
Method Article
Séquençage de nouvelle génération d’ARN et pipeline de bioinformatique pour identifier les lignes exprimées au niveau spécifique au locus
Dans cet article
Résumé
Ici, nous présentons une approche et des analyses bioinformatiques pour identifier l’expression LINE-1 au niveau spécifique du locus.
Résumé
Les éléments longs INterspersed-1 (LINEs/N1) sont des éléments répétitifs qui peuvent être copiés et insérer aléatoirement dans le génome, ce qui entraîne une instabilité génomique et une mutagenèse. La compréhension des schémas d’expression des loci L1 au niveau individuel prêtera à la compréhension de la biologie de cet élément mutagène. Cet élément autonome constitue une portion importante du génome humain avec plus de 500 000 exemplaires, bien que 99% soient tronqués et défectueux. Cependant, leur abondance et leur nombre dominant de copies défectueuses rendent difficile l’identification authentiquement exprimée N1 des séquences liées à la L1 exprimées dans d’autres gènes. Il est également difficile d’identifier quel locus L1 spécifique est exprimé en raison de la nature répétitive des éléments. En surmontant ces défis, nous présentons une approche bioinformatique de l’ARN-SEQ pour identifier l’expression L1 au niveau spécifique du locus. En résumé, nous recueillons l’ARN cytoplasmique, sélectionnons les transcriptions polyadénylées et utilisons des analyses ARN-SEQ spécifiques aux brins pour cartographier de façon unique les lectures sur des loci L1 dans le génome de référence humain. Nous avons visuellement curé chaque locus L1 avec des lectures mappées de façon unique pour confirmer la transcription de son propre promoteur et ajuster les lectures de transcription mappées pour tenir compte de la mappabilité de chaque locus L1 individuel. Cette approche a été appliquée à une lignée de cellules tumorales de la prostate, DU145, pour démontrer la capacité de ce protocole à détecter l’expression à partir d’un petit nombre d’éléments L1 pleine longueur.
Introduction
Les rétrotransposons sont des éléments d’ADN répétitifs qui peuvent «sauter» dans le génome dans un mécanisme de copier-coller via des intermédiaires d’ARN. Un sous-ensemble de rétrotransposons est connu sous le nom de long INterspersed elements-1 (LINEs/N1) et constitue un sixième du génome humain avec plus de 500, 0000 copies1. En dépit de leur abondance, la plupart de ces copies sont défectueuses et tronquées avec seulement une estimation de 80-120 éléments L1 considérés comme actifs2. Une L1 pleine longueur est d’environ 6 Ko de longueur avec 5 'et 3 'régions non traduites, un promoteur interne et associé anti-sens promoteur, deux non-chevauchement des cadres de lecture ouverte (ORFS), et un signal et la queue polyA3,4,5 . Chez l’homme, les N1 sont des sous-familles distinguées par l’âge évolutionnaire avec les familles plus âgées ayant accumulé des mutations de séquences plus uniques au fil du temps par rapport à la plus jeune sous-famille, L1HS6,7. Les N1 sont les seuls rétrotransposons autonomes et humains et leurs ORFs encodent une transcriptase inverse, une endonucléase et des RNPs avec des activités de liaison à l’ARN et de chaperon requises pour rétrotransposer et insérer dans le génome dans un processus dénommé «cible-amorcée» transcription inversée8,9,10,11,12.
La rétrotransposition de N1 a été signalée comme causant des maladies germinales humaines par divers mécanismes, dont la mutagenèse insertionnelle, les suppressions de sites cibles et les réarrangements13,14,15, 16. récemment, il a été émis l’hypothèse que N1 peut jouer un rôle dans l’oncogenèse et/ou la progression tumorale comme l’augmentation de l’expression et des événements d’insertion de cet élément mutagène ont été observés dans une variété de cancers épithéliaux17,18 . On estime qu’il y a une nouvelle insertion L1 dans chaque 200 naissances19. Par conséquent, il est impératif de mieux comprendre la biologie de l’expression active N1. La nature répétitive et l’abondance des copies défectueuses trouvées dans les transcriptions d’autres gènes ont rendu ce niveau d’analyse difficile.
Heureusement, avec l’avènement des technologies de séquençage à haut débit, des progrès ont été faits pour analyser et identifier authentiquement exprimant N1 au niveau de locus-specific. Il existe différentes philosophies sur la façon de mieux identifier exprimée N1 en utilisant l’ARN séquençage de prochaine génération. Il n’y a eu que deux approches raisonnables suggérées pour cartographier les transcriptions L1 au niveau spécifique au locus. On ne se concentre que sur la transcription potentielle qui se lit à travers le signal de polyadénylation L1 et dans les séquences flanquant20. Notre approche tire parti des petites différences de séquence entre les éléments L1 et seulement les cartes que ces ARN-SEQ lisent qui mappe de façon unique à un locus21. Ces deux méthodes ont des limites en termes de quantification des niveaux de transcription. La quantification peut être améliorée potentiellement en ajoutant une correction pour la «mappabilité unique» de chaque locus L121, ou en utilisant des algorithmes plus complexes qui redistribuent les lectures multimappées qui ne peuvent pas être mappées de manière unique à un locus spécifique22. Ici, nous détaillons de manière étape par étape l’extraction d’ARN et le protocole de séquençage et de bioinformatique de nouvelle génération pour identifier les éléments L1 exprimés au niveau spécifique au locus. Notre approche profite au maximum de notre connaissance de la biologie des éléments fonctionnels L1. Cela inclut la connaissance que les éléments fonctionnels L1 doivent être générés à partir du promoteur L1, initié au début de l’élément L1, doivent être traduits dans le cytoplasme et que leurs transcriptions doivent être co-linéaires avec le génome. Brièvement, nous recueillons l’ARN frais, cytoplasmique, sélectionnons pour les transcriptions polyadénylées, et utilisons des analyses ARN-SEQ spécifiques aux brins pour cartographier de façon unique les lectures sur les loci L1 dans le génome de référence humain. Ces lectures alignées requièrent alors encore une grande conservation manuelle pour déterminer si les lectures de transcription proviennent du promoteur L1 avant de désigner un locus comme un L1 authentiquement exprimé. Nous appliquons cette approche sur l’échantillon de la lignée de cellules tumorales de la prostate DU145 pour démontrer comment il identifie un relativement peu de membres de L1 transcrits activement à partir de la masse des copies inactives.
Protocole
1. extraction de l’ARN cytoplasmique
- Obtenez des cellules via les méthodes suivantes.
- Recueillir des cellules vivantes de 2,75% – 100% confluent, T-75 flacons.
- Laver le flacon 2 fois dans 5 mL de PBS froid, et dans le dernier lavage, gratter les cellules et les transférer dans un tube conique de 15 mL. Centrifugez pendant 2 min à 1 000 x g et 4 ° c, puis enlevez et jetez délicatement le surnageant (table des matières).
- Collecter des cellules de spécimens de tissus.
- Préparez le tissu pour l’extraction de l’ARN cytoplasmique dans une heure d’être disséqué et toujours garder sur la glace. Pour le stockage à long terme, utilisez des solutions d’inhibiteur d’ARN pour stocker le tissu jusqu’à 72 heures après la dissection suivant le protocole du fabricant (table des matières).
- Découpez un échantillon de 10 μM3 et homogénéisez l’échantillon frais avec 5 ml de PBS froid dans un homogénéisateur de dounce stérile, transférez-le dans un tube conique de 15 ml, Centrifugez pendant 2 min à 1 000 x g à 4 ° c, et enlevez et jetez délicatement le surnageant (table des matières < /C8 >).
- Recueillir des cellules vivantes de 2,75% – 100% confluent, T-75 flacons.
- Ajouter 2 mL de tampon de lyse au mélange de granulés cellulaires et incuber sur la glace pendant 5 min.
- Préparer le tampon de lyse frais avec 150 mM de NaCl, 50 mM de HEPES (pH 7,4) et 25 μg/mL de digitonine (table des matières).
- Comme la concentration minimale de digitonine dans le tampon de lyse nécessaire pour pénétrer la membrane plasmatique peut varier selon le type de cellule, confirmer au microscope que les cellules traitées avec le tampon de lyse perdent la membrane plasmatique et conservent la membrane nucléaire intacte.
- Juste avant l’utilisation, ajouter 1 000 U/mL inhibiteur de RNase (table des matières).
- Centrifugez pendant 1 min à 1 000 x g et 4 ° c, et récupérez le surnageant.
- Ajouter le surnageant au pré-réfrigéré 7,5 mL de Trizol et 1,5 mL de chloroforme. Toutes les étapes nécessitant le chloroforme doivent être faites à l’intérieur d’une hotte chimique propre (tableau des matériaux).
- Centrifuger pour 35 min à 3 220 x g et 4 ° c.
- Transférer la portion aqueuse (couche supérieure) dans un tube frais pré-refroidi de 15 mL.
- Ajouter 4,5 mL de chloroforme et de Vortex.
- Centrifuger pendant 10 min à 3 220 x g et 4 ° c.
- Transférer la portion aqueuse dans un tube frais pré-refroidi.
- Ajouter 4,5 mL d’isopropanol, bien agiter et incuber à-80 ° c pendant la nuit (table des matières).
- Centrifuger à 3 220 x g et 4 ° c pendant 45 minutes.
- Retirer l’isopropanol, ajouter 15 mL d’éthanol à 100% (table des matières).
- Centrifuger à 3 220 x g pendant 10 min.
- Retirer l’éthanol, égoutter et sécher pendant environ 1 h.
- Utiliser un coton-tige stérile pour éponger l’éthanol restant (table des matières).
- Resuspendre l’échantillon en 100 à 200 μL d’eau libre de RNase en fonction de la granulométrie (tableau des matériaux).
- Fractionner les échantillons à l’aide de la technologie d’électrophorèse pour déterminer la qualité et la concentration des échantillons selon les intructions du fabricant23 (tableau des matériaux).
- Les échantillons sont admissibles à l’analyse de l’ARN-SEQ si RIN > 824.
2. séquençage de nouvelle génération
- Soumettre des échantillons d’ARN cytoplasmique à séquencer en utilisant la plate-forme de séquençage de nouvelle génération visant à générer au moins 50 millions paires-fin 100 BP lectures.
- Sélectionnez pour les ARN poly-adénylés et le séquençage spécifique aux brins.
3. créer des annotations (facultatif si on a une annotation existante)
- Créez une annotation L1 pleine longueur ou téléchargez l’annotation L1 pleine longueur (fichier supplémentaire 1a-b).
- Téléchargez les annotations répétition masker pour les éléments LINE-1 du navigateur du génome UCSC avec l’outil de navigateur de table (https://genome.ucsc.edu/cgi-bin/hgTables). Spécifiez le clade mammifère, le génome humain, l’assembly hg19 (ou hg38 pour un génome plus mis à jour) et le filtre pour «LINE1» sous nom de classe. Télécharger en tant que fichier. GTF et l’étiquette comme FL-L1-BLAST. GTF.
- Exécutez une recherche locale BLAST de la première 300 BP de l’élément L1 de L 1.3 pleine longueur englobant la région promotrice dans le génome humain et ajoutez 6 000 BP en aval pour créer une extrémité des coordonnées L1 dans le fichier d’annotation. Enregistrer dans un fichier GTF et l’étiquette comme FL-L1-RM. GTF.
- Intersectez l’annotation RepeatMasker et l’annotation L1 basée sur le promoteur à l’aide des outils de lit et étiquetez FL-L1-BLAST_RM. txt (packages logiciels).
- Utilisez cette commande dans le terminal Linux: bedtools Intersect-a FL-L1-Blast. GTF-b FL-L1-RM. gtf > FL-L1-BLAST_RM. txt.
- Séparez l’annotation FL-L1 intersectée par le brin supérieur et inférieur.
- Copiez sur le FL-L1-BLAST_RM. txt dans le logiciel de feuille de calcul et trier par le "moins" et "plus" brin et puis Trier par emplacement du chromosome.
- Créez deux nouveaux documents de feuille de calcul, l’un avec les coordonnées intersectées pour la longueur totale N1 sur le brin moins et l’autre sur le brin inférieur, et enregistrez comme FL-L1-BLAST_RM_minus. xls et FL-L1-BLAST_RM_plus. xls.
- Enregistrez les deux nouveaux documents sous forme de fichiers. txt.
- Utilisez le programme mac2unix pour convertir les fichiers. txt en fichiers d’annotation corrects (progiciels).
- Utilisez cette commande dans le terminal: Mac2unix.sh FL-L1-BLAST_RM_minus. GFF.
- Utilisez cette commande dans le terminal: Mac2unix.sh FL-L1-BLAST_RM_plus. GFF.
- Enregistrez de nouveaux fichiers avec l’extension. GFF.
- Vous pouvez également utiliser AWK pour filtrer les lignes associées au brin + et –.
- Utilisez la commande suivante pour obtenir le + Strand: awk'/+/'FL-L1_BLAST_RM. gtf > FL-L1_BLAST_RM_plus. GTF.
- Utilisez la ligne de commande suivante pour obtenir le-Strand: awk'/-/'FL-L1_BLAST_RM. gtf > FL-L1_BLAST_RM_minus. GTF.
4. lire le pipeline d’alignement pour identifier les N1 exprimés
| option | description |
| – p | Cela détaille le nombre de threads que l’ordinateur doit utiliser pour exécuter l’alignement. La mémoire d’ordinateur plus grande permettra plus de threads et devrait être empiriquement d. |
| – m 1 | Cela indique au programme d’accepter uniquement les lectures qui ont une correspondance dans le génome qui est mieux que n’importe quel autre génome match. |
| – y | Il s’agit du commutateur TryHard qui effectue la recherche de mappage pour toutes les correspondances possibles et ne lui permet pas de quitter après un nombre fixe de correspondances est atteint. |
| – v 3 | Cela permet uniquement au programme d’utiliser la mémoire pour les lectures mappées avec 3 ou moins de décorrespondances au génome. |
| – X 600 | Cela ne permet que des lectures appariées que la carte dans 600 bases de l’autre. Cela permet de s’assurer que les paires de lecture sont co-linéaires dans le génome et sélectionne contre s impliquant des molécules d’ARN traitées. |
| – chunkmbs 8184 | Cette commande attribue une mémoire supplémentaire pour gérer la grande quantité d’alignements possibles pour chaque lecture liée à L1. |
Tableau 1: options de ligne de commande pour noeud papillon.
- Exécutez l’alignement des fichiers FastQ de séquençage de fin apparié avec l’échantillon d’ARN-SEQ d’intérêt utilisant le noeud papillon.
Remarque: Bowtie1 doit être utilisé et non Bowtie2 parce que les paramètres requis pour l’alignement unique sont spécifiquement trouvés dans cette version du noeud papillon (progiciels). Le noeud papillon est utilisé sur des aligneurs de Splice-Aware comme le STAR dans l’ordre évaluent concordant, les lectures contiguës plus pertinentes à la biologie et à l’expression L1.- Utilisez cette ligne de commande dans le terminal Linux: noeud papillon-p 10-m 1-S-y-v 3-X 600--chunkmbs 8184 hg_X_Y_M_index-1 hg_sample_1. FQ-2 hg_sample_2. FQ | vue samtools-hbuS-| samtools Trier-hg_sample_sorted. BAM. Reportez-vous au tableau 1 pour une description des options de ligne de commande pour Bowtie.
- Strand sépare le fichier BAM de sortie en utilisant samtools (progiciels) et les commandes Linux suivantes. Notez que les valeurs réelles de l’indicateur peuvent varier si on n’utilise pas les protocoles de séquençage de prochaine génération standard.
- Utilisez cette ligne de commande pour sélectionner pour le brin supérieur: vue samtools-h hg_sample_sorted. BAM | awk’substr ($ 0, 1, 1) = = "@" | | $2 = = 83 | | $2 = = 163 {Print} ' | vue samtools-BS-> hg_sample_sorted_topstrand. BAM.
- Utilisez cette ligne de commande pour sélectionner pour le brin inférieur: vue samtools-h hg_sample_sorted. BAM | awk’substr ($ 0, 1, 1) = = "@" | | $2 = = 99 | | $2 = = 147 {Print} ' | vue samtools-BS-> hg_sample_sorted_bottomstrand. BAM.
- Générez des comptes de lecture par rapport aux annotations pour les loci L1 à l’aide de bedtools (packages logiciels).
- Utilisez cette ligne de commande pour générer des nombres de lecture pour N1 dans la direction de sens sur le brin supérieur: couverture de bedtools-Abam FL-L1-BLAST_RM_plus. GFF-b hg_sample_sorted_topstrand. bam > hg_sample_sorted_bowtie_tryhard_plus_top. txt.
- Utilisez cette ligne de commande pour générer des nombres de lecture pour N1 dans la direction de sens sur le brin inférieur: couverture de bedtools-Abam FL-L1-BLAST_RM_minus. GFF-b hg_sample_sorted_bottomstrand. bam > hg_sample_sorted_bowtie_tryhard_minus_bottom. txt.
- Index BAM fichier à partir de l’étape 5.1.1 pour le rendre visible dans le visualiseur de génomique intégrative (IGV)25 (progiciels).
- Utilisez cette ligne de commande: samtools index hg_sample_sorted. BAM
- Pour utiliser un mode batch pour augmenter le nombre d’échantillons d’ARN-SEQ canped à la fois, utilisez un script de supercalculateur pour terminer l’étape 4,1 appelée human_bowtie. sh, un script pour terminer les étapes 4.2 à 4.3 a été créé appelé human_L1_pipeline. sh, et un script pour terminer l’étape 4,4 a été créée, appelée bam_index. sh. Ces scripts peuvent être trouvés dans le fichier supplémentaire 2 avec les commandes de superordinateur associées pour exécuter les scripts.
5. conservation manuelle
- Créez une feuille de calcul pour les lectures mappées à chaque locus L1 annotée.
- Copiez sur hg_sample_sorted_bowtie_tryhard_minus_bottom. txt créé à l’étape 4.3.2 et la page d’étiquette comme "moins-Bottom."
- Trier toutes les colonnes en fonction du plus haut au plus petit nombre de lectures trouvées dans la colonne J.
- Copiez sur hg_sample_sorted_bowtie_tryhard_plus_top. txt créé à l’étape 4.3.1 et l’étiquette comme "Top-plus" dans une autre feuille de calcul.
- Trier toutes les colonnes en fonction du plus haut au plus petit nombre de lectures trouvées dans la colonne J.
- Créez une troisième page étiquetée comme "combinée" et ajoutez tous les loci avec dix lectures ou plus à partir des pages "moins-Bottom" et "plus-Top".
- Trier toutes les colonnes en fonction du plus haut au plus petit nombre de lectures trouvées dans la colonne J.
- Chargez les fichiers suivants dans IGV25 (progiciels): 1) génome de référence d’intérêt pour visualiser les gènes annotés, 2) FL-L1-BLAST_RM. GFF pour visualiser l’annotation L1, 3) hg_sample_sorted. BAM pour visualiser les transcriptions mappées de échantillon d’intérêt, et 4) hg_genomicDNA_sorted. BAM pour évaluer la mappabilité des régions génomiques.
- Supprimez les lignes de couverture et de jonction associées à chaque fichier BAM.
- Compressez hg_sample_sorted. BAM et hg_genomicDNA_sorted. BAM pour que toutes les pistes IGV s’adaptent à un seul écran.
- Copiez sur hg_sample_sorted_bowtie_tryhard_minus_bottom. txt créé à l’étape 4.3.2 et la page d’étiquette comme "moins-Bottom."
- Curate manuellement.
- Utilisation des coordonnées des loci répertoriés sur la feuille de calcul "combiné", vue appelée loci dans IGV25 (progiciels).
- Curate un locus pour être authentiquement exprimé hors de lui-même s’il n’y a pas de lectures en amont dans la direction L1 jusqu’à 5 KB.
- Étiqueter la ligne verte en couleur et noter pourquoi il s’agit d’un L1 authentiquement exprimé.
Remarque: une exception à cette règle existe si la région en amont du L1 n’est pas mappable. Si tel est le cas, étiqueter la ligne rouge en couleur et noter que l’expression de la région en amont du promoteur L1 ne peut pas être évaluée et donc l’expression N1 n’est pas en mesure d’être déterminée en toute confiance.
- Étiqueter la ligne verte en couleur et noter pourquoi il s’agit d’un L1 authentiquement exprimé.
- Curate un locus pour ne pas être authentiquement exprimé hors de son propre promoteur s’il ya des lectures en amont jusqu’à 5 KB.
- Étiquetez la ligne rouge en couleur et notez pourquoi il n’est pas un L1 authentiquement exprimé.
- Curate un locus comme faux si elle est exprimée dans un intron d’un gène exprimé dans la même direction avec des lectures en amont de la L1, si elle est en aval d’un gène exprimé dans la même direction avec des lectures en amont de la L1, ou pour des modèles d’expression non annotés avec re annonces en amont de la L1.
Remarque: une exception à cette règle s’applique lorsqu’il y a des lectures minimales qui chevauchent directement le site de démarrage du promoteur L1, mais légèrement en amont du L1. S’il n’y a pas d’autres lectures en amont d’un cas L1 comme celui-ci, considérez cette L1 comme authentiquement exprimée. Étiquetez la couleur verte de ligne et notez pourquoi elle est authentiquement exprimée L1.
- Curate un locus L1 comme susceptible d’être faux si le modèle de lectures mappées au locus ne correspondent pas aux régions N1 spécifiques de la mappabilité.
NOTE: par exemple, si un L1 est hautement mappable mais n’a qu’une pile de lectures dans une région condensée au sein de la L1, il est moins susceptible d’être lié à l’expression L1 de son propre promoteur et plus susceptibles d’être des sources non annotées comme exons ou LTRs. Dans des cas comme celui-ci, curé les loci comme orange et notez pourquoi le locus est suspect. Vérifiez les sources de piles suspectes en vérifiant l’emplacement L1 dans le navigateur du génome UCSC. - Curate un locus pour ne pas être authentiquement exprimé s’il se trouve dans un environnement génomique de régions non annotées sporadiquement exprimées
Remarque: par exemple, les lectures peuvent être exprimées à 10 Ko en amont du L1, mais toutes les 10 Ko ou alors il y a des lectures mappées et certaines de ces lectures s’alignent avec le L1. Ces N1 sont moins susceptibles d’être exprimés à partir de leur propre promoteur, et plus susceptibles d’avoir des lectures cartographiées en raison de modèles non annotés de l’expression génomique. Dans des cas comme celui-ci, curé les loci comme orange et notez pourquoi le locus est suspect.
6. lire la stratégie d’alignement pour évaluer la mappabilité dans le génome de référence (facultatif si on a un jeu de données d’ADN génomique aligné existant)
- Télécharger les fichiers de séquences d’ADN du génome entier et convertir en fichiers. FQ
- Rendez-vous sur le site Web du NCBI: https://www.ncbi.nlm.nih.gov/sra
- Tapez l' extrémité appariée de WGS HeLa.
- Sélectionnez pour Homo sapiens sous résultats par taxon.
- Sélectionnez un exemple qui est couplé fin et a lectures avec 100 ou plus BP comme l’exemple suivant: https://www.ncbi.nlm.nih.gov/sra/ERX457838 [ACCN]
- Confirmez la longueur de lecture en sélectionnant exécuter , puis métadonnées comme illustré ici: https://trace.ncbi.nlm.nih.gov/traces/SRA/?Run=ERR492384
- Pour télécharger les données de la séquence d’ADN du génome entier, entrez cette commande dans le terminal Linux: sratoolkit. mac64/bin/prefetch-X 100G ERR492384
Remarque: la fonction de prérécupération de la boîte à outils SRA télécharge le numéro d’accession «ERR492384» trouvé sur le site NCBI (progiciels). Le «100G» limite la quantité de données téléchargées à 100 gigaoctets. - Entrez cette commande dans le terminal Linux: FastQ-dump--Split-Files ERR492384
Remarque: cela divise le jeu de données d’ADN génomique téléchargé en deux fichiers FastQ.
- Exécutez l’alignement à l’aide de Bowtie.
- Utilisez cette commande sous Linux pour l’alignement: noeud papillon-p 10-m 1-S-y-v 3-X 600--chunkmbs 8184 hg_X_Y_M_index-1 hg_genomicDNA_1. FQ-2 hg_genomicDNA_2. FQ | vue samtools-hbuS-| samtools Trier-hg_genomicDNA_sorted. BAM.
- Référez-vous à l’étape 4,1 pour comprendre les paramètres utilisés dans l’alignement de noeud papillon (progiciels).
- Téléchargez le fichier BAM aligné génomiquement pour évaluer la mappabilité disponible sur demande de l’auteur.
- Utilisez cette commande sous Linux pour l’alignement: noeud papillon-p 10-m 1-S-y-v 3-X 600--chunkmbs 8184 hg_X_Y_M_index-1 hg_genomicDNA_1. FQ-2 hg_genomicDNA_2. FQ | vue samtools-hbuS-| samtools Trier-hg_genomicDNA_sorted. BAM.
- Indexer le fichier BAM à partir de l’étape 4.2.1 à l’aide de samtools pour le rendre visible dans IGV25 (progiciels) pour en savoir plus sur la curation manuelle.
- Utilisez cette ligne de commande sous Linux: samtools index hg_genomicDNA_sorted. BAM
- Evaluer la mappabilité de chaque locus L1
- Déterminez le nombre de lectures mappées de façon unique sur les loci L1 à l’aide du programme d’outils de lit, de l’annotation FL-L1 et des données de séquence génomique alignées (progiciels).
- Utilisez cette ligne de commande sous Linux: bedtools Coverage-Abam FL-L1-BLAST_RM. GTF – b hg_genomicDNA_sorted. bam ≫ L1_Mappability_hg_genomicDNA. txt.
- Désigner un locus L1 pour avoir une mappabilité complète de la couverture lorsque 400 lectures uniques sont alignées.
- Déterminer le facteur nécessaire à l’échelle vers le haut ou vers le bas l’ADN génomique aligné lectures à 400 pour chaque L1 individuel.
- Pour avoir une mesure d’expression à l’échelle selon la mappabilité de locus L1 individuelle, multipliez le facteur déterminé à l’étape 1 à 1 par le nombre de lectures de transcription de l’ARN qui s’alignent sur des N1 authentiquement exprimés, déterminés dans les sections 4 à 5.
- Déterminez le nombre de lectures mappées de façon unique sur les loci L1 à l’aide du programme d’outils de lit, de l’annotation FL-L1 et des données de séquence génomique alignées (progiciels).
Résultats
Les étapes décrites ci-dessus et décrites graphiquement dans la figure 1 ont été appliquées à une lignée de cellules de tumeur de la prostate humaine DU145. L’échantillon d’ARN a été préparé dans le cytoplasme et a été séquencé à la prochaine génération dans un protocole poly-A sélectionné, spécifique à un brin, couplé à la fin. À l’aide de Bowtie, les fichiers de séquençage de fin appariés ont été alignés, ce qui permet uniquement des correspondances uniques dans lesquelles la lecture de fin appariée correspond mieux à un emplacement génomique comparé à n’importe quel autre emplacement génomique. Les fichiers de séquence DU145 ont été alignés sur le génome de référence humain créant un fichier BAM, qui est disponible sur demande de l’auteur. À l’aide de bedtools, les données ont été extraites des fichiers BAM séparés par des brins DU145 sur le nombre de lectures mappées sur la longueur totale N1. Ces lectures ont été triées dans une feuille de calcul de la plus grande à la plus petite et préparées manuellement en examinant l’environnement génomique autour de chaque locus L1 dans IGV pour confirmer son authenticité (tableau supplémentaire 1). Si un échantillon a été sélectionné pour être authentiquement exprimé, il a été codé en couleur vert avec une explication pour son acceptation dans la colonne la plus à droite. On trouvera dans la figure 2a-bdes exemples de loci L1 acceptés pour être authentiquement exprimés suivant les directives décrites dans la section des méthodes. Si un échantillon a été rejeté pour être authentiquement exprimé, il a été codé en couleur rouge avec la raison du rejet sur la colonne la plus à droite. Des exemples de loci L1 rejetés en raison de l’expression d’un promoteur autre que leurs propres directives suivantes décrites dans la section des méthodes sont détaillés dans la figure 2c-e.
Ici, seuls les N1 pleine longueur avec une région de promoteur intact ont été étudiés. Si cette distinction n’est pas faite, une grande source de bruit transcriptionnel provenant de la N1 tronquée est introduite. Des exemples de N1 tronqués dans DU145 sont montrés dans la figure 3a-b où ils ont été identifiés comme ayant des lectures d’ARN-SEQ mappées de façon unique. Dans l’IGV, cependant, il est évident que ces transcriptions n’ont pas été initiées à partir de la L1 tronquée, mais de l’inclusion de la séquence L1 dans un gène ou en aval d’un gène exprimé.
Globalement, en DU145, le pourcentage de loci L1 de longueur totale et de lectures qui sont rejetés comme authentiquement exprimés N1 après la conservation manuelle est d’environ 50% (tableau supplémentaire 2) démontrant le niveau élevé des lectures de transcription cartographiées L1 qui seraient autrement être enregistrés comme faux positifs sans curation manuelle. Plus précisément, en DU145, il y avait 114 loci totaux de longueur totale de L1 pour avoir des lectures cartographiées de façon unique dans la direction du sens avec un total de 3 152 lectures, mais il n’y avait que 60 loci identifiés pour être exprimés au large de leur propre promoteur après la conservation manuelle avec 1 879 lectures ( Tableau 1 supplémentaire). C’est le cas même lorsque des mesures ont été prises pour réduire l’expression non pertinente à la biologie L1 en sélectionnant pour l’ARNm cytoplasmique. Notez que le locus avec le plus haut niveau de transcriptions mappées dans DU145 a été rejeté parce qu’il n’était pas un L1 authentiquement exprimé (figure 4). Dans l’ensemble, le nombre de transcriptions mappées à des loci L1 spécifiques varie de la même façon entre les loci L1 acceptés et rejetés comme authentiquement exprimés après la conservation manuelle (figure 4).
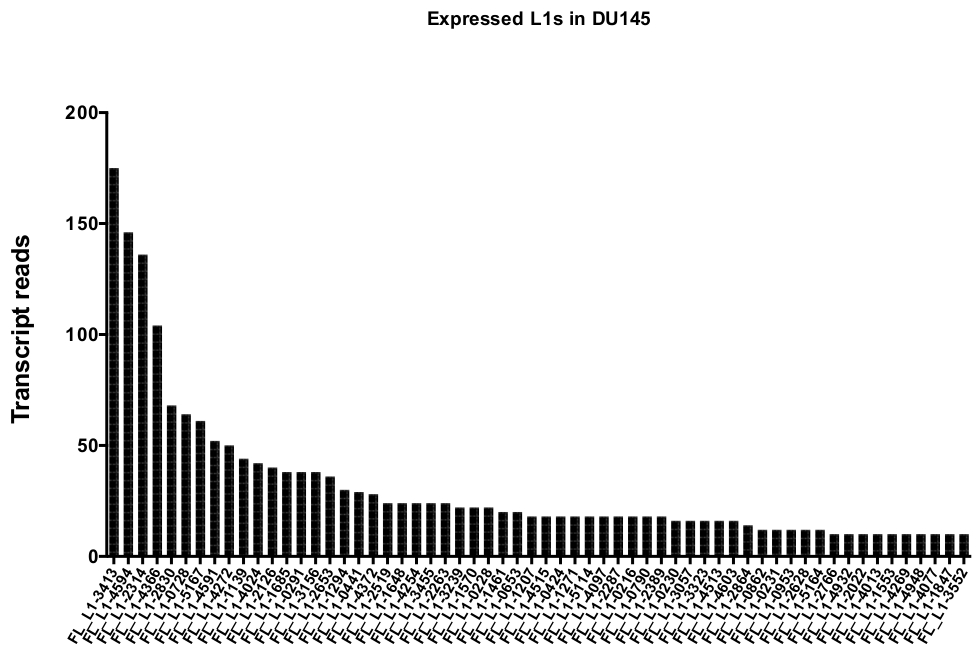
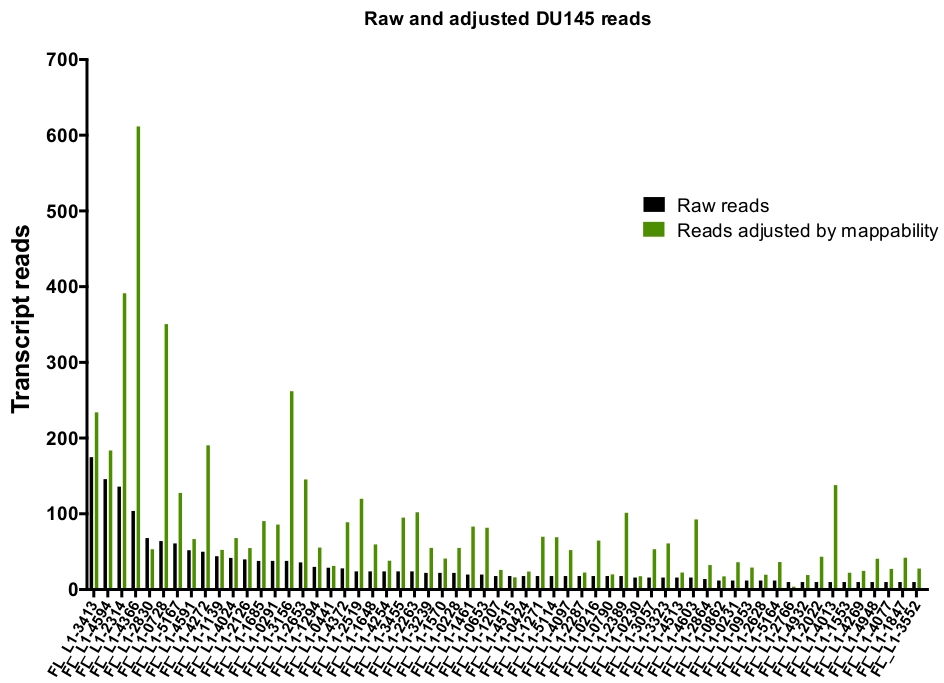
Après la curation manuelle, le nombre de lectures qui mappaient de façon unique à des loci L1 spécifiques authentiquement exprimés dans DU145 varient de 175 lectures à une coupure minimale choisie arbitrairement de 10 lectures (figure 5). Cette approche consistant à identifier des lectures de transcription mappées de manière unique à N1 limite la capacité de quantifier précisément l’expression. Pour tenir compte de cela, un facteur de correction pour chaque locus basé sur sa mappabilité a été créé. Pour créer ce facteur de correction, les premiers outils de lit ont été utilisés pour extraire le nombre de lectures mappées de façon unique à partir du fichier BAM génomique HeLa qui s’alignait sur tous les loci L1 de longueur totale et a représenté ces loci du plus élevé au plus bas des lectures de transcriptions cartographiées (supplément Figure 1). Il a été arbitrairement désigné que N1 avec 400 lectures ont eu la mappability pleine couverture. Le nombre de lectures pouvant être mappées à un locus L1 dans l’échantillon de séquençage génomique HeLa a été mis à l’échelle par rapport à 400 lectures et ce nombre à l’échelle a ensuite été multiplié par le nombre de lectures qui ont été mappés à chaque locus L1 authentiquement exprimé dans DU145 (tableau supplémentaire 2) . Comme prévu, les éléments L1 qui avaient des scores de correction plus importants pour la mappabilité provenaient de sous-familles plus jeunes comme L1PA2 (tableau supplémentaire 2). Une fois que les lectures ont été ajustées pour les scores de mappabilité dans chaque locus, la quantification pour l’expression de la plupart des loci a augmenté (figure 6). Le nombre de lectures qui ont été mappées de façon unique à des loci L1 spécifiques authentiquement exprimés avec des corrections de mappabilité dans DU145 variait de 612 à 4 lectures et il y avait une réorganisation des loci les plus élevés aux plus faibles (figure 6).

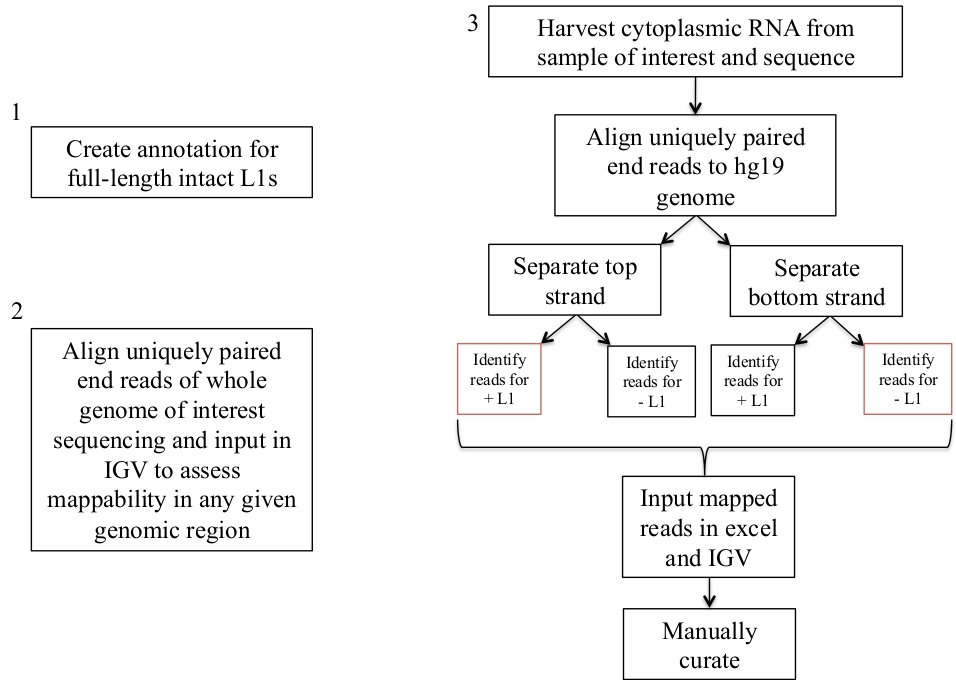
Figure 1: schéma de Workflow.
Les étapes pour identifier les N1 exprimés dans un échantillon humain sont décrites graphiquement. Notez que les étapes 1 et 2 n’ont pas besoin d’être répétées si les fichiers appropriés sont déjà disponibles. Ces fichiers appropriés peuvent être téléchargés à partir du supplément de fichier 1a-b et le fichier de supplément 2. Les cases en rouge indiquent les étapes où le programme de couverture des outils de lit est utilisé pour compter le nombre de lectures de mappage à N1 dans la même direction sens. Ces loci avec des lectures de cartographie orientées sens sont les N1 qui doivent être sélectionnés manuellement. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}

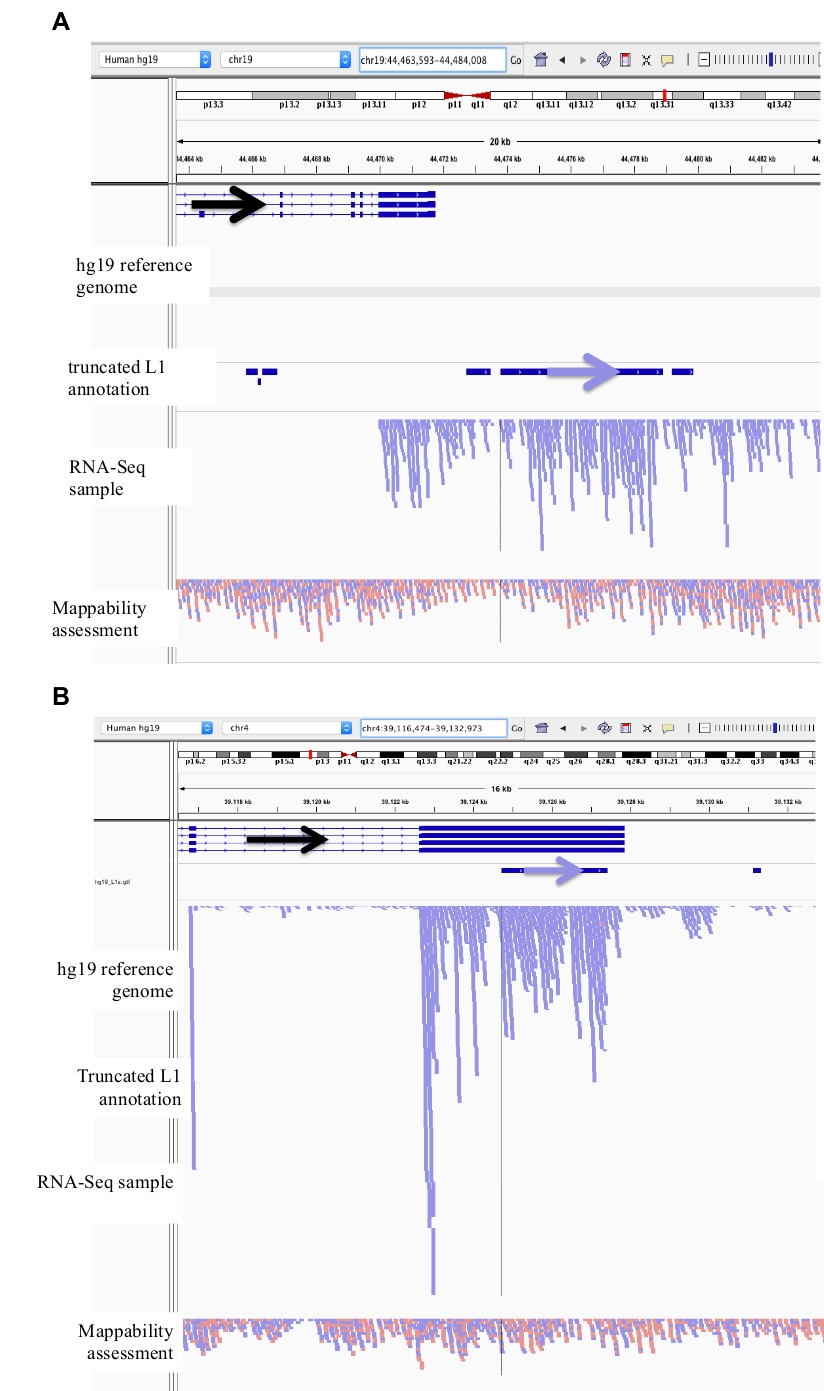
Figure 2: exemples de loci L1 organisés en DU145.
Chargé dans IGV sont le génome de référence, le fichier d’annotation L1 GFF pleine longueur correspondant à la version du génome de référence (supplément file 1), le fichier BAM DU145, et enfin le fichier génomique hela BAM pour évaluer la mappabilité, qui sont tous disponibles sur l’auteur demande. Des flèches ont été ajoutées pour faciliter la visualisation de la direction de la L1 annotée. Les flèches et les lectures en rouge sont orientées en séquence de droite à gauche. Les flèches et les lectures en bleu sont orientées en séquence de gauche à droite. a) dans IgV, ce locus L1 semble être exprimé au large de son propre promoteur car il n’y a pas de lectures en amont de la L1 dans l’orientation sens pour plus de 5 KB. Cette L1 a une faible mappabilité, elle n’est pas dans un gène, et a la preuve de l’activité de promoteur antisens attendue26. b) dans IgV, ce locus L1 semble être exprimé au large de son propre promoteur car il n’y a pas de lectures en amont de la L1 dans l’orientation sens pour plus de 5 KB. Cette L1 a une faible mappabilité et est dans un gène de direction opposée. c) dans IgV, ce locus L1 a été rejeté comme un L1 exprimé car il y a des lectures en amont dans la même orientation dans les 5 KB. Cette L1 est dans un gène de la même direction de sorte que les lectures de transcription sont très probablement originaires du promoteur du gène exprimé. d) dans IgV, ce locus L1 a été rejeté comme un L1 exprimé car il y a des lectures en amont dans la même orientation dans les 5 KB. Cette L1 est en aval d’un gène fortement exprimé dans la même direction de sorte que les lectures de transcription sont très probablement originaires du promoteur de ce gène exprimé et s’étendant au-delà de la terminaison de gène normale. e) en IgV, ce locus L1 a été rejeté comme un L1 exprimé car il y a des lectures en amont dans la même orientation dans les 5 KB. Cette L1 n’est pas à l’intérieur ou à proximité d’un gène annoté dans le gène de référence de sorte que l’origine de ces transcriptions à l’intérieur et en amont de l’élément L1 suggèrent un promoteur non annotée. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 3: le bruit de fond provient du N1 tronqué aussi bien.
Notre annotation L1 n’inclut pas les N1 tronquées car elles constituent une source majeure de bruit de fond. Des flèches ont été ajoutées pour faciliter la visualisation de la direction de la L1 annotée. Les flèches et les lectures en bleu sont orientées en séquence de gauche à droite. a) démontré est un exemple d’un L1 tronqué dans la SUFAMILLE L1MB5 qui est 2706 bps. Dans l’IGV, il est évident que les lectures proviennent de l’extension en aval d’un gène exprimé. b) montré est un autre exemple d’un L1 tronqué. Ce L1 est un L1PA11 qui est 4767 BPS long. Dans l’IGV, il est évident que les lectures de mappage de façon unique à la L1 proviennent de l’exon exprimé, dont le L1 est à l’intérieur. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 4: la transcription lit cette carte de façon unique à toutes les N1 intactes de longueur totale dans le génome humain exprimée en DU145 lignée de cellules tumorales de la prostate.
En noir sont les loci spécifiques à identifier comme authentiquement exprimés après la conservation manuelle et en rouge sont les loci spécifiques à rejeter comme authentiquement exprimée lectures après la conservation manuelle. En gris sont des loci avec moins de dix lectures de mappage à chacun. Comme ces loci représentent une petite fraction des lectures de transcription, ils n’étaient pas curate manuellement. Les graduations de l’axe x désignent chaque 100 pleine longueur, intacte N1. environ 4 500 loci ne sont pas représentés graphiquement car ils n’avaient pas de lectures mappées. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 5: la transcription lit cette carte de manière unique pour une N1 intacte pleine longueur dans la lignée de cellules tumorales de la prostate DU145.
Montré sont les nombres de transcription lit cette carte à des loci spécifiques dans les cellules DU145 après la curation manuelle. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 6: lit le mappage de façon authentiquement exprimée L1 lorsqu’il est ajusté par mappability.
Les nombres de lectures de transcription sont ajustés par des scores de mappabilité spécifiques aux loci qui mappés aux loci L1 manuellement sélectionnés dans les cellules DU145. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}
Fichier supplémentaire 1: annotations pour la N1 humaine pleine longueur, intacte selon l’orientation. a) FL-L1-BLAST_RM_minus. GFF. b) FL-L1-BLAST_RM_plus. GFF. Veuillez cliquer ici pour télécharger ce fichier.
Fichier supplémentaire 2: scripts de supercalculateur utilisés pour automatiser le pipeline de bioinformatique détaillé dans la section 4. Veuillez cliquer ici pour télécharger ce fichier.
Figure supplémentaire 1: échantillon d’ADN génomique utilisé pour déterminer la mappabilité L1.
Le nombre de relevés génomiques est indiqué à partir de l’échantillon de la lignée de cellules HeLa qui est unique à tous les 5 000 locus L1 de longueur totale dans le génome. Il a été désigné qu’un L1 a la mappability pleine couverture quand 400 lit la carte au L1. Veuillez cliquer ici pour télécharger ce chiffre.
Tableau supplémentaire 1: curation manuelle de N1 en DU145. Veuillez cliquer ici pour télécharger ce tableau.
Tableau supplémentaire 2: N1 en DU145 avec ajustement de la mappabilité. Veuillez cliquer ici pour télécharger ce tableau.
Discussion
Il a été démontré que l’activité de L1 provoque des dommages génétiques et une instabilité contribuant à la maladie27,28,29. Sur les environ 5 000 copies L1 complètes, seules quelques dizaines de jeunes N1 évolutivement représentent la majorité de l’activité de rétrotransposition2. Cependant, il est prouvé que même certains plus âgés, rétrotranspositionnellement-incompentent N1 sont encore en mesure de produire de l’ADN des protéines dommageables30. Pour apprécier pleinement le rôle du N1 dans l’instabilité génomique et la maladie, l’expression L1 au niveau spécifique au locus doit être comprise. Cependant, l’arrière-plan élevé des séquences liées à la L1 incorporées dans d’autres ARN sans rapport avec la rétrotransposition L1 pose un défi important dans l’interprétation de l’expression L1 authentique. Un autre défi dans l’identification et donc la compréhension des schémas d’expression des loci L1 individuels se produit en raison de leur nature répétitive qui ne permet pas de nombreuses séquences de lecture courtes pour mapper à un seul locus unique. Pour surmonter ces difficultés, nous avons développé l’approche décrite ci-dessus pour identifier l’expression des loci L1 individuels à l’aide de données ARN-Seq.
Notre approche filtre le niveau élevé (plus de 99%) du bruit transcriptionnel généré à partir de séquences L1 qui ne sont pas liées à la rétrotransposition L1 en prenant un certain nombre d’étapes. La première étape consiste à préparer l’ARN cytoplasmique. En sélectionnant pour l’ARN cytoplasmique, les lectures liées à la L1 trouvées dans l’ARNm intronique exprimée dans le noyau sont significativement épuisées. Dans la préparation de la bibliothèque de séquençage, une autre étape prise pour réduire le bruit transcriptionnel sans rapport avec N1 inclut la sélection des transcriptions polyadénylées. Cela élimine les bruits de transcription liés à la L1 trouvés chez les espèces non mRNA. Une autre étape comprend le séquençage spécifique aux brins afin d’identifier et d’éliminer les transcriptions liées à l’antisens L1. L’utilisation d’une annotation pour N1 pleine longueur avec des régions promotrices fonctionnelles lors de l’identification du nombre de transcriptions d’ARN-SEQ qui mappent à N1 élimine également les bruits de fond qui proviennent autrement de N1 tronquées. Enfin, la dernière étape critique pour éliminer le bruit transcriptionnel des séquences L1 sans rapport avec la rétrotransposition L1 est la conservation manuelle de la N1 pleine longueur identifiée pour avoir mappé des transcriptions de l’ARN-Seq. La conservation manuelle implique la visualisation de chaque locus L1, identifié par bioinformatisation, dans le contexte de son environnement génomique environnant, afin de confirmer que cette expression provient du promoteur L1. Cette approche a été appliquée à DU145, une lignée de cellules tumorales de la prostate. Même avec toutes les mesures de préparation prises pour réduire le bruit de fond, environ 50% des loci L1 identifiés bioinformatiquement dans DU145 ont été rejetés comme bruit de fond L1 provenant d’autres sources transcriptionnelles (figure 4), en insistant sur la rigueur requise pour produire des résultats fiables. Cette approche utilisant la conservation manuelle est laborieuse, mais nécessaire dans le développement de ce pipeline pour évaluer et comprendre l’environnement génomique entourant une L1 pleine longueur. Les prochaines étapes comprennent la réduction de la quantité de la conservation manuelle nécessaire en automatisant certaines des règles de conservation, mais en raison de la nature toujours pas complètement connue de l’expression génomique, des sources d’expression non annotées dans le génome de référence, les régions de faible la mappabilité, et même les facteurs de complication impliqués dans la construction d’un génome de référence, il n’est pas possible d’automatiser entièrement la conservation L1 à ce moment.
Le deuxième défi dans l’identification de l’expression des loci L1 individuels avec le séquençage se rapporte à la cartographie des transcriptions répétées de L1. Dans cette stratégie d’alignement, il est nécessaire qu’une transcription soit alignée de façon unique et co-linéairement sur le génome de référence afin d’être cartographiée. En sélectionnant pour les séquences couplées qui mappent concordalement, la quantité de transcriptions qui s’alignent de façon unique sur les loci L1 trouvés dans le génome de référence augmente. Cette stratégie de cartographie unique fournit la confiance dans l’appel de la cartographie de lectures spécifiquement à un locus L1 unique, bien qu’elle sous-estime potentiellement la quantité d’expression de chaque exprimée-à-être-authentiquement exprimé, répétitif L1. À peu près correct pour cette sous-estimation, un score de «mappability» pour chaque locus L1 basé sur sa mappabilité a été développé et appliqué au nombre de lectures de transcription mappées de façon unique (figure 6). Il est à noter que, idéalement, la mappabilité doit être notée à la couverture complète lit sur l’ensemble de la longueur L1 en fonction de l’échantillon WGS appariés. Ici, nous utilisons WGS de cellules HeLa pour déterminer les scores de la mappabilité de chaque locus L1 afin de gonfler ou de dégonfrer les lectures de mappage à des loci L1 dans les lignées cellulaires de tumeur de la prostate DU145. Ce calcul de la mappabilité est un score de correction brut, mais la «mappabilité complète de la couverture» de 400 lectures a été déterminée avec la nature dynamique des lignées cellulaires tumorales à l’esprit. Il peut être observé dans la figure 1 supplémentaire, qu’il y a quelques loci L1 avec hela WGS avec le nombre extrêmement élevé de lectures cartographiées. Ceux-ci proviennent probablement de séquences chromosomiques dupliquées au sein d’HeLa qui ne se trouvent pas dans le génome de référence, raison pour laquelle ces loci n’ont pas été choisis pour être représentatifs de la couverture de la mappabilité complète. Au lieu de cela, il a été déterminé que la moyenne de 100% de la couverture de lecture se produit autour de 400 lectures selon la figure supplémentaire 1 et a ensuite supposé que cette moyenne s’applique à la lignée de cellules de la prostate DU145 tumeur aussi bien.
Cette stratégie d’alignement avec 100-200 BP lectures de la technologie de l’ARN-SEQ sélectionne également préférentiellement pour les N1 évolutivement plus âgés dans le génome de référence que les N1 plus âgés ont accumulé au fil du temps des mutations uniques qui les rendent plus mappables. Cette approche, par conséquent, a une sensibilité limitée quand il s’agit d’identifier le plus jeune de N1 ainsi que non-référence, N1 polymorphique. Pour identifier le plus jeune de N1, nous vous suggérons d’utiliser 5 'RACE sélection de transcriptions L1 et la technologie de séquençage comme PacBio qui utilisent des lectures plus longues21. Cela permet une cartographie plus unique et donc une identification sûre des jeunes N1. l’utilisation des approches ARN-SEQ et PacBio peut aboutir à une liste plus complète des N1 authentiquement exprimés. Pour identifier les N1 polymorphiques authentiquement exprimés, les premières étapes suivantes comprennent la construction et l’insertion de séquences polymorphes dans le génome de référence.
Les défis biologiques et techniques dans l’étude des séquences répétées sont grands, mais avec la procédure ci-dessus rigoureuse pour éliminer le bruit transcriptionnel des séquences L1 non liées à la rétrotransposition en utilisant la technologie de séquençage de l’ARN, nous commençons à passer au crible les grands niveaux de bruit de fond transcriptionnel et d’être en confiance et de manière rigoureuse identifier les modèles d’expression L1 et la quantité au niveau de locus individuel.
Déclarations de divulgation
Les auteurs n’ont rien à divulguer.
Remerciements
Nous aimerions remercier le Dr Yan Dong pour les cellules tumorales de la prostate DU145. Nous aimerions remercier le Dr Nathan Ungerleider pour ses conseils et son Conseil dans la création de scripts de supercalculateur. Une partie de ce travail a été financée par des subventions NIH R01 GM121812 à la police, R01 AG057597 à VPB, et 5TL1TR001418 aux savoirs traditionnels. Nous aimerions également souligner le soutien des croisés du cancer et du centre de bioinformatique du Tulane Cancer Center.
matériels
| Name | Company | Catalog Number | Comments |
| 1 M HEPES | Affymetrix | AAJ16924AE | |
| 5 M NaCl | Invitrogen | AM9760G | |
| Agilent bioanalyzer 2100 | Agilent technologies | ||
| Agilent RNA 6000 Nano Kit | Agilent technologies | 5067-1511 | |
| bedtools.26.0 | https://bedtools.readthedocs.io/en/latest/content/installation.html | ||
| bowtie-0.12.8 | https://sourceforge.net/projects/bowtie-bio/files/bowtie/0.12.8/ | ||
| Cell scraper | Olympus plastics | 25-270 | |
| Chloroform | Fisher | C298-500 | |
| Digitonin | Research Products International Corp | 50-488-644 | |
| Ethanol | Fisher | A4094 | |
| Gibco (Phosphate Buffered Saline) | Invitrogen | 10-010-049 | |
| Homogenizer | Thomas Scientific | BBI-8541906 | |
| IGV 2.4 | https://software.broadinstitute.org/software/igv/download | ||
| Isopropanol | Fisher | A416-500 | |
| mac2unix | https://sourceforge.net/projects/cs-cmdtools/files/mac2unix/ | ||
| Q-tips | Fisher | 23-400-122 | |
| RNAse later solution | Invitrogen | AM7022 | |
| RNaseZap RNase Decontamination Solution | Invitrogen | AM9780 | |
| samtools-1.3 | https://sourceforge.net/projects/samtools/files/ | ||
| sratoolkit.2.9.2 | https://github.com/ncbi/sra-tools/wiki/Downloads | ||
| SUPERase·In RNase Inhibitor | Invitrogen | AM2694 | |
| Trizol | Invitrogen | 15-596-018 | |
| Water (DNASE, RNASE free) | Fisher | BP2484100 |
Références
- International Human Genome Sequencing. Initial sequencing and analysis of the human genome. Nature. 409, 860(2001).
- Brouha, B., et al. Hot L1s account for the bulk of retrotransposition in the human population. Proceedings of the National Academy of Sciences of the United States of America. 100 (9), 5280-5285 (2003).
- Dombroski, B. A., Mathias, S. L., Nanthakumar, E., Scott, A. F., Kazazian, H. H. Isolation of an active human transposable element. Science. 254 (5039), 1805(1991).
- Swergold, G. D. Identification, characterization, and cell specificity of a human LINE-1 promoter. Molecular and Cellular Biology. 10 (12), 6718-6729 (1990).
- Speek, M. Antisense promoter of human L1 retrotransposon drives transcription of adjacent cellular genes. Molecular and Cellular Biology. 21 (6), 1973-1985 (2001).
- Deininger, L., Batzer, M. A., Hutchison, C. A., Edgell, M. H. Master genes in mammalian repetitive DNA amplification. Trends in Genetics. 8 (9), 307-311 (1992).
- Boissinot, S., Chevret, P., Furano, A. L1 (LINE-1) Retrotransposon Evolution and Amplification in Recent Human History. Molecular Biology and Evolution. 17 (6), 915-918 (2000).
- Khazina, E., Weichenrieder, O. Non-LTR retrotransposons encode noncanonical RRM domains in their first open reading frame. Proceedings of the National Academy of Sciences of the United States of America. 106 (3), 731-736 (2009).
- Martin, S. L., Bushman, F. D. Nucleic acid chaperone activity of the ORF1 protein from the mouse LINE-1 retrotransposon. Molecular and Cellular Biology. 21 (2), 467-475 (2001).
- Feng, Q., Moran, M. H., Kazazian, H. H., Boeke, J. D. Human L1 Retrotransposon Encodes a Conserved Endonuclease Required for Retrotransposition. Cell. 87 (5), 905-916 (1996).
- Mathias, S. L., Scott, A. F., Kazazian, H. H., Boeke, J. D., Gabriel, A. Reverse transcriptase encoded by a human transposable element. Science. 254 (5039), 1808(1991).
- Luan, D. D., Korman, M. H., Jakubczak, J. L., Eickbush, T. H. Reverse transcription of R2Bm RNA is primed by a nick at the chromosomal target site: A mechanism for non-LTR retrotransposition. Cell. 72 (4), 595-605 (1993).
- van den Hurk, J. A. J. M., et al. Novel types of mutation in the choroideremia (CHM) gene: a full-length L1 insertion and an intronic mutation activating a cryptic exon. Human Genetics. 113 (3), 268-275 (2003).
- Miné, M., et al. A large genomic deletion in the PDHX gene caused by the retrotranspositional insertion of a full-length LINE-1 element. Human Mutation. 28 (2), 137-142 (2007).
- Solyom, S., et al. Pathogenic orphan transduction created by a nonreference LINE-1 retrotransposon. Human Mutation. 33 (2), 369-371 (2012).
- Hancks, D. C., Kazazian, H. H. Roles for retrotransposon insertions in human disease. Mobile DNA. Mobile DNA. 7, 9-9 (2016).
- Tubio, J. M. C., et al. Mobile DNA in cancer. Extensive transduction of nonrepetitive DNA mediated by L1 retrotransposition in cancer genomes. Science. 345 (6196), 1251343-1251343 (2014).
- Ewing, A. D., et al. Widespread somatic L1 retrotransposition occurs early during gastrointestinal cancer evolution. Genome Research. 25 (10), 1536-1545 (2015).
- Beck, C. R., Garcia-Perez, J. L., Badge, R. M., Moran, J. V. LINE-1 elements in structural variation and disease. Annual Review of Genomics and Human Genetics. 12, 187-215 (2011).
- Philippe, C., et al. Activation of individual L1 retrotransposon instances is restricted to cell-type dependent permissive loci. eLife. 5, e13926(2016).
- Deininger, P., et al. A comprehensive approach to expression of L1 loci. Nucleic Acids Research. 45 (5), e31-e31 (2017).
- Jin, Y., Tam, O. H., Paniagua, E., Hammell, M. TEtranscripts: a package for including transposable elements in differential expression analysis of RNA-seq datasets. Bioinformatics. 31 (22), 3593-3599 (2015).
- Agilent RNA 6000 Nano Kit Guide. , Agilent. (2017).
- Mueller, O. L., Schroeder, A. RNA Integrity Number (RIN) –Standardization of RNA Quality Control. , Agilent Technologies. (2016).
- Robinson, J. T., et al. Integrative genomics viewer. Nature Biotechnology. 29, 24(2011).
- Speek, M. Antisense promoter of human L1 retrotransposon drives transcription of adjacent cellular genes. Molecular Cellular Biology. 21 (6), 1973-1985 (2001).
- Belancio, V. P., Deininger, L., Roy-Engel, A. M. LINE dancing in the human genome: transposable elements and disease. Genome Medicine. 1 (10), 97-97 (2009).
- Iskow, R. C., et al. Natural Mutagenesis of Human Genomes by Endogenous Retrotransposons. Cell. 141 (7), 1253-1261 (2010).
- Scott, E. C., et al. A hot L1 retrotransposon evades somatic repression and initiates human colorectal cancer. Genome Research. 26 (6), 745-755 (2016).
- Kines, K. J., Sokolowski, M., deHaro, D. L., Christian, C. M., Belancio, V. P. Potential for genomic instability associated with retrotranspositionally-incompetent L1 loci. Nucleic Acids Research. 42 (16), 10488-10502 (2014).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.