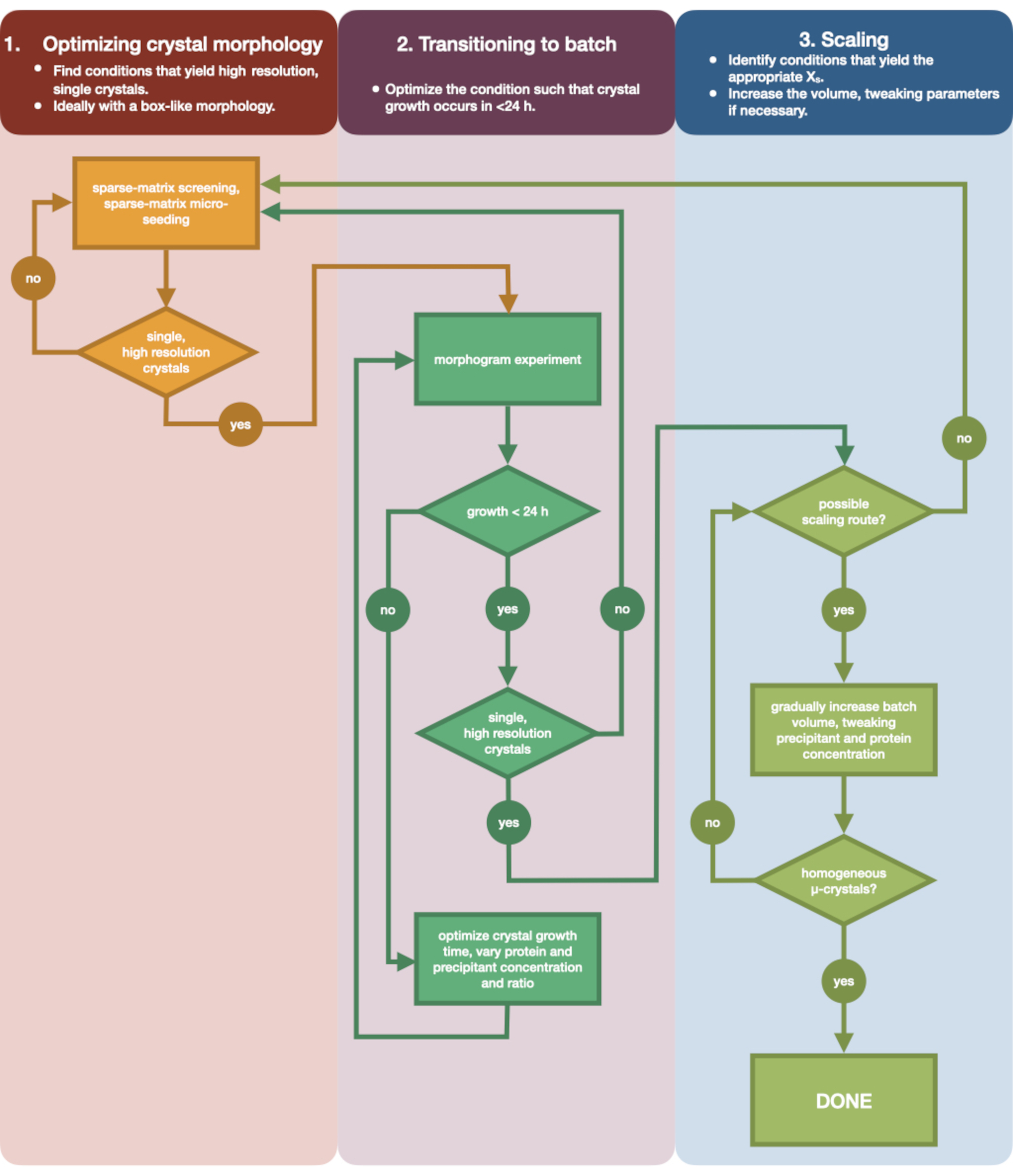
La Figura 3 muestra un diagrama de flujo del proceso de optimización completo desde la cristalización inicial por difusión de vapor de pequeño volumen hasta el lote a gran escala. Para la mayoría de los proyectos de cristalografía en serie, este protocolo comenzará en el Paso 2: 'transición a lote', ya que la proteína diana ya habrá sido cristalizada. Sin embargo, el Paso 1 se ha incluido para completar y recordar a los lectores su importancia. Encontrar una condición que dé lugar a un cristal grande, único y difractor es el mejor punto de partida para la optimización de microcristales. En el Paso 2, esta condición se puede optimizar de la difusión de vapor al lote, y se puede trazar un morfograma de la nucleación y las regiones metaestables. Una vez hecho esto, se puede escalar la condición del lote a volúmenes más grandes en el paso 3. Al final del diagrama de flujo, un cristalógrafo habrá creado un protocolo de microcristalización repetible (> 100 μL), de microcristalización, por lotes para la endotiapepsina. Este método se puede aplicar a su proteína particular de interés.
Protocolo
NOTA: Todos los experimentos de cristalización de gota sentada de 96 pocillos se configuraron utilizando placas de 2 o 3 gotas. Se utilizó un robot de manejo de líquidos y un generador de imágenes de cristalización / hotel para facilitar la preparación y el monitoreo de todas las pantallas de 96 pocillos. Todas las concentraciones de reactivos para los experimentos de cristalización se dan en sus concentraciones iniciales antes de la mezcla.
1. Optimización de la morfología cristalina
NOTA: Pasos 1.1.1. y 1.1.6. Describir cómo se encontraron las condiciones de cristalización de endotiapepsina y cómo se optimizaron estas condiciones para encontrar una sola condición que produjera cristales únicos y bien difractantes.
- Optimización de matrices dispersas
- Prepare una solución fresca de endotiapepsina.
NOTA: La endotiapepsina, cuando se adquiere como Superan 600, debe transferirse tampón fuera de su solución de almacenamiento y concentrarse.- Preparar 3 L de 0,1 m de acetato de na pH 4,6 a 4 °C.
- Corte 20 cm del tubo de diálisis y lave brevemente el tampón. Selle un extremo del tubo con un clip, coloque 50 ml de la solución de endotiapepsina en el tubo y luego selle el otro extremo.
- Dejar la solución dializar durante al menos 4 h (o durante la noche) a 4 °C en 1 L del tampón acetato de Na. Debido a los componentes del tampón de almacenamiento, la solución en la bolsa de diálisis ahora será de aproximadamente 100 ml.
- Transfiera la bolsa de diálisis que contiene la endotiapepsina a un litro fresco de 4 °C, 0,1 M Na acetato pH 4.6. Repita este paso una vez más para que el tampón original se haya diluido 2000x contra el acetato de Na.
- La endotiapepsina ahora estará en aproximadamente 10 mg / ml. Concentrar a 100 mg/ml utilizando un concentrador centrífugo de 10 kDa y una centrífuga.
- Enfriar rápidamente la solución de endotiapepsina en nitrógeno líquido en alícuotas de 50 μL y almacenar a -80 °C.
- Prepare una pantalla de matriz dispersa PACT Premier de 96 pocillos.
- Usando un robot de manejo de líquidos, dispensar 100 nL de 70 mg/ml de endotiapepsina y 100 nL de solución de pozo en un solo subpozo por pocillo. Mezcle la proteína y la solución del pozo 3 veces después de agregar el tampón de cristalización.
- Sellar la placa y dejar durante 28 días a 20 °C tomando imágenes todos los días durante la primera semana y luego cada semana a partir de entonces durante 4 semanas.
- Análisis de matriz dispersa
- Identificar los golpes que producen cristales individuales de endotiapepsina. A partir de la pantalla PACT, las condiciones que contenían MgCl2 crecieron como singletons en lugar de grupos de agujas.
- Optimización de matrices dispersas
- A partir de las condiciones que contienen MgCl2 identificadas en el Paso 1.1.3.1, cree una pantalla de 96 pocillos que combine y varie aleatoriamente los diferentes componentes del pocillo.
- Usando un robot de manejo de líquidos, dispensar 100 nL de 70 mg/ml de endotiapepsina y 100 nL de solución de pozo en un solo subpozo por pocillo. Mezcle la proteína y la solución del pozo 3 veces después de agregar el tampón de cristalización.
- Sellar la placa y dejar durante 28 días a 20 °C tomando imágenes todos los días durante la primera semana y luego cada semana a partir de entonces durante 4 semanas.
- Análisis de optimización
- Utilizando un software de hoja de cálculo adecuado, clasifique las condiciones de cristalización que dan lugar a cristales en función de la calidad del cristal y el nivel de precipitación, sin cristales (0) a ideales (5) y bajos (0) a altos (5), respectivamente. Con respecto a la calidad del cristal, los criterios generales son cristales individuales con una morfología similar a una caja.
- Realice un análisis de correlación de Pearson entre el contenido de la condición de cristalización y la cantidad de cristales y el nivel de precipitación.
- Trazar estos datos como un mapa de calor. Busque componentes y condiciones que se correlacionaron con los resultados preferidos.
- Análisis de difracción.
- Confirmar que los cristales cultivados a partir de las condiciones identificadas en el Paso 1.1.5 son adecuados para la cristalografía en serie realizando un experimento de difracción de rayos X.
- Cargue una muestra de los cristales de endotiapepsina de cada una de las condiciones identificadas en soportes que permitan la recopilación de datos a 100 o 293 K y realice un experimento de difracción de rayos X. Si trabaja bajo criocrio, use 25% de etilenglicol como crioprotector.
- Procesar estos datos a través de un paquete de software adecuado. Los cristales de endotiapepsina deben difractarse más allá de 1,5 Å. Compruebe si hay maclado, ya que los cristales maclados pueden complicar significativamente el procesamiento de datos cristalográficos en serie.
- Si los cristales son singletons y difractan a 1.5 Å, continúe con el Paso 2. Si no es así, vuelva al Paso 1.1.2 y pruebe más pantallas de matriz dispersa para identificar condiciones prometedoras. Después de los análisis realizados en los pasos 1.1.5. y 1.1.6., una condición de cristalización de 25% (p/v) PEG 6,000, 0.1 M Tris-HCl pH 7.0 y 0.15 M MgCl2 debería haberse encontrado como el ideal aproximado.
- Prepare una solución fresca de endotiapepsina.
2. Transición a lote
- Experimento de morfograma
- Crea un stock de semillas de microcristales.
NOTA: Es una buena práctica al hacer existencias de semillas, hacer las semillas de cristales específicamente cultivados para la tarea. Esto ayuda enormemente con la reproducibilidad. Otras ideas presentadas en los pasos 2.1.1.1 a 2.1.1.11 son utilizar siempre los cristales cultivados a partir de un número estándar de pozos - aquí 5 - y alícuotas las existencias una vez que se hacen para anular los ciclos de congelación-descongelación.- Preparar una placa de cristalización de 96 pocillos con pocillos que contengan el tampón de cristalización: 25% (p/v) PEG 6.000, 0,1 M Tris-HCl pH 7,0 y 0,15 M MgCl2.
- Usando un robot de manejo de líquidos, dispensar 200 nL de 70 mg/ml de endotiapepsina descongelada y 200 nL de solución de pozo en un solo subpocillo por pocillo. Mezcle la proteína y la solución del pozo 3 veces después de agregar el tampón de cristalización.
- Sellar el plato y dejar actuar durante 24 h.
- Llene un tubo de centrífuga de 1,5 ml con 250 μL de tampón de cristalización y 10-15 perlas de vidrio de 1 mm. Deje enfriar el tubo de la centrífuga en hielo durante 5-10 minutos.
- Seleccione 5 pocillos con cristales, abra los pocillos con un bisturí y, con una punta de pipeta, triture los cristales en los pocillos.
- Aspirar 1 μL de tampón del tubo de centrífuga helado y utilizar para homogeneizar la suspensión de cristal triturada. Una vez homogéneo, aspirar toda la suspensión y recoger en el tubo de centrífuga enfriado.
- Repita el paso 2.1.2.6 para cada uno de los 5 subpozos.
- Vortex el tubo de centrífuga que contiene el tampón, lodos agrupados y perlas a 1000 rpm durante 30 s.
- Devolver el tubo de la centrífuga al hielo durante 30 s.
- Repita los pasos 2.1.2.8 y 2.1.2.9 dos veces más.
- El material de semillas ya está listo y puede ser alicitado en lotes de 10 μL y almacenado a -20 °C.
- Realizar experimento de morfograma.
- Prepare una pantalla de cuadrícula de 96 pocillos de 2 gotas. Varíe la concentración de PEG 6,000 de 5 a 40% (p/v) a lo largo de las columnas de la placa, manteniendo el tampón y la sal a 0.1 M Tris-HCl pH 7.0 y 0.15 M MgCl2, respectivamente.
- Preparar una dilución secuencial de endotiapepsina en 0.1 M Na Acetato pH 4.6 de 100 a 12.5 mg / ml en 8 pasos. Se utilizará una concentración diferente de endotiapepsina para cada fila de la placa.
- Usando un robot de manejo de líquidos, dispensar 150 nL de endotiapepsina en los subpocillos 1 y 2. En el subpozo 1, dispensar 150 nL de la solución del pozo. En el subpocillo 2, multiaspirar 50 nL de semilla descongelada y 100 nL de solución de pozo, y luego dispensar ambos en la solución de proteína. Mezclar las soluciones 3 veces tras la adición del tampón de cristalización.
- Sellar la placa y dejar a 20 °C tomando imágenes cada 0, 3, 6, 12, 18, 24 h, luego todos los días durante la primera semana, y cada semana durante las cuatro siguientes. Si las imágenes automáticas no son posibles, no se preocupe por las imágenes por hora el día 1.
- Crea un stock de semillas de microcristales.
- Análisis de morfograma
- Mirando las imágenes tomadas después de 24 h, estime el número de cristales que están presentes en cada pozo y registre estas estimaciones en la hoja de trabajo "generador de morfograma" proporcionada. Estas estimaciones no tienen que ser precisas; Contar individualmente miles de microcristales, si están presentes, no es práctico ni necesario. Principalmente trate de asegurarse de que las estimaciones sean consistentes en todo el plato.
NOTA: La regla de las 24 horas se basó en las observaciones realizadas en Beale et al . (2019)26. Las condiciones de cristalización por difusión de vapor pueden tardar días o semanas en equilibrarse. Los cristales que aparecen rápidamente tienen más probabilidades de haber crecido a través de un proceso por lotes en lugar de por el equilibrio gradual de los componentes de la gota. El criterio de 24 h es, por lo tanto, algo arbitrario y un tiempo de corte exacto entre un experimento de difusión de vapor y por lote dependerá de la mezcla específica de la condición [ver Beale et al. (2019) 26 para más detalles]. - Introducir las concentraciones iniciales de endotiapepsina y PEG 6.000 en las casillas indicadas.
- La hoja de trabajo trazará automáticamente los resultados en el formato tradicional de diagrama de fase con concentración de precipitante y proteína en los ejes x e y , respectivamente. Las condiciones de pozo que solo dan lugar a cristales en sus gotas sembradas indican la región metaestable del diagrama (azul transparente), mientras que las condiciones que tienen cristales tanto en las gotas sembradas como en las no sembradas indican la zona de nucleación (verde sólido).
NOTA: Idealmente, la mayoría de la zona de nucleación debe estar presente en el diagrama (es decir, hay algunos pozos claros en la parte inferior del diagrama y algo de precipitado debe ser visible a altas concentraciones de proteínas y precipitantes). Si este no es el caso, tal vez, repita el experimento pero aumente la concentración de proteína y / o precipitante (si es posible). - Si el cristal ha aparecido en menos de 24 h, continúe con el Paso 2.3.1. Si no es así, continúe con el paso 2.4 y continúe optimizando hacia lotes.
- Mirando las imágenes tomadas después de 24 h, estime el número de cristales que están presentes en cada pozo y registre estas estimaciones en la hoja de trabajo "generador de morfograma" proporcionada. Estas estimaciones no tienen que ser precisas; Contar individualmente miles de microcristales, si están presentes, no es práctico ni necesario. Principalmente trate de asegurarse de que las estimaciones sean consistentes en todo el plato.
- Análisis de cristales
- Como se dijo al final del Paso 1, antes de pasar al siguiente paso, asegúrese de que estos cristales tengan la morfología y la calidad de difracción deseadas. Con respecto a la morfología, ¿los cristales están observablemente desenmaclados y formándose como singletons en lugar de estructuras en forma de bola de aguja o en forma de abanico? Con respecto a la difracción, recopile datos de difracción de los cristales si es posible. Si estos cristales no se difractan, es improbable que los cristales cultivados en un volumen mayor se difracten.
- Cargue una muestra de los cristales de endotiapepsina del experimento del morfograma en soportes que permitan la recopilación de datos a 100 o 293 K y realice un experimento de difracción de rayos X. Si trabaja bajo criocrio, use 25% de etilenglicol como crioprotector.
- Procesar estos datos a través de un paquete de software adecuado. Los cristales de endotiapepsina deben difractarse más allá de 1,5 Å. A través de la muestra de cristales, observe el tamaño de la célula, el número total de observaciones y la mosaicidad; Estas medidas darán una indicación de la homogeneidad de los cristales difractantes.
- Si la morfología del cristal y la calidad de la difracción son suficientes, continúe con el paso 3.
- Optimizar el tiempo de crecimiento de cristales.
NOTA: El análisis del morfograma (Paso 2.2) habrá dado una indicación del punto de partida de la cristalización (es decir, la región del diagrama de fases donde se encuentra la gota cuando se mezclaron las soluciones precipitante y proteica). ¿La caída está en la región metaestable o por debajo de la línea de solubilidad? La cristalización por lotes comienza en la zona de nucleación (Figura 1C). El objetivo de este paso es mover este punto de partida desde debajo de la línea de solubilidad o la región metaestable, hacia la zona de nucleación (Figura 1D). Si la semilla cae del paso 2.2. han producido cristales rápidamente, esto es una indicación de que la mezcla de gotas ya está en la región metaestable, si no, entonces es probable que la gota no esté sobresaturada.- Optimización del tiempo de crecimiento de cristales.
- Usando la misma pantalla que en el Paso 2.1.3, prepare un experimento de cristalización por difusión de vapor de 96 pocillos en una placa de 3 gotas.
- Aumentar la concentración proteica inicial de endotiapepsina en el eje y (es decir, concentrar la proteína aún más, tal vez 120 mg/ml para la endotiapepsina).
- Realice una dilución en serie, como en el paso 2.1.3.2, de modo que cada fila de la placa contenga una concentración de proteína secuencialmente menor.
- Use diferentes proporciones de gota en cada una de las tres gotas en el plato: 1: 1, 1: 2 y 2: 1, proteína: precipitante.
- Vea o imagine el plato el primer día a las 0, 3, 6, 12, 18, 24 h y luego todos los días durante la primera semana, y cada semana durante las siguientes cuatro. Si las imágenes automáticas no son posibles, no se preocupe por las imágenes por hora el día 1.
- Identifique las gotas que producen los cristales que aparecen más rápidamente y los convierte en los puntos de partida de optimizaciones repetidas hasta que se produce el crecimiento del cristal dentro de las 24 h.
- Cuando se haya identificado una condición cristalina que aparece rápidamente, regrese al Paso 2.1 para volver a trazar el morfograma como preludio para comenzar a escalar.
- Optimización del tiempo de crecimiento de cristales.
3. Escalado
- Clasificar rutas de escalado. En esta etapa, no es necesario decidir una sola ruta de escalado, solo identificar y clasificar las opciones para que puedan explorarse a su vez. A medida que aumenta el volumen de la mezcla por lotes durante el procedimiento de escalado, se producirán cambios en la velocidad de nucleación y el rango de tamaños de cristal. Sin embargo, estos pueden superarse ajustando cuidadosamente las concentraciones de los componentes a medida que aumenta el volumen escalado.
NOTA: Los pasos 3.1.1 y 3.1.2 describen cómo discernir, a partir del morfograma, si un protocolo de lote o lote sembrado es más apropiado.- Protocolo de lote directo
- ¿Es elX n en la zona de nucleación proporcional a la concentración de proteína y/o precipitante? Es decir, ¿aumenta Xn en función de la concentración de precipitantes y/o proteínas? -¿Sí? Vaya al paso 3.1.1.2. ¿No? Vaya al paso 3.1.2.
- Localice las condiciones que producen cristales del tamaño requerido y vaya al Paso 3.2.
- Protocolo de lote semilla
- ¿Es el Xn plano a través de la zona de nucleación? Es decir, Xn no aumenta en función de la concentración de precipitante y/o proteína.
- Localice las condiciones de siembra que producen cristales del tamaño requerido y vaya al Paso 3.2. Si todos los cristales son demasiado grandes, vaya al Paso 3.1.2.3.
- Repita el experimento del morfograma (Paso 2.1) pero esta vez aumente la concentración del stock de semillas utilizado en los pozos sembrados. El stock de semillas se puede aumentar utilizando más cristales en su creación. Por ejemplo, en lugar de 5 pozos en el Paso 2.1.1.5, use 10 pozos.
- Ver o imagen de la placa durante las primeras 0, 3, 6, 12, 18, 24 h.
- El Xn debería haber aumentado y el Xs disminuido en las gotas sembradas. Repita este ciclo si se necesitan cristales más pequeños y luego siga un protocolo de lote sembrado.
- Protocolo de lote directo
- Escalado gradual
- Escalado en placas de 96 pocillos. A partir del morfograma de endotiapepsina, se seleccionó inicialmente para el escalado un método de lote recto que utiliza la condición de cristalización 0.1 M Tris-HCl pH 7.0, 0.15 M MgCl2 y 30% (p/v) PEG 6,000. 100 mg/ml de endotiapepsina mezclada con el tampón de cristalización en una proporción de 1:1.
- Prepare 2-3 pocillos en una placa de caída sentada de 96 pocillos de 2 pocillos con 100 μL de 0.1 M Tris-HCl pH 7.0, 0.15 M MgCl2 y 30% (p/v) PEG 6,000.
- Con una solución de endotiapepsina de 100 mg/ml recién descongelada, dispensar 0,5 μL de proteína y 0,5 μL de precipitante por pocillo, sellar y almacenar a 20°C.
- Ver o imagen de la placa durante las primeras 0, 3, 6, 12, 18, 24 h. Tenga en cuenta cualquier cambio en el rango de Xs y Xn.
- Si se han producido cambios, repita los pasos 3.2.1.1 a 3.2.1.2, pero aumente o disminuya la concentración de proteína, precipitante y/o semilla para restaurar cualquier cambio en el rango de Xs y Xn.
- Cuando el rango de Xs y Xn sea aceptable, continúe con el paso 3.2.2.
- Escalado en placas colgantes de 24 pocillos
- Prepare un solo pocillo de una placa colgante de 24 pocillos engrasando los bordes del pozo con grasa de vacío.
- Prepare 0.5 mL de 0.1 M Tris-HCl pH 7.0, 0.15 M MgCl2 y 30% (p/v) PEG 6,000 y llene el pozo engrasado.
- Con una solución de endotiapepsina recién descongelada, pipetear 1 μL de proteína sobre la superficie de un cubreobjetos de vidrio. Pipetear 1 μL de tampón de cristalización sobre la gota de proteína y mezclar con la pipeta.
- Ver o imagen de la placa durante las primeras 0, 3, 6, 12, 24 h. Tenga en cuenta cualquier cambio en el rango de Xs y Xn.
- Si se han producido cambios, repita los pasos 3.2.2.1 a 3.2.2.4, pero aumente o disminuya la concentración de proteínas, precipitantes y/o semillas para restaurar cualquier cambio en el rango de Xs y Xn.
- Cuando/si el rango de Xs y Xn es aceptable, continúe con el Paso 3.2.2.7.
- Repita los pasos 3.2.2.1 a 3.2.2.5, aumentando gradualmente el volumen total del experimento a 10 μL.
- Una vez que tenga un volumen de 10 μL o más, proceda a los tubos de centrifugación en el paso 3.2.3.
- Incrustación en tubos centrífugos
NOTA: El refinamiento de la condición de lote de endotiapepsina ocurrió principalmente en el punto de volúmenes de 200 μL (ver Resultados, Escala). El proceso comenzó con una condición de cristalización de 0.1 M Tris-HCl pH 7.0, 0.15 M MgCl2 y 30% (p/v) PEG 6,000. Sin embargo, la concentración de PEG finalmente cambió a 40% (p/v). También se requerían semillas para controlar el Xn, y para evitar que los cristales crecieran demasiado grandes, el crecimiento de cristales tenía que ser apagado. Los pasos 3.2.3.1 a 3.2.3.7 detallan el proceso de optimización de condiciones. Paso 3.2.4. Describir el protocolo de lote final.- Prepare 1 ml de tampón de cristalización: 0.1 M Tris-HCl pH 7.0, 0.15 M MgCl2 y 30% (p/v) PEG 6,000.
- Usando 100 mg/ml de endotiapepsina recién descongelada, agregue 25 μL de proteína a un tubo de centrífuga de 1,5 ml.
- Mezcle bien el tampón de cristalización con la solución de proteína en una proporción de 1: 1 con una punta de pipeta. Colocar el tubo en un revólver/rotador con alta agitación a 20 °C.
- Tomar alícuotas regulares (5, 10, 30, 60 min, 2, 5, 10, 24 h) de 2,5 μL y ver en un hemocitómetro. Registre el rango Xn yX s .
- Si se han producido cambios, repita los pasos 3.2.3.1. a 3.2.3.4. pero aumentar o disminuir la concentración de proteína, precipitante y/o semilla para restaurar cualquier cambio en el rango de Xs y Xn
- Cuando el rango de Xs y Xn sea aceptable, continúe con el Paso 3.2.3.7.
- Repita los pasos 3.2.2.1 a 3.2.2.5, aumentando gradualmente el volumen total del experimento a 200 μL o más, según sea necesario.
- Protocolo final de lote sembrado
- Preparar el stock de semillas.
- Preparar 2 mL de tampón de cristalización: 0.1 M Tris-HCl pH 7.0, 0.15 M MgCl2 y 40% (p/v) PEG 6,000.
- Usando 100 mg/ml de endotiapepsina recién descongelada, agregue 100 μL de proteína a un tubo de centrífuga de 1,5 ml.
- Mezcle bien el tampón de cristalización con la solución de proteína en una proporción de 1: 1 con una punta de pipeta. Coloque el tubo en un revólver/rotador con alta agitación a 20 °C durante 24 h para permitir que crezcan cristales de 50 μm.
- Añadir 10-15 perlas de vidrio de 1 mm a la suspensión de cristal de 50 μm.
- Vortex el tubo de centrífuga que contiene la suspensión y las perlas a 1000 rpm durante 30 s.
- Devolver el tubo de la centrífuga al hielo durante 30 s.
- Repita los pasos 3.2.4.1.5 y 3.2.4.1.6 10 veces más.
- Esto es ahora 200 μL de un stock de semillas 1x. Diluir el stock de semillas 10 veces mediante la adición de 1,8 ml de tampón de cristalización. Alícuota el material semilla 10x en lotes de 50 μL y almacenar a -20 °C.
- Protocolo de lote sembrado.
- Preparar tampón de cristalización: 0.1 M Tris-HCl pH 7.0, 0.15 M MgCl2 y 40% (p/v) PEG 6,000.
- En un tubo de centrífuga, mezclar 100 μL de tampón de cristalización con los 50 μL de material de semilla 10x recién descongelado.
- Usando 100 mg/ml de endotiapepsina recién descongelada, agregue 150 μL de proteína a un tubo de centrífuga de 1,5 ml.
- Mezclar bien la mezcla tampón de cristalización/semilla con la solución de endotiapepsina con una punta de pipeta y colocar el tubo en un revólver/rotador con alta agitación a 20 °C.
- Controle la cristalización tomando alícuotas regulares de 2,5 μL y vea los cristales en un hemocitómetro. Registre el rango Xn yX s .
- Después de aproximadamente 80 min, cuando los cristales hayan alcanzado un Xs de 15 μm, apagar la reacción mediante la adición de 150 μL de 0,05 M de acetato de Na pH 4,6, 0,05 M Tris-HCl pH 7,0, 0,075 M MgCl2, y 20% (p/v) PEG 6.000 (una solución compuesta de tampón de endotiapepsina y tampón de cristalización, mezclado 1:1).
- Conservar los cristales a 20 °C.
- ¿Ha producido el protocolo un rango de tamaño y número de cristales aceptables para el experimento previsto? Sí - HECHO- No - volver al paso 3.1. e intente una opción de escalado alternativa. Por ejemplo, una relación proteína:precipitante diferente puede ser posible o agregar semillas si esto no se hizo anteriormente. Cuando todos estos se agotan, puede ser necesario encontrar una nueva condición en el Paso 1.
- Preparar el stock de semillas.
- Escalado en placas de 96 pocillos. A partir del morfograma de endotiapepsina, se seleccionó inicialmente para el escalado un método de lote recto que utiliza la condición de cristalización 0.1 M Tris-HCl pH 7.0, 0.15 M MgCl2 y 30% (p/v) PEG 6,000. 100 mg/ml de endotiapepsina mezclada con el tampón de cristalización en una proporción de 1:1.
Resultados
Optimización de la morfología cristalina
El paso 1, optimizar la morfología del cristal, se ha incluido para recordar al lector su importancia. Puede ser posible crear microcristales perfectos a partir de bolas de aguja de difracción deficiente; Sin embargo, los autores sugieren que es mejor optimizar los dos por separado. Primero, encuentre condiciones que den lugar a una buena difracción de un solo cristal a través de la difusión de vapor, y luego convierta estas condiciones en lotes en lugar de tratar de combinar los dos pasos juntos. Descubrir condiciones altamente nucleantes, en esta etapa, no es necesario; La morfología y la calidad de la difracción son los objetivos principales.
Antes de comenzar la microcristalización de la endotiapepsina, se realizó un análisis de las condiciones de cristalización de la estructura depositada a partir del PDB. Se pudieron obtener condiciones de cristalización y protocolos aproximados para 47 de las 48 deposiciones de entotiapepsina. Todos estos se basaron ampliamente en la primera cristalización de endotiapepsina realizada por Moews y Bunn (1970)46. Dadas las similitudes de estas condiciones y su origen "clásico", se realizó una pantalla de matriz dispersa de difusión de vapor de 96 pocillos para explorar una variedad más amplia de condiciones de cristalización. La endotiapepsina se concentró a 70 mg/ml y se realizó un cribado de matriz dispersaPACT 47 en una placa de 96 pocillos a 20 °C mezclando 100 nL de proteína con 100 nL de solución de pocillos. Cada condición de este experimento después de 36 h dio lugar a cristales. Sin embargo, un análisis de la morfología del cristal indicó que algunas condiciones podrían resultar mejores para la optimización de la microcristalización.
La Figura 4A muestra una caída de la pantalla PACT que fue ampliamente representativa de las observadas en la mayoría de la placa. A primera vista, puede ser tentador pensar que estos cristales podrían valer la pena optimizarlos aún más para la microcristalización. Los cristales son grandes y parece haber una nucleación significativa. Sin embargo, la morfología general del cristal no es ideal. En primer lugar, los cristales no son observablemente singletons, ya que parece que múltiples cristales están creciendo desde puntos de nucleación individuales. En segundo lugar, el tamaño del cristal es altamente asimétrico con un crecimiento que ocurre principalmente en un solo eje. Tales cristales son teóricamente más propensos a alinearse preferentemente cuando se entregan al haz de rayos X. Ambas características presentan problemas durante la recopilación y el procesamiento de datos cristalográficos en serie.
La figura 4B, sin embargo, muestra cristales de endotiapepsina cultivados en presencia de MgCl2. Esta morfología fue consistente en todas las condiciones que contenían MgCl 2 y, por lo tanto, sugirió que su morfología se debía a MgCl2. Las condiciones de MgCl2 produjeron cristales individuales, más en forma de caja que representaban un mejor objetivo para los experimentos en serie finales.
Había cuatro condiciones dentro de la pantalla PACT que contenían MgCl2. Para comprender mejor la influencia de todos los diferentes componentes de estas condiciones en la cristalización de endotiapepsina, se realizó una optimización aleatoria. Se creó una pantalla que contenía una combinación aleatoria de los tampones y precipitantes en un rango de concentraciones y pHs. La concentración de MgCl2 también se varió y luego las gotas resultantes se clasificaron arbitrariamente de 0 a 5 (0 sin cristales ni precipitación) en términos de su calidad visual de cristales y nivel de precipitación.
La Figura 5A muestra un mapa de calor de los resultados de un análisis de correlación de Pearson entre el nivel de precipitación y la calidad del cristal, y las variables de pantalla (ejemplos de las gotas de este experimento se muestran en las Figuras 5B, C y D). Los resultados indicaron que el pH de la solución estaba altamente correlacionado con el nivel de precipitación, con tampones alcalinos que resultaban en más precipitación. La concentración de MgCl2 se correlacionó ligeramente con el nivel de precipitación, al igual que el pH y la concentración de precipitante a la calidad del cristal.
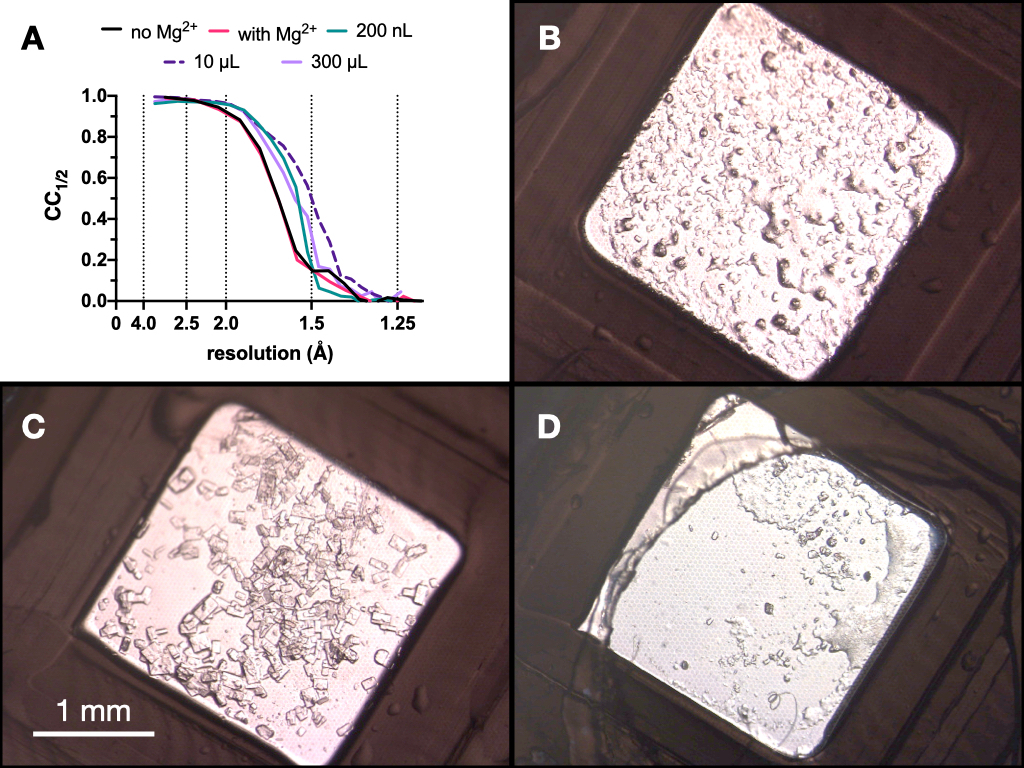
Sobre la base de estos resultados, se tomó la decisión de llevar los cristales cultivados en 0.1 M Tris-HCl pH 7.0, 0.15 M MgCl2, 20% (p/v) PEG 6,000 al siguiente paso del protocolo: Transición a lote. La morfología de los cristales fue aceptable y un análisis de la difracción de rayos X y las métricas de calidad de los datos de estos cristales sugirieron que no había diferencias significativas entre los cristales cultivados dentro y fuera de la presencia de Mg2+ (Figura 9).
Transición a lote
Para muchas optimizaciones de microcristalización de cristalografía en serie, el Paso 2 será el punto de partida. La proteína de interés ya habrá sido cristalizada para criocristalografía y el protocolo de cristalización ahora necesitará transformarse para crear lodos de microcristales. Este protocolo sólo ha utilizado placas de difusión de vapor de 96 pocillos para realizar la transformación a lote, ya que la difusión de vapor es el método de cristalización utilizado por el 95% de las entradas PDB26. El protocolo ha evitado pasar al microlote34,35,37 ya que esta transición aún podría incurrir en una optimización similar. Esto no quiere decir que este protocolo solo se pueda hacer en placas de difusión de vapor. Todos los pasos presentados, también funcionarían en microlotes si este fuera el método de cristalización original.
Para evaluar la cristalización de la endotiapepsina en la condición elegida, se creó un morfograma, o un diagrama de fase aproximada. El propósito del experimento del morfograma es triple. En primer lugar, un análisis del morfograma es de gran utilidad al evaluar las rutas de escala en el Paso 3 - Escala. En segundo lugar, el morfograma actúa como una herramienta de optimización, ayudando a descubrir las condiciones de difusión de vapor que dan lugar a cristales a través de lotes [es decir, cristales que aparecen rápidamente (< 24 h)]. En tercer lugar, si los cristales no han aparecido rápidamente, un análisis de las gotas sembradas puede dar al cristalógrafo una idea de la ubicación aproximada de la condición actual en el diagrama de fase. Por ejemplo, si las condiciones sembradas dan cristales pero los no sembrados no, es probable que esas condiciones estén en la región metaestable.
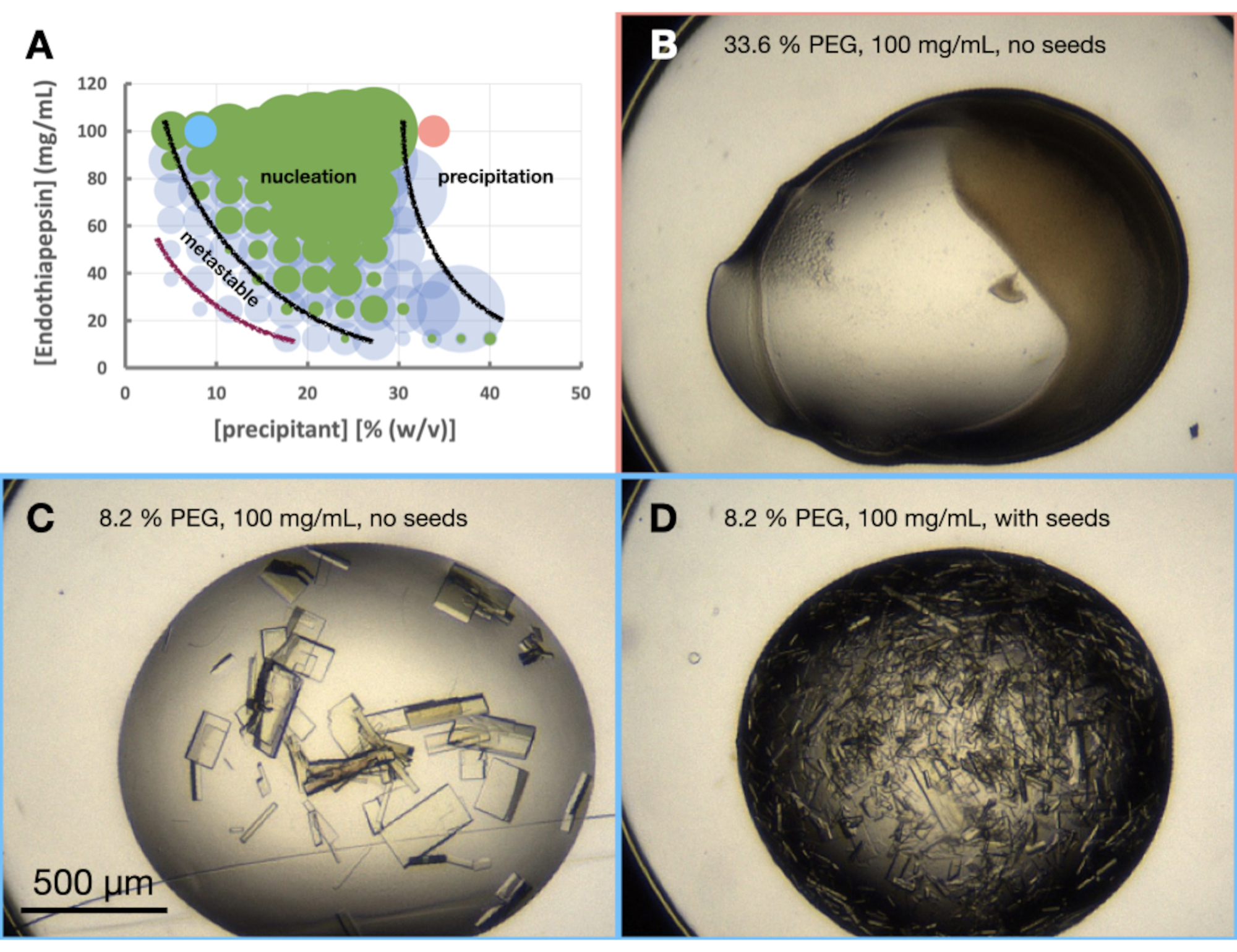
El experimento morfograma de endotiapepsina se realizó basado en la condición 0.1 M Tris-HCl pH 7.0, 0.15 M MgCl2, 20% (p/v) PEG 6,000. Las concentraciones de proteína y PEG variaron de 100 a 12,5 mg/mL y 5 a 40% (p/v), respectivamente. Las gotas se analizaron y los resultados se trazaron utilizando la hoja de trabajo proporcionada (Figura 6A).
También estaba claro desde la etapa de optimización de la morfología cristalina que el crecimiento de cristales de endotiapepsina en esta condición, y en estas concentraciones de proteínas, daría como resultado cristales cultivados en menos de 24 h. Esto indicaba que la cristalización se producía a través de un lote en lugar de un proceso impulsado por difusión de vapor. El cristal cultivado en estas condiciones era, por lo tanto, adecuado para escalar a volúmenes más grandes.
Si los cristales no hubieran sido visibles en las gotas sin semillas después de 24 h, entonces habría sido probable que la cristalización todavía dependiera de una transición (Figura 1B) y, por lo tanto, no del lote. En este caso, los resultados del experimento del morfograma siguen siendo de interés. Dan una indicación del punto de partida probable para la cristalización en el diagrama de fases y, por lo tanto, cómo debe proceder la optimización posterior. Mira las gotas sembradas. Las semillas permitirán el crecimiento de cristales en la región metaestable independientemente de la nucleación. Por ejemplo, si aparecen cristales dentro de las 24 h en las gotas sembradas pero no en las gotas sin sembrar, esto indica que se puede observar parte de la región metaestable. Si no se observan cristales en las gotas sembradas o no sembradas, todos los pozos permanecen subsaturados.
Escalada
Mirando el morfograma (Figura 6A), se podrían hacer varias observaciones. La cantidad de nucleación pareció verse afectada tanto por la proteína como por las concentraciones precipitantes. También hubo una demarcación muy clara de las gotas que conducen a la precipitación de proteínas, con gotas que contienen: nada, cristales o precipitado (Figura 6B). La adición de semillas (Figura 6D) también aumentó considerablemente Xn en comparación con las gotas sin semillas (Figura 6C). Tomando todos estos resultados juntos, se decidió intentar escalar tanto un protocolo de lote como un protocolo de lote sembrado al 30% (p/v) PEG 6,000 y 100 mg / ml de endotiapepsina.
El escalado de prueba inicial se realizó en placas de caída colgantes de 24 pocillos. Los volúmenes de gota se incrementaron gradualmente para que se pudiera observar cualquier cambio en el comportamiento de cristalización (Figura 7). Como se puede ver, tanto en las gotas sin semillas como en las sembradas se ha producido un crecimiento cristalino. Todas las gotas sin semillas crecieron en una gama de tamaños de cristal, pero predominantemente cristales grandes (100-200 μm - dimensión más larga). Las gotas sembradas, sin embargo, produjeron cristales más pequeños (5 - 50 μm - dimensión más larga). Estas pruebas iniciales sugirieron que se requeriría que las semillas disminuyeran Xs, pero también, que esta condición debería ser adecuada para volúmenes más grandes.
Cuando el volumen se incrementó en 200 μL, el volumen de cristalización se agitó continuamente durante el crecimiento del cristal. La razón principal de esta agitación fue asegurar que la solución de cristalización permanezca homogénea y que los cristales en crecimiento no se depositen en el fondo o los lados de los tubos. El asentamiento de cristales puede conducir a una población de cristales heterogéneos con cristales muy grandes y pequeños. La agitación de la solución de cristalización también puede promover la nucleación44,45.
Desafortunadamente, el 30% (p/v) PEG 6.000 sin sembrar no produjo cristales, por lo que la concentración de PEG se incrementó al 35% (p/v). Este aumento mejoró notablemente la cristalización, con un rango final de Xn y Xs de 3,6 ± 1,2 x 106 cristales·mL-1 y 42 ± 4,1 μm, respectivamente (Figura 8A y B - negro). Aunque fue una mejora significativa y una concentración de cristal aceptable, los cristales finales eran demasiado grandes para el experimento planeado, por lo que se llevaron a cabo optimizaciones adicionales. Para reducir el tamaño de los cristales finales se exploraron dos vías (Figura 1E): disminuir la concentración de proteínas para tratar de limitar el crecimiento final del cristal (Figura 8A y B - rosa caliente), y aumentar la concentración de PEG para tratar de aumentar la nucleación (Figura 8A y B - verde).
Desafortunadamente, la reducción de la concentración de proteína también redujo drásticamente elX n, que finalmente produjo cristales aún más grandes. El aumento de la concentración de PEG al 40% produjo un rango final de Xn y Xs de 3.1 ± 0.7 x 106 cristales·mL-1 y 39 ± 2.3 μm, respectivamente. Estos no fueron significativamente diferentes al 35%, pero dado que el tamaño final del cristal se redujo, esta condición se continuó con las optimizaciones adicionales.
Para aumentar elX n, se agregaron semillas. Esto aumentó dramáticamente el Xn (1.1 ± 1.8 x 10 8 cristales·mL-1) y condujo a un Xs más pequeño (4.2 ± 4.0 μm) (Figura 8A y B - púrpura discontinuo). Estos cristales, aunque muy adecuados para algunos experimentos de cristalografía en serie, se consideraron demasiado pequeños, por lo que se cambió la concentración de las semillas añadidas.
Sin embargo, este ajuste del stock de semillas añadido resultó difícil de repetir de manera confiable; Por lo tanto, se intentó el enfriamiento. Después de la adición de un stock de semillas, se monitoreó el tamaño del cristal y una vez que se logró un tamaño de cristal adecuado (aproximadamente 10 - 20 μm), se apagó la cristalización del lote (Figura 8C y D). El enfriamiento fue propuesto, con respecto a la microcristalización, en Kupitz et al. (2014)25. Aunque tal vez no sea un método ideal, ya que la solución de proteína finalmente se desperdiciará26, la técnica fue muy útil en esta situación ya que el crecimiento de cristales era difícil de controlar. La idea detrás del enfriamiento es devolver rápidamente la mezcla de cristalización a un punto justo por encima de la línea de solubilidad (Figura 1F). Una vez que la solución ha regresado a la línea de solubilidad, la solución ha regresado a una solución saturada estable y no se producirá ningún crecimiento adicional de cristales.
Intentar apagar una reacción de cristalización no está exento de riesgos. Si se agrega una solución de enfriamiento demasiado grande, la proteína en solución podría diluirse tanto que se pase la línea de solubilidad. En este caso, la solución se subsaturará y los cristales comenzarán a disolverse. Para evitar esto, es posible estimar la cantidad de solución de enfriamiento requerida en función de los resultados del morfograma. En el momento de enfriamiento, tome la concentración de la solución de proteína. Al comparar la concentración de proteína en la línea de solubilidad y la concentración de proteína en solución, se puede hacer una estimación de la dilución requerida.
La versión apagada del experimento de semilla diluida PEG 6,000 al 40% (p/v) dio una concentración final de cristal y un rango de tamaño de 2.6 ± 3.1 x 106 cristales·mL-1 y 15 ± 3.9 μm, respectivamente.
A lo largo de todo el proceso, las colecciones de datos de rayos X de prueba de los cristales de endotiapepsina se recolectaron en la línea de luz PXII de Swiss Light Source utilizando un foco de 10 x 30 μm, una energía de 12.4 keV atenuada en un 80%, y bajo condiciones criogénicas. Los datos fueron procesados usando diales y la Figura 9 muestra una comparación de CC1/2. No se observó ningún cambio dramático en CC1/2 en el transcurso de la optimización.

Figura 1: Una visión general de la cristalización transicional y por lotes, y métodos de escala mapeados en un diagrama de fase. R. Las zonas y límites del diagrama de fase de cristalización de proteínas arquetípicas. Las concentraciones de precipitante y proteína se representan en los ejes x e y , respectivamente, con el punto de agua pura en el origen. La línea púrpura indica el límite de sobresaturación de proteínas, y las zonas metaestables, de nucleación y precipitación se muestran en azul, verde y rosa, respectivamente. B. Un ejemplo de los límites de penetración de la zona de nucleación de un método de cristalización de "fase de transición", como la difusión de vapor. En este experimento teórico, las concentraciones de precipitante de caída y proteína comienzan justo debajo de la línea de solubilidad, aún no sobresaturadas. Mientras la gota se equilibra, las concentraciones de componentes de gota aumentan de tal manera que la gota se sobresatura y continúa moviéndose, o transición, hacia la zona de nucleación. Tras la nucleación cristalina, la concentración de proteína en solución comienza a disminuir. La concentración continúa disminuyendo a medida que los cristales crecen hasta que finalmente se detiene en la línea de solubilidad. La línea punteada azul marca un límite teórico de la transición a la zona de nucleación. Tan pronto como comience la nucleación, la concentración de proteína disminuirá, evitando una mayor penetración. C. Ejemplos de trayectorias de cristalización por lotes y lotes sembrados. En lote, la mezcla de la proteína y el precipitante debe crear una solución sobresaturada dentro de la zona de nucleación para que pueda ocurrir el crecimiento de cristales. En el lote sembrado, no es estrictamente necesario estar en la zona de nucleación debido a la adición de microsemillas, por lo que también se pueden explorar ubicaciones en la región metaestable. D. Una optimización hipotética del experimento de cristalización mostrado en B desde la difusión de vapor hasta el lote. El punto de partida de difusión de vapor original ha pasado, a través del vector de optimización resultante, a la nueva posición inicial; dentro de la zona de nucleación. El vector resultante es el producto de dos optimizaciones: un aumento en las concentraciones de proteínas y precipitantes. E. Ejemplos de optimizaciones al escalar condiciones de lote para adaptar los Xn y Xs finales. F. Apagar el experimento de cristalización mediante la adición de tampón de cristalización. Es esencial que el enfriamiento no saque la concentración de proteínas de la región metaestable y, por lo tanto, por debajo del punto de sobresaturación de proteínas. De lo contrario, los cristales comenzarán a disolverse nuevamente en solución. B. y C . han sido adaptados de Beale et al. (2019)26 con el permiso de los autores. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2: Aumento de Xn y disminuciónde X s. La relación idealizada entre el número de cristales producidos a partir de un experimento de cristalización y su dimensión media más larga. Para crear este gráfico, se utilizó la cristalización de una proteína modelo hipotética de 10 kDa. La proteína cristalizó a una concentración de 10 mg/mL y produjo cristales de P2 12 1 21 con dimensiones de 49x50x51 Å. Se suponía que cada evento de nucleación producía un cristal. Se asumió que el crecimiento de cristales era homogéneo en cada cara. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3: Un diagrama de flujo que muestra los pasos para optimizar un cristal cultivado en un experimento de difusión de vapor de pequeño volumen (<500 nL) en un experimento por lotes de gran volumen (> 100 μL). La optimización de cristales se divide en tres etapas: (1) Optimización de la morfología cristalina. (2) Transición a lote. (3) Escalado. En la Etapa 1 es importante identificar cristales adecuados para la microcristalización. Algunas proteínas solo están presentes en una morfología monocristalina independientemente de la condición de cristalización. Sin embargo, vale la pena buscar condiciones que den lugar a cristales individuales, en forma de cubo, o tan cerca de estos como sea humanamente posible. Los cristales individuales, en forma de cubo, hipotética y anecdóticamente, generalmente darán lugar a mejores resultados de los experimentos de cristalografía en serie. Una vez que se ha seleccionado una morfología cristalina y se ha confirmado la difracción, es necesario mover el experimento de cristalización de la difusión de vapor al lote (Etapa 2). Aquí, los cristales deben ser optimizados por su tiempo de nucleación. El objetivo es encontrar condiciones que produzcan cristales de aparición rápida (> 24 h), ya que es probable que estas condiciones lleguen a la zona de nucleación inmediatamente y, por lo tanto, sean por lotes. Una vez que se ha encontrado una condición en la zona de nucleación, se puede crear un morfograma. El morfograma permite mapear la mayor parte de la zona de nucleación y las posibles rutas de escalamiento identificadas para la Etapa 3. El volumen de una condición de lote identificada puede entonces escalarse gradual o rápidamente en tamaño para producir un volumen final de >100 μL. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 4: Un análisis de las condiciones de cristalización de endotiapepsina a partir de una pantalla de matriz dispersa PACT. A . y B. son fotos después de 24 h de los pozos A4 y C10, respectivamente, de la pantalla PACT . Los componentes del tampón de cristalización se resaltan en la figura. El tampón SPG es ácido succínico, fosfato de dihidrógeno de sodio y glicina mezclados en una proporción molar de 2: 7: 7. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 5: Un análisis de la optimización de la cristalización de endotiapepsina a partir de las condiciones PACT MgCl2. R. Un mapa de calor de los resultados de un análisis de correlación de Pearson entre el pH amortiguador, la concentración de MgCl2 y la concentración de precipitante y el nivel de precipitación y la calidad del cristal. El nivel de precipitación y la calidad del cristal se evaluaron arbitrariamente en una escala de 0-5 (siendo 0 sin cristales o precipitación) después de 24 h. a. C. y D. muestran ejemplos de cristalización y precipitación en tres gotas diferentes. También se muestran la condición de cristalización y las evaluaciones del nivel de precipitación y la calidad del cristal. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 6: Morfograma de endotiapepsina cristalizado en 0,1 M Tris-HCl pH 7,0, 0,15 M MgCl2 y PEG 6.000. Se proporciona un morfograma creado a partir de la hoja de cálculo "diagrama de fase-generador". El número relativo de cristales en cada gota se denota por el tamaño de los círculos, y los resultados de la gota 1 (proteína y precipitante) y la gota 2 (proteína, precipitante y semillas) se resaltan en verde y azul, respectivamente. Los valores de las concentraciones de proteína y precipitante, en los ejes x e y, respectivamente, denotan valores premezclados de cada uno en lugar de volúmenes finales. Con base en los resultados, se han dibujado líneas negras y una línea púrpura para mostrar los límites de la zona de nucleación y la zona metaestable, respectivamente. B. C. y D. muestran algunos resultados de ejemplo del experimento. Los puntos rojos y azules marcados en A. indican las ubicaciones de B. y C. y D., respectivamente. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 7: Ensayos iniciales de escalado de endotiapepsina en placas colgantes de 24 pocillos. Se utilizaron las mismas concentraciones de proteína y precipitante para todos los ensayos: 100 mg/ml de endotiapepsina en 0,1 M de acetato de Na pH 4,6 y 0,1 M Tris-HCl pH 7,0, 0,15 M MgCl2 y 30% (p/v) PEG 6.000, respectivamente. Todas las imágenes mostradas se tomaron después de 24 h y los volúmenes de gota final están etiquetados en cada imagen. El panel izquierdo (A, D y G) es una mezcla 1:1 de proteína y precipitante, el panel central (B, E y H) es una mezcla 1:2:3 de semillas, precipitante y proteína y el panel derecho (C, F e I) son imágenes ampliadas del panel central. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 8: Análisis de la microcristalización de endotiapepsina en volúmenes de 200-300 μL. A. y C. muestran cómo Xn cambió durante el tiempo del experimento. B. y D. muestran cómoX s (dimensión más larga) cambió con el tiempo. Los resultados de los experimentos se han separado para mayor claridad. La línea punteada roja en C. y D. muestra el punto en el que se realizó el enfriamiento. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 9: Resultados CC1/2 e imágenes de cristales obtenidos en cada etapa del proceso de microcristalización para evaluar la calidad de la difracción. A. CC1/2 trazado contra la resolución de los datos recogidos de cristales cultivados: con y sin Mg - parte de la optimización de la Etapa 1, en un volumen de 200 nL, un volumen de 10 μL y el volumen final de 300 μL. B.C. y D. muestran los cristales del volumen de 200 nL, 10 μL y 300 μL, respectivamente. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
| Información sobre proteínas | |
| Proteína | Endotiapepsina |
| Peso molecular (kDa) | 33.8 |
| Grupo espacial | P12 11 |
| a, b, c (Å) | 45.2, 73.3, 52.7 |
| α, β, ɣ (°) | 90.0, 109.2, 90.0 |
| Parámetros de destino fijo | |
| Volumen cargado por chip (μL) | 150 |
| Aperturas por chip | 25,600 |
| Concentración de cristal requerida (cristales/ml) | 500,000 |
| Información de muestra | |
| Masa proteica utilizada para producir 200 μL de muestra (mg) | 10 |
| Dimensión más larga del cristal (μm) | 15 |
| Concentración de cristales (cristales/ml) | 2,500,000 |
| Variables experimentales | |
| Número de puntos de tiempo requeridos | 5 |
| Número de imágenes requeridas por punto de tiempo | 50,000 |
| Tasa de aciertos (patrones integrados/imágenes recopiladas) | 0.3 |
| Objetivos fijos requeridos por punto de tiempo (redondeado al alza) | 7 |
| Requisitos de la muestra | |
| Volumen de muestra requerido por punto de tiempo (μL) | 1,050 |
| Volumen total de muestra requerido para el experimento (ml) | 5.25 |
| Masa total de proteína requerida (mg) | 52.5 |
Tabla 1: Un ejemplo de los requisitos de muestra para un experimento hipotético de bomba-sonda óptica realizado utilizando objetivos fijos. La proteína utilizada en este experimento teórico fue la endotiapepsina. Los parámetros de objetivo fijo se basaron en experimentos reportados en Ebrahim et al. (2019)48 y Davy et al. (2019)49. La información de la muestra provino del protocolo reportado en este artículo de video y las variables experimentales fueron estimaciones conservadoras basadas en la experiencia vivida. Los siguientes requisitos de muestra se calcularon posteriormente dadas las hipótesis anteriores.
Discusión
El método presentado muestra cómo optimizar la cristalización de endotiapepsina de cristales grandes (≥ dimensión más larga de 100 μm), cultivados en pantallas de 96 pocillos de matriz dispersa, a microcristales, cultivados en tubos de centrífuga (volumen de 300 μL) por lote. La idea detrás del protocolo es que los pasos tomados para optimizar la endotiapepsina también podrían usarse para otras proteínas. En última instancia, respondiendo al problema de crear grandes volúmenes (>100 μL) de microcristales (10-20 μm) para experimentos de cristalografía en serie en XFELs y sincrotrones.
El protocolo divide la tarea de microcristalización de gran volumen en tres pasos: (1) Optimización de la morfología del cristal, (2) Transición a lote y (3) Escalado. En el Paso 1, la gama de formas cristalinas que una proteína puede crear debe explorarse en placas de difusión de vapor. Las condiciones que dan lugar a cristales individuales, en forma de caja, que se difractan a la resolución requerida deben ser el objetivo. En el paso 2, las condiciones seleccionadas se pueden transformar de difusión de vapor en lote. Aquí, el criterio de optimización es el tiempo de crecimiento del cristal y encontrar condiciones que den lugar a cristales de proteínas dentro de las 24 h. También se puede trazar un morfograma dando al experimentador una idea de la ubicación de la línea de solubilidad y los límites de la zona de nucleación. Este morfograma es de gran utilidad en el Paso 3, Escala. El morfograma dará una indicación de si la nucleación sola puede aumentar Xn y reducir Xs. A medida que aumenta el volumen del experimento, Xn y Xs pueden evaluarse continuamente como los criterios clave para escalar el éxito.
En el caso de la endotiapepsina, el Paso 1 desenterró lo que potencialmente era una morfología cristalina previamente desconocida para la endotiapepsina. Esta morfología tenía el mismo grupo espacial que los reportados anteriormente, pero, lo que es importante para la cristalografía en serie, una forma más parecida a una caja. Los cristales individuales también parecían crecer a partir de puntos de nucleación individuales, a diferencia de los ventiladores creados a partir de otras condiciones (Figura 4). Para la condición seleccionada, el Paso 2 ya estaba parcialmente satisfecho ya que el crecimiento del cristal ocurrió en < 24 h. El morfograma indicó que tanto un protocolo de lote recto como de lote sembrado podría tener éxito al escalar en el paso 3. La escala inicial en lote recto, creó una condición que produjo cristales con un rango Xn y Xs de 3.6 ± 1.2 x 106 cristales·mL-1 y 42 ± 4.1 μm, respectivamente. Estos cristales, aunque aceptables para algunos experimentos de cristalografía en serie, se consideraron demasiado grandes. Por lo tanto, se realizaron optimizaciones adicionales. El protocolo final produjo cristales con un rango de concentración y tamaño de 3,1 x 106 cristales·mL-1 y 15 ± 3,9 μm, respectivamente. Esto era más que ideal para los experimentos planeados.
El método se centra en la transformación de cristales de proteínas "solubles" cultivados en placas de difusión de vapor en lotes. La razón de este enfoque es que la gran mayoría de los cristales de proteínas solubles se cultivan a través de la difusión de vapor26. Sin embargo, los conceptos presentados también podrían aplicarse a cristales de proteínas solubles cultivados utilizando otros métodos, como el microlote. Los conceptos también pueden ser aplicables a los cristales de proteína de membrana cultivados en LCP; ya que esto también es un proceso de cristalización por lotes.
Un aspecto clave del protocolo es el proceso de transformación de las condiciones de los cristales cultivados en placas de difusión de vapor de tal manera que puedan cultivarse en lotes. Para esta transformación, el método utiliza el criterio propuesto por Beale et al. (2019)26. Los cristales cultivados a través de un proceso discontinuo, incluso en placas de difusión de vapor, se formarán rápidamente (< 24 h). Este criterio es una aproximación basada en la velocidad del equilibrio de la gota de difusión de vapor, y es más cierto para condiciones precipitantes basadas en PEG. Sin embargo, las condiciones de cristalización contendrán una amplia variedad de compuestos que influirán en el tiempo de equilibrio. El equilibrio de las condiciones de cristalización a base de sal, por ejemplo, cloruro de amonio altamente concentrado, puede ocurrir en 1-2 días. Por lo tanto, el criterio de 24 h puede no ser cierto para las condiciones a base de sal. Las condiciones basadas en sal también pueden tener diagramas de fase más complejos26,30 que pueden no ajustarse al arquetipo presentado en este protocolo. Puede ser necesario reducir el criterio de tiempo para las condiciones basadas en la sal a 12 o 6 h si resulta imposible escalar a volúmenes más grandes.
Otra limitación de este método es su aparente complejidad. El protocolo que se siguió para optimizar la microcristalización de la endotiapepsina en realidad cambió relativamente poco la condición original de la pantalla de matriz dispersa. El primer impacto observado en la pantalla PACT fue 0.1 HEPES pH 7.0, 0.2 M MgCl2 y 20% (p/v) PEG 6,000. El tampón de cristalización a escala final fue 0.1 Tris-HCl pH 7.0, 0.15 M MgCl2 y 40% (p/v) PEG 6,000. También es muy posible que el cambio en el tampón de HEPES a Tris-HCl, y la concentración de MgCl2 , contribuyeran poco al éxito del proceso. Dejando el aumento en la concentración de PEG 6,000 como la única optimización, y una que podría haberse logrado de manera bastante simple.
Esta evaluación, sin embargo, también es demasiado simplista. No solo descarta los problemas encontrados durante el escalado (es decir, el uso de semillas y el enfriamiento), sino también el hecho de que solo porque esta proteína resultó sencilla, no hay garantía de que la siguiente también lo sea. Los pasos aconsejados en el protocolo, fueron ideados porque optimizar el escalado de los volúmenes de cristalización de proteínas puede ser muy costoso en proteínas. Durante los siete ensayos de raspado de endotiapepsina que se muestran, se consumieron 100 mg de proteína. Es cierto que algunos de estos pasos se realizaron para mostrar sus consecuencias a la luz de este protocolo. Aun así, 100 mg de una proteína, más potencialmente otros 50 mg para la proteína consumida durante un experimento (Tabla 1), puede ser una inversión significativa en tiempo o dinero.
Afortunadamente, no está claro que esta masa de muestra requerida sea ubicua en todas las proteínas. La endotiapepsina era altamente soluble y, por lo tanto, requería una gran concentración de proteínas para alcanzar la sobresaturación. En otros (actualmente en optimización), la sobresaturación se puede alcanzar a 10 o incluso 5 mg / ml. Tales variables son específicas de proteínas y deben ser aceptadas cuando aparecen.
Otras limitaciones del método incluyen su dependencia de equipos complejos, como robots de manejo de líquidos para la creación de pantallas y placas, y generadores de imágenes para obtener imágenes automáticas de placas cuando sea necesario. Se han ofrecido rutinas alternativas para limitar la necesidad de algunos de estos equipos, pero el protocolo llevará más tiempo seguirlo sin ellas. El protocolo también sugiere probar la difracción de cristales optimizados. Para los cristalógrafos sin acceso regular a un sincrotrón, estas pruebas podrían resultar desafiantes. Los controles en cada paso pueden no ser necesarios, pero estas pruebas se recomiendan encarecidamente una vez que se ha identificado un golpe y antes y después del escalado. Los cristales no difractores en un XFEL no son, desafortunadamente, una ocurrencia infrecuente. Dado esto, es mejor errar por el lado de la precaución con respecto a las suposiciones sobre la difracción de cristales.
En última instancia, este protocolo y los resultados presentados aquí ofrecerán una guía, ideas y un ejemplo para aquellos que luchan con la producción de muestras para experimentos de cristalografía en serie. Con suerte, a medida que la cristalografía en serie se desarrolle aún más, las demandas de muestra de la técnica se reducirán de tal manera que se reducirá la necesidad de protocolos como este. Sin embargo, incluso en este caso, las estrategias presentadas aquí seguirán siendo útiles para aquellos que deseen explorar el espacio de cristalización de su proteína.
Divulgaciones
Los autores no tienen conflictos de intereses que revelar.
Agradecimientos
Este proyecto ha recibido financiación del programa de investigación e innovación Horizonte 2020 de la Unión Europea en virtud del acuerdo de subvención Marie Skłodowska-Curie nº 701647. Muchas gracias por la asistencia y el apoyo de los científicos de la línea de luz suiza X10SA-PXII.
Materiales
| Name | Company | Catalog Number | Comments |
| Swissci 96-well 2-Drop plates | Molecular Dimensions | MD11-002 | 96-well 2-drop crystallisation plate |
| Swissci 96-well 3-Drop plates | Molecular Dimensions | MD11-003 | 96-well 3-drop crystallisation plate |
| mosquito LCP liquid handling robot | sptlabtech | mosquito LCP | Crystallisation robot |
| ClearVue Sheets | Molecular Dimensions | MD6-015 | 96-well crystallization plate seals |
| Safe-Tube 1.5 mL | Eppendorf | 30120086 | 1.5 mL centrifuge tubes |
| Scaple | Swan and Morton | No. 3 scalple and No. 3 handle | Scalple for cutting open plate seals |
| MS 3 Vortex | IKA | 3319000 | Vortex for mixing solution and making seed stocks |
| 24-well XRL Plate | Molecular Dimensions | MD3-11 | 24-well hanging-drop plates |
| Tube revolver/rotator | Thermo Fischer Scientific | 88881001 | Tube revolver for mixing solution during scaling |
| Eppendorf Research plus pipettes | Eppendorf | Range of manual pipettes, 0.5-10, 1-20, 10-100, 100-1000 µL | |
| Eppendorf pipette tips | Eppendorf | Range of tip sizes for manual pipettes | |
| Suparen 600 | Prochem AG | Suparen 600 | Endothiapepsin solution |
| Sodium Acetate | Sigma-Aldrich | 241245-1KG | Sodium Acetate |
| Tris | Merck | 8382T014 | Tris |
| Magnesium Chloride | Sigma-Aldrich | M2670-1kg | Magnesium Chloride |
| PEG 6,000 | Sigma-Aldrich | 81255-1kg | PEG 6,000 |
| Ethelyene glycol | Sigma-Aldrich | 324558-1L | Ethelyene glycol for cyro-protecting the crystals |
| PACT Premier HT screen | Molecular Dimensions | MD1-36 | PACT Premier 96-well crystal screen |
| DOW CORNING high vacuum grease | Molecular Dimensions | MD6-02 | Grease for sealing 24-well plates |
| Hirschmann 22 x 22 mm glaser cover slides | Hirschmann | 8000104 | Cover slides for sealing 24-well sitting drop plates |
| Crystal pins | PSI | Manufactured inhouse | Thin-film supports for micro-crystals. |
| 1-1.3 mm SiLibeads Type S | Faust | 6239547 | Glass beads for making mico-seed stocks |
| Macbook Pro | Apple | Macbook Pro | Computer for performing data analysis |
| CCP4 software suite | CCP4 | Diffraction pattern data processing software | |
| Excel | Microsoft | Microsoft Office | Plotting tool for phase diagram |
| Hausser Scientific Bright-Line counting chamber | Thermo Fischer Scientific | 02-671-51B | Tool to calculate crystal concentration |
| PACT Premier | Molecular Dimensions | MD1-29-ECO | Sparse-matrix crystallization screen |
| Rock Imager | Formulatrix | Rock Imager | Temperature controlled crystal plate storage and imager |
| Rock MakerWeb | Formulatrix | Rock MakerWeb | Crystal plate creation and image storage stoftware |
| Formulator | Formulatrix | Formulator | 96-well crystal screen creation liquid handling robot |
| Leica MZ16 Microscope | Leica | Leica MZ16 | Light microscope |
| LAS V4.6 | Leica | LAS V4.6 | Software for Leica microscopes |
| Spectra/Por 3.5 kDa dialysis tubing | Spectrumlabs | Spectra/Por 3 Dialysis Membrane | 3.5 kDa dialysis membrane |
| Dialysis tubing closures | Spectrumlabs | Spectra/Por 3 Duniversal Closures | Clips to seal the dialysis tubing ends |
| Amicon 10 kDa centrifugal concentrator | Merck-Millipore | Amicon Ultra-15 10 kDa centrifugal concentrator | 10 kDa centrifugal filter |
| 5810 R swing bucket centrifuge | Eppendorf | 5810 R Centrifuge | Swing bucket centrifuge |
Referencias
- DePonte, D. P., et al. Gas dynamic virtual nozzle for generation of microscopic droplet streams. Journal of Physics D: Applied Physics. 41 (19), 195505 (2008).
- Hunter, M. S., et al. Fixed-target protein serial microcrystallography with an x-ray free electron laser. Scientific Reports. 4 (1), 6026 (2014).
- Weierstall, U., et al. Lipidic cubic phase injector facilitates membrane protein serial femtosecond crystallography. Nature Communications. 5 (1), 1-6 (2014).
- Roessler, C. G. G., et al. Acoustic Injectors for Drop-On-Demand Serial Femtosecond Crystallography. Structure. 24 (4), 631-640 (2016).
- Sherrell, D. A., et al. A modular and compact portable mini-endstation for high-precision, high-speed fixed target serial crystallography at FEL and synchrotron sources. Journal of Synchrotron Radiation. 22 (6), 1372-1378 (2015).
- Roedig, P., et al. A micro-patterned silicon chip as sample holder for macromolecular crystallography experiments with minimal background scattering. Scientific Reports. 5 (1), 1-11 (2015).
- Botha, S., et al. Room-temperature serial crystallography at synchrotron X-ray sources using slowly flowing free-standing high-viscosity microstreams. Acta Crystallographica Section D Biological Crystallography. 71 (2), 387-397 (2015).
- Weinert, T., et al. Serial millisecond crystallography for routine room-temperature structure determination at synchrotrons. Nature Communications. 8 (1), 542 (2017).
- Tenboer, J., et al. Time-resolved serial crystallography captures high-resolution intermediates of photoactive yellow protein. Science. 346 (6214), 1242-1246 (2014).
- Nango, E., et al. A three-dimensionalmovie of structural changes in bacteriorhodopsin. Science. 354 (6319), 1552-1557 (2016).
- Suga, M., et al. Light-induced structural changes and the site of O=O bond formation in PSII caught by XFEL. Nature. 543 (7643), 131-135 (2017).
- Mehrabi, P., et al. Liquid application method for time-resolved analyses by serial synchrotron crystallography. Nature Methods. 16 (10), 979-982 (2019).
- Halle, B. Biomolecular cryocrystallography: structural changes during flash-cooling. Proceedings of the National Academy of Sciences of the United States of America. 101 (14), 4793-4798 (2004).
- Fraser, J. S., et al. Hidden alternative structures of proline isomerase essential for catalysis. Nature. 462 (7273), 669-673 (2009).
- Fenwick, R. B., van den Bedem, H., Fraser, J. S., Wright, P. E. Integrated description of protein dynamics from room-temperature X-ray crystallography and NMR. Proceedings of the National Academy of Sciences of the United States of America. 111 (4), 445-454 (2014).
- Keedy, D. A., et al. Mapping the conformational landscape of a dynamic enzyme by multitemperature and XFEL crystallography. Elife. 4, (2015).
- Thomaston, J. L., et al. XFEL structures of the influenza M2 proton channel: Room temperature water networks and insights into proton conduction. Proceedings of the National Academy of Sciences of the United States of America. 114 (51), 13357-13362 (2017).
- Haas, D. J., Rossmann, M. G. Crystallographic studies on lactate dehydrogenase at -75 °C. Acta Crystallographica Section B Structural Crystallography and Crystal Chemistry. 26 (7), 998-1004 (1970).
- Hope, H. Cryocrystallography of biological macromolecules: a generally applicable method. Acta Crystallographica Section B Structural Science. 44 (1), 22-26 (1988).
- Wu, W., et al. Batch crystallization of rhodopsin for structural dynamics using an X-ray free-electron laser. Acta Crystallographica Section:F Structural Biology Communications. 71 (7), 856-860 (2015).
- Ishchenko, A., Cherezov, V., Liu, W. Preparation and delivery of protein microcrystals in lipidic cubic phase for serial femtosecond crystallography. Journal of Visualized Experiments. (115), e54463 (2016).
- Andersson, R., et al. Well-based crystallization of lipidic cubic phase microcrystals for serial X-ray crystallography experiments. Acta Crystallographica Section D: Structural Biology. 75 (10), 937-946 (2019).
- Luft, J. R., et al. The detection and subsequent volume optimization of biological nanocrystals. Structural Dynamics. 2 (4), 041710 (2015).
- Lee, D. B., et al. Supersaturation-controlled microcrystallization and visualization analysis for serial femtosecond crystallography. Scientific Reports. 8 (1), 1-10 (2018).
- Kupitz, C., et al. Microcrystallization techniques for serial femtosecond crystallography using photosystem II from Thermosynechococcus elongatus as a model system. Philosophical transactions of the Royal Society of London. Series B, Biological Sciences. 369 (1647), 20130316 (2014).
- Beale, J. H., et al. Successful sample preparation for serial crystallography experiments. Journal of Applied Crystallography. 52, 1385-1396 (2019).
- Rayment, I. Small-scale batch crystallization of proteins revisited: An underutilized way to grow large protein crystals. Structure. 10 (2), 147-151 (2002).
- García-Ruiz, J. M. Nucleation of protein crystals. Journal of Structural Biology. 142 (1), 22-31 (2003).
- McPherson, A., Kuznetsov, Y. G. Mechanisms, kinetics, impurities and defects: Consequences in macromolecular crystallization. Acta Crystallographica Section F:Structural Biology Communications. 70 (4), 384-403 (2014).
- Rupp, B. Origin and use of crystallization phase diagrams. Acta crystallographica. Section F, Structural biology communications. 71, 247-260 (2015).
- Luft, J. R., DeTitta, G. T. A method to produce microseed stock for use in the crystallization of biological macromolecules. Acta Crystallographica Section D Biological Crystallography. 55 (5), 988-993 (1999).
- Ireton, G. C., Stoddard, B. L. Microseed matrix screening to improve crystals of yeast cytosine deaminase. Acta crystallographica. Section D, Biological crystallography. 60 (3), 601-605 (2004).
- Forsythe, E. L., Maxwell, D. L., Pusey, M. Vapor diffusion, nucleation rates and the reservoir to crystallization volume ratio. Acta Crystallographica Section D Biological Crystallography. 58 (10), 1601-1605 (2002).
- Chayen, N. E., Shaw Stewart, P. D., Maeder, D. L., Blow, D. M. IUCr An automated system for micro-batch protein crystallization and screening. Journal of Applied Crystallography. 23 (4), 297-302 (1990).
- Chayen, N. E., Shaw Stewart, P. D., Blow, D. M. Microbatch crystallization under oil - a new technique allowing many small-volume crystallization trials. Journal of Crystal Growth. 122 (1-4), 176-180 (1992).
- Chayen, N. E. Comparative studies of protein crystallization by vapour-diffusion and microbatch techniques. Acta Crystallographica Section D: Biological Crystallography. 54 (1), 8-15 (1998).
- D'Arcy, A., Mac Sweeney, A., Stihle, M., Haber, A. The advantages of using a modified microbatch method for rapid screening of protein crystallization conditions. Acta Crystallographica - Section D Biological Crystallography. 59 (2), 396-399 (2003).
- Darmanin, C., et al. Protein crystal screening and characterization for serial femtosecond nanocrystallography. Scientific Reports. 6, 25345 (2016).
- Gati, C., et al. Atomic structure of granulin determined from native nanocrystalline granulovirus using an X-ray free-electron laser. Proceedings of the National Academy of Sciences of the United States of America. 114 (9), 2247-2252 (2017).
- Cheng, R. K. Y. Towards an optimal sample delivery method for serial crystallography at XFEL. Crystals. 10 (3), 215 (2020).
- Schmidt, M. Mix and Inject: Reaction Initiation by Diffusion for Time-Resolved Macromolecular Crystallography. Advances in Condensed Matter Physics. 2013, 1-10 (2013).
- Oberthuer, D., et al. Double-flow focused liquid injector for efficient serial femtosecond crystallography. Scientific Reports. 7 (1), 44628 (2017).
- McPherson, A., Gavira, J. A. Introduction to protein crystallization. Acta crystallographica. Section F, Structural biology communications. 70, 2-20 (2014).
- Yaoi, M., et al. Effect of stirring method on protein crystallization. Japanese Journal of Applied Physics, Part 2: Letters. 43 (10), 1318 (2004).
- Castro, F., Ferreira, A., Teixeira, J. J., Rocha, F. Influence of Mixing Intensity on Lysozyme Crystallization in a Meso Oscillatory Flow Reactor. Crystal Growth & Design. 18 (10), 5940-5946 (2018).
- Moews, P. C., Bunn, C. W. An X-ray crystallographic study of the rennin-like enzyme of Endothia parasitica. Journal of Molecular Biology. 54 (2), 395-397 (1970).
- Newman, J., et al. Towards rationalization of crystallization screening for small- to medium-sized academic laboratories: the PACT/JCSG+ strategy. Acta crystallographica. Section D, Biological. 61, 1426-1431 (2005).
- Ebrahim, A., et al. Resolving polymorphs and radiation-driven effects in microcrystals using fixed-target serial synchrotron crystallography. Acta Crystallographica Section D Structural Biology. 75, 151-159 (2019).
- Davy, B., et al. Reducing sample consumption for serial crystallography using acoustic drop ejection. Journal of Synchrotron Radiation. 26 (5), (2019).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados