
Method Article
RNA Next-Generation Sequencing and a Bioinformatics Pipeline to Identify Expressed LINE-1s at the Locus-Specific Level
In This Article
Summary
Here, we present a bioinformatic approach and analyses to identify LINE-1 expression at the locus specific level.
Abstract
Long INterspersed Elements-1 (LINEs/L1s) are repetitive elements that can copy and randomly insert in the genome resulting in genomic instability and mutagenesis. Understanding the expression patterns of L1 loci at the individual level will lend to the understanding of the biology of this mutagenic element. This autonomous element makes up a significant portion of the human genome with over 500,000 copies, though 99% are truncated and defective. However, their abundance and dominant number of defective copies make it challenging to identify authentically expressed L1s from L1-related sequences expressed as part of other genes. It is also challenging to identify which specific L1 locus is expressed due to the repetitive nature of the elements. Overcoming these challenges, we present an RNA-Seq bioinformatic approach to identify L1 expression at the locus specific level. In summary, we collect cytoplasmic RNA, select for polyadenylated transcripts, and utilize strand-specific RNA-Seq analyses to uniquely map reads to L1 loci in the human reference genome. We visually curate each L1 locus with uniquely mapped reads to confirm transcription from its own promoter and adjust mapped transcript reads to account for mappability of each individual L1 locus. This approach was applied to a prostate tumor cell line, DU145, to demonstrate the ability of this protocol to detect expression from a small number of the full-length L1 elements.
Introduction
Retrotransposons are repetitive DNA elements that can “jump” in the genome in a copy-and-paste mechanism via RNA intermediates. One subset of retrotransposons is known as Long INterspersed Elements-1 (LINEs/L1s) and makes up a sixth of the human genome with over 500,0000 copies1. Despite their abundance, most of these copies are defective and truncated with only an estimated 80-120 L1 elements thought to be active2. A full-length L1 is about 6 kb in length with 5’ and 3’ untranslated regions, an internal promoter and associated anti-sense promoter, two non-overlapping open-reading frames (ORFs), and a signal and polyA tail3,4,5. In humans, L1s are made up of subfamilies distinguished by evolutionary age with the older families having accumulated more unique sequence mutations over time compared to the youngest subfamily, L1HS6,7. L1s are the only autonomous, human retrotransposons and their ORFs encode a reverse transcriptase, endonuclease, and RNPs with RNA-binding and chaperone activities required to retrotranspose and insert in the genome in a process referred to as target-primed reverse transcription8,9,10,11,12.
Retrotransposition of L1s has been reported to cause human germline diseases by a variety of mechanisms including insertional mutagenesis, target-site deletions, and rearrangements13,14,15,16. Recently it has been hypothesized that L1s may play a role in oncogenesis and/or tumor progression as increased expression and insertion events of this mutagenic element have been observed in a variety of epithelial cancers17,18. It is estimated that there is one new L1 insertion in every 200 births19. Therefore, it is imperative to better understand the biology of the actively expressing L1s. The repetitive nature and abundance of defective copies found within transcripts of other genes have made this level of analysis challenging.
Fortunately, with the advent of high throughput sequencing technologies, strides have been made to parse out and identify authentically expressing L1s at the locus-specific level. There are differing philosophies on how to best identify expressed L1s using RNA next-generation sequencing. There have been only two reasonable approaches suggested for mapping L1 transcripts at the locus-specific level. One focuses only on the potential transcription that reads through the L1 polyadenylation signal and into flanking sequences20. Our approach takes advantage of small sequence differences between L1 elements and only maps those RNA-Seq reads that uniquely map to one locus21. Both of these methods have limitations in terms of quantitation of transcript levels. Quantitation can be improved potentially by adding a correction for the ‘unique mappability’ of each L1 locus21, or using more complex algorithms that redistribute the multi-mapped reads that could not be uniquely mapped to a specific locus22. Here, we will detail in a step-by-step manner the RNA extraction and next-generation sequencing and bioinformatics protocol to identify expressed L1 elements at the locus-specific level. Our approach takes maximal advantage of our knowledge of the biology of functional L1 elements. This includes knowing that functional L1 elements must be generated from the L1 promoter, initiated at the beginning of the L1 element, must be translated in the cytoplasm and that their transcripts should be co-linear with the genome. Briefly, we collect fresh, cytoplasmic RNA, select for polyadenylated transcripts, and utilize strand-specific RNA-Seq analyses to uniquely map reads to L1 loci in the human reference genome. These aligned reads then still require extensive manual curation to determine if transcript reads originate from the L1 promoter before designating a locus as an authentically expressed L1. We apply this approach on the DU145 prostate tumor cell line sample to demonstrate how it identifies a relatively few actively transcribed L1 members from the mass of inactive copies.
Protocol
1. Cytoplasmic RNA extraction
- Obtain cells via the following methods.
- Collect live cells from 2.75%–100% confluent, T-75 flasks.
- Wash the flask 2 times in 5 mL of cold PBS, and in the last wash scrape off cells and transfer to a 15 mL conical tube. Centrifuge for 2 min at 1,000 x g and 4 °C, and carefully remove and discard supernatant (Table of Materials).
- Collect cells from tissue specimens.
- Prepare tissue for cytoplasmic RNA extraction within an hour from being dissected and always keep on ice. For long-term storage, use RNA inhibitor solutions to store tissue for up to 72 hours after dissection following the manufacturer’s protocol (Table of Materials).
- Dice a 10 µm3 sample and homogenize the fresh sample with 5 mL of cold PBS in a sterile dounce homogenizer, transfer to a 15 mL conical tube, centrifuge for 2 min at 1,000 x g at 4 °C, and carefully remove and discard supernatant (Table of Materials).
- Collect live cells from 2.75%–100% confluent, T-75 flasks.
- Add 2 mL of lysis buffer to cellular pellet- mix and incubate on ice for 5 min.
- Prepare fresh lysis buffer with 150 mM NaCl, 50 mM HEPES (pH 7.4), and 25 μg/mL digitonin (Table of Materials).
- As the minimum concentration of digitonin in the lysis buffer required to penetrate the plasma membrane may vary by cell type, microscopically confirm that cells treated with lysis buffer lose the plasma membrane and retain the intact nuclear membrane.
- Just before use, add 1,000 U/mL RNase inhibitor (Table of Materials).
- Centrifuge for 1 min at 1,000 x g and 4 °C, and collect the supernatant.
- Add supernatant to pre-chilled 7.5 mL of Trizol and 1.5 mL of chloroform. All the steps that require chloroform must be done inside a clean chemical hood (Table of Materials).
- Centrifuge for 35 min at 3,220 x g and 4 °C.
- Transfer the aqueous portion (top layer) to a fresh pre-chilled 15 mL tube.
- Add 4.5 mL of chloroform and vortex.
- Centrifuge for 10 min at 3,220 x g and 4 °C.
- Transfer the aqueous portion to fresh pre-chilled tube.
- Add 4.5 mL of isopropanol, shake well, and incubate at -80 °C overnight (Table of Materials).
- Centrifuge at 3,220 x g and 4 °C for 45 minutes.
- Remove isopropanol, add 15 mL of 100% ethanol (Table of Materials).
- Centrifuge at 3,220 x g for 10 min.
- Remove ethanol, drain and dry for approximately 1 h.
- Use a sterile cotton swab to blot out any remaining ethanol (Table of Materials).
- Re-suspend sample in 100 to 200 μL of RNase free water depending on pellet size (Table of Materials).
- Fractionate samples using electrophoresis technology to determine quality and concentration of samples according to manufacturer’s intructions23 (Table of Materials).
- Samples qualify for RNA-Seq analysis if RIN > 824.
2. Next-Generation sequencing
- Submit cytoplasmic RNA samples to be sequenced using next-generation sequencing platform aimed at generating at least 50 million paired-end 100 bp reads.
- Select for poly-adenylated RNAs and strand-specific sequencing.
3. Create annotations (optional if one has an existing annotation)
- Create full-length L1 annotation or download the full-length L1 annotation (Supplemental File 1a-b).
- Download Repeat Masker annotations for LINE-1 elements from the UCSC genome browser with the table browser tool (https://genome.ucsc.edu/cgi-bin/hgTables). Specify the mammal clade, the human genome, the hg19 assembly (or hg38 for a more updated genome), and filter for “LINE1” under Class Name. Download as a .gtf file and label as FL-L1-BLAST.gtf.
- Run a local BLAST search of the first 300 bp of the L1.3 full-length L1 element encompassing the promoter region in the human genome and add 6,000 bp downstream to create an end of the L1 coordinates to the annotation file. Save in a gtf file and label as FL-L1-RM.gtf.
- Intersect the RepeatMasker annotation and the promoter-based L1 annotation using bedtools, and label as FL-L1-BLAST_RM.txt (Software Packages).
- Use this command in the Linux terminal: bedtools intersect -a FL-L1-BLAST.gtf -b FL-L1-RM.gtf > FL-L1-BLAST_RM.txt.
- Separate the intersected FL-L1 annotation by the top and bottom strand.
- Copy over the FL-L1-BLAST_RM.txt into spreadsheet software and sort by the “minus” and “plus” strand and then sort by chromosome location.
- Create two new spreadsheet documents, one with the intersected coordinates for full length L1s on the minus strand and one on the bottom strand, and save as FL-L1-BLAST_RM_minus.xls and FL-L1-BLAST_RM_plus.xls.
- Save the two new documents as .txt files.
- Use the mac2unix program to convert the .txt files to the correct annotation files (Software Packages).
- Use this command in the terminal: mac2unix.sh FL-L1-BLAST_RM_minus.gff.
- Use this command in the terminal: mac2unix.sh FL-L1-BLAST_RM_plus.gff.
- Save new files with the .gff extension.
- Alternatively, use AWK to filter rows associated with the + and – strand.
- Use the following command to get the + strand: awk ‘/+/’ FL-L1_BLAST_RM.gtf > FL-L1_BLAST_RM_plus.gtf.
- Use the following command line to get the - strand: awk ‘/-/’ FL-L1_BLAST_RM.gtf > FL-L1_BLAST_RM_minus.gtf.
4. Read alignment pipeline to identify expressed L1s
| Option | Description |
| –p | This details the number of threads the computer should use running the alignment. Larger computer memory will allow more threads and should be empirically d. |
| –m 1 | This tells the program to only accept reads that have one match in the genome that is better than any other genome match. |
| – y | This is the tryhard switch which makes the mapping search for all possible matches and not allow it to quit after a fixed number of matches is reached. |
| –v 3 | This only allows the program to utilize memory for mapped reads with 3 or less mismatches to the genome. |
| –X 600 | This only allows paired reads that map within 600 bases of one another. This makes sure the read pairs are co-linear in the genome and selects against s involving processed RNA molecules. |
| –chunkmbs 8184 | This command assigns extra memory for handling the large amount of alignments possible for each L1-related read. |
Table 1: Command line options for Bowtie.
- Run alignment paired-end sequencing fastq files with the RNA-Seq sample of interest using Bowtie.
NOTE: Bowtie1 must be used and not Bowtie2 because the parameters required for unique alignment are specifically only found in this version of bowtie (Software Packages). Bowtie is used over splice-aware aligners like STAR in order evaluate concordant, contiguous reads more relevant to L1 biology and expression.- Use this command line in the Linux terminal: bowtie -p 10 -m 1 -S -y -v 3 -X 600 --chunkmbs 8184 hg_X_Y_M_index -1 hg_sample_1.fq -2 hg_sample_2.fq | samtools view -hbuS - | samtools sort – hg_sample_sorted.bam. See Table 1 for a description of command line options for Bowtie.
- Strand separate the output bam file using samtools (Software Packages) and the following Linux commands. Note that the actual flag values may vary if one is not using standard next generation sequencing protocols.
- Use this command line to select for the top strand: samtools view -h hg_sample_sorted.bam | awk 'substr($0,1,1) == "@" || $2 == 83 || $2 == 163 {print}' | samtools view -bS - > hg_sample_sorted_topstrand.bam.
- Use this command line to select for the bottom strand: samtools view -h hg_sample_sorted.bam | awk 'substr($0,1,1) == "@" || $2 == 99 || $2 == 147 {print}' | samtools view -bS - > hg_sample_sorted_bottomstrand.bam.
- Generate read counts against annotations for L1 loci using bedtools (Software Packages).
- Use this command line to generate read counts for L1s in the sense direction on the top strand: bedtools coverage -abam FL-L1-BLAST_RM_plus.gff -b hg_sample_sorted_topstrand.bam > hg_sample_sorted_bowtie_tryhard_plus_top.txt.
- Use this command line to generate read counts for L1s in the sense direction on the bottom strand: bedtools coverage -abam FL-L1-BLAST_RM_minus.gff -b hg_sample_sorted_bottomstrand.bam > hg_sample_sorted_bowtie_tryhard_minus_bottom.txt.
- Index bam file from step 5.1.1 to make it viewable in the Integrative Genomics Viewer (IGV)25 (Software Packages).
- Use this command line: samtools index hg_sample_sorted.bam
- To use a batch mode to increase the number of RNA-Seq samples piped through at a time, use a supercomputer script to complete step 4.1 called human_bowtie.sh, a script to complete steps 4.2-4.3 has been created called human_L1_pipeline.sh, and a script to complete step 4.4 has been created called bam_index.sh. These scripts may be found in Supplemental File 2 with associated supercomputer commands to run the scripts.
5. Manual curation
- Create a spreadsheet for reads mapped to each annotated L1 locus.
- Copy over hg_sample_sorted_bowtie_tryhard_minus_bottom.txt created in step 4.3.2 and label page as “minus-bottom.”
- Sort all columns based on highest to lowest number of reads found in column J.
- Copy over hg_sample_sorted_bowtie_tryhard_plus_top.txt created in step 4.3.1 and label as “top-plus” in another spreadsheet.
- Sort all columns based on highest to lowest number of reads found in column J.
- Create a third page labeled as “combined” and add all loci with ten or more reads from “minus-bottom” and “plus-top” pages.
- Sort all columns based on highest to lowest number of reads found in column J.
- Load the following files into IGV25 (Software Packages): 1) reference genome of interest to visualize annotated genes, 2) FL-L1-BLAST_RM.gff to visualize the L1 annotation, 3) hg_sample_sorted.bam to visualize mapped transcripts from sample of interest, and 4) hg_genomicDNA_sorted.bam to assess mappability of genomic regions.
- Remove coverage and junction rows associated with each bam file.
- Compress hg_sample_sorted.bam and hg_genomicDNA_sorted.bam so all the IGV tracks fit on one screen.
- Copy over hg_sample_sorted_bowtie_tryhard_minus_bottom.txt created in step 4.3.2 and label page as “minus-bottom.”
- Manually curate.
- Using coordinates from loci listed on the spreadsheet “combined” page, view called loci in IGV25 (Software Packages).
- Curate a locus to be authentically expressed off its own if there are no reads upstream in the L1 direction up to 5 kb.
- Label the row green in color and note why it is an authentically expressed L1.
NOTE: An exception to this rule exists if the region upstream of the L1 is not mappable. If this is the case, label the row red in color and note that the expression of the region upstream of the L1 promoter cannot be evaluated and therefore the L1’s expression is not able to be confidently determined.
- Label the row green in color and note why it is an authentically expressed L1.
- Curate a locus to not be authentically expressed off its own promoter if there are reads upstream up to 5 kb.
- Label the row red in color and note why it is not an authentically expressed L1.
- Curate a locus as false if it is expressed within an intron of an expressed gene in the same direction with reads upstream of the L1, if it is downstream of an expressed gene in the same direction with reads upstream of the L1, or for un-annotated expression patterns with reads upstream of the L1.
NOTE: An exception to this rule applies when there are minimal reads directly overlapping the L1 promoter start site, but slightly upstream of the L1. If there are no other reads upstream of an L1 case like this, consider this L1 to be authentically expressed. Label the row green color and note why it is an authentically expressed L1.
- Curate an L1 locus as likely to be false if the pattern of mapped reads to the locus do not correlate with the specific L1’s regions of mappability.
NOTE: For example, if an L1 is highly mappable but only has a pile up of reads in a condensed region within the L1, it is less likely to be related to L1 expression off its own promoter and more likely to be from un-annotated sources like exons or LTRs. In cases like this, curate the loci as orange and note why the locus is suspicious. Verify sources of suspicious pile-ups by checking the L1 location in UCSC genome browser. - Curate a locus to not be authentically expressed if it is within a genomic environment of sporadically expressed un-annotated regions
NOTE: For example, reads may be expressed 10 kb upstream of the L1, but every 10 kb or so there are mapped reads and some of those reads align with the L1. These L1s are less likely to be expressed off its own promoter, and more likely to have mapped reads due to un-annotated patterns of genomic expression. In cases like this, curate the loci as orange and note why the locus is suspicious.
6. Read alignment strategy to assess mappability in reference genome (optional if one has an existing aligned genomic DNA dataset)
- Download whole genome DNA sequence files and convert to .fq files
- Go to the NCBI website found here: https://www.ncbi.nlm.nih.gov/sra
- Type in WGS HeLa paired end.
- Select for Homo sapiens under Results by taxon.
- Select a sample that is paired end and has reads with 100 or more bp like the following sample: https://www.ncbi.nlm.nih.gov/sra/ERX457838[accn]
- Confirm read length by selecting Run and then Metadata as shown here: https://trace.ncbi.nlm.nih.gov/Traces/sra/?run=ERR492384
- To download the whole genome DNA sequence data, enter this command in the Linux terminal: sratoolkit.2.9.2-mac64/bin/prefetch -X 100G ERR492384
NOTE: The SRA toolkit prefetch function downloads the accession number “ERR492384” found in the NCBI site (Software Packages). The “100G” limits amount of downloaded data to 100 gigabytes. - Enter this command in the Linux terminal: fastq-dump --split-files ERR492384
NOTE: This splits the downloaded genomic DNA dataset into two fastq files.
- Run alignment using Bowtie.
- Use this command in Linux for alignment: bowtie -p 10 -m 1 -S -y -v 3 -X 600 --chunkmbs 8184 hg_X_Y_M_index -1 hg_genomicDNA_1.fq -2 hg_genomicDNA_2.fq | samtools view -hbuS - | samtools sort – hg_genomicDNA_sorted.bam.
- Refer to step 4.1 to understand parameters used in the Bowtie alignment (Software Packages).
- Download the genomically aligned bam file to assess mappability available upon author request.
- Use this command in Linux for alignment: bowtie -p 10 -m 1 -S -y -v 3 -X 600 --chunkmbs 8184 hg_X_Y_M_index -1 hg_genomicDNA_1.fq -2 hg_genomicDNA_2.fq | samtools view -hbuS - | samtools sort – hg_genomicDNA_sorted.bam.
- Index bam file from step 4.2.1 using samtools to make it viewable in IGV25 (Software Packages) to further inform manual curation.
- Use this command line in Linux: samtools index hg_genomicDNA_sorted.bam
- Assess mappability of each L1 loci
- Determine the number of uniquely mapped reads to L1 loci using the bedtools program, the FL-L1 annotation, and the aligned genomic sequence data (Software Packages).
- Use this command line in Linux: bedtools coverage -abam FL-L1-BLAST_RM.gtf –b hg_genomicDNA_sorted.bam > L1_Mappability_hg_genomicDNA.txt.
- Designate an L1 locus to have full coverage mappability when 400 unique reads are aligned to it.
- Determine the factor required to scale up or down genomic DNA aligned reads to 400 for each individual L1.
- To have a scaled measure of expression according to individual L1 locus mappability, multiply the factor determined in step 6.4.3 to the number of RNA transcript reads that align to authentically expressed L1s determined in sections 4–5.
- Determine the number of uniquely mapped reads to L1 loci using the bedtools program, the FL-L1 annotation, and the aligned genomic sequence data (Software Packages).
Results
The steps described above and described graphically in Figure 1 were applied to a human prostate tumor cell line DU145. The RNA sample was cytoplasmically prepped and was next-gen sequenced in a poly-A selected, strand-specific, paired-end protocol. Using Bowtie, the paired-end sequencing files were aligned allowing only unique matches in which the paired-end read matched better to one genomic location compared to any other genomic location. The DU145 sequence files were aligned to the human reference genome creating a bam file, which is available upon author request. Using bedtools, data was extracted from the DU145 strand-separated bam files on the number of reads that mapped to full length L1s. Those reads were sorted in a spreadsheet from largest to smallest and manually curated by examining the genomic environment around each L1 locus in IGV to confirm its authenticity (Supplemental Table 1). If a sample was curated to be authentically expressed, it was color-coded green with an explanation for its acceptance in the right most column. Examples of L1 loci accepted to be authentically expressed following guidelines described in the methods section are shown in Figure 2a-b. If a sample was rejected to be authentically expressed, it was color-coded as red with the reason for rejection on the right most column. Examples of L1 loci rejected because of expression from a promoter other than their own following guidelines described in the methods section are detailed in Figure 2c-e.
Here, only full-length L1s with an intact promoter region were studied. If this distinction is not made, a large source of transcriptional noise originating from truncated L1s is introduced. Examples of truncated L1s in DU145 are shown in Figure 3a-b where they were identified as having uniquely mapped RNA-Seq reads. In IGV, however, it is apparent that those transcripts were not initiated from the truncated L1, but from the inclusion of the L1 sequence in a gene or downstream from an expressed gene.
Overall in DU145, the percentage of full-length L1 loci and reads that are rejected as authentically expressed L1s after manual curation is approximately 50% (Supplemental Table 2) demonstrating the high level of L1 mapped transcript reads that would otherwise be recorded as false positives without manual curation. Specifically, in DU145 there were 114 total full-length L1 loci to have uniquely mapped reads in the sense direction with a total of 3,152 reads, but there were only 60 loci identified to be expressed off their own promoter after manual curation with 1,879 reads (Supplemental Table 1). This is the case even when steps were taken to reduce expression irrelevant to L1 biology by selecting for cytoplasmic mRNA. Note that the locus with the highest level of mapped transcripts in DU145 was rejected because it was not an authentically expressed L1 (Figure 4). Overall the number of mapped transcripts to specific L1 loci ranges similarly between the accepted and rejected L1 loci as authentically expressed after manual curation (Figure 4).
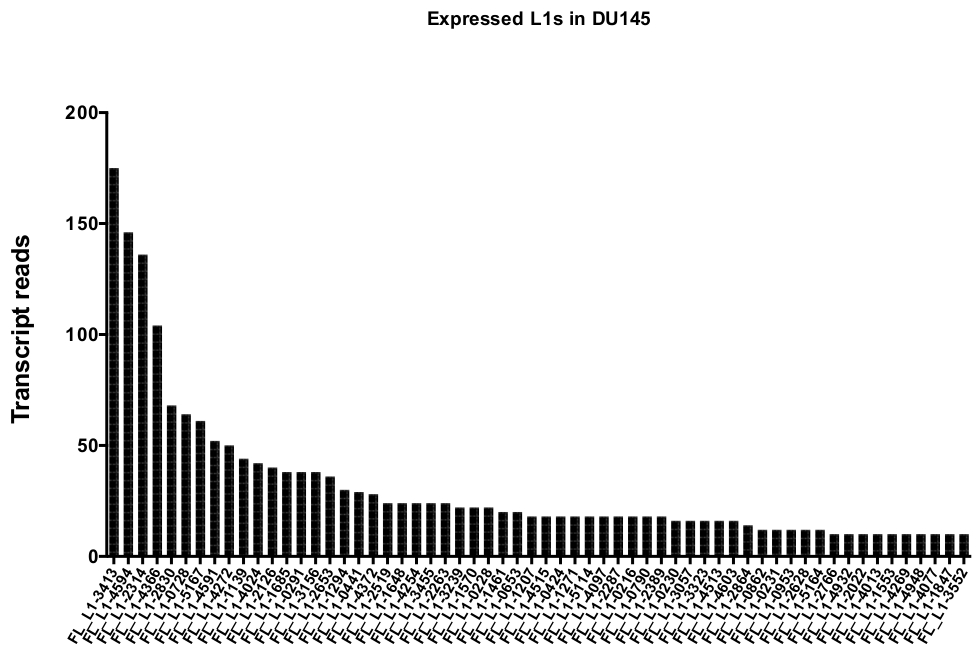
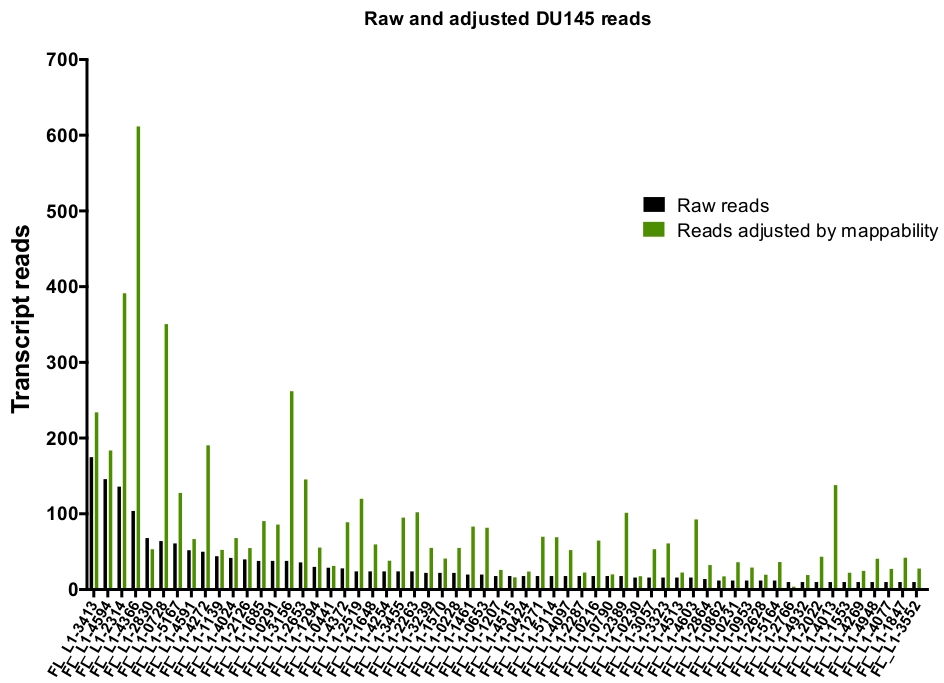
After manual curation, the number of reads that map uniquely to authentically expressed specific L1 loci in DU145 range from 175 reads to an arbitrarily chosen minimum cut off of 10 reads (Figure 5). This approach of identifying uniquely mapped transcript reads to L1s limits the ability to accurately quantify expression. To account for this, a correction factor for each locus based on its mappability was created. To create this correction factor, first bedtools was used to extract the number of uniquely mapped reads from the HeLa genomic bam file that aligned to all full-length L1 loci and graphed those loci from highest to lowest mapped transcript reads (Supplemental Figure 1). It was arbitrarily designated that L1s with 400 reads had full coverage mappability. The number of reads able to map to a L1 locus in HeLa genomic sequencing sample was scaled relative to 400 reads and that scaled number was then multiplied to the number of reads that mapped to each authentically expressed L1 loci in DU145 (Supplemental Table 2). As expected, the L1 elements that had larger correction scores for mappability came from younger subfamilies like L1PA2 (Supplemental Table 2). Once reads were adjusted for mappability scores in each locus, the quantitation for expression for most loci increased (Figure 6). The number of reads that mapped uniquely to authentically expressed specific L1 loci with mappability corrections in DU145 ranged from 612 to 4 reads and there was a re-ordering of highest to lowest expressing loci (Figure 6).

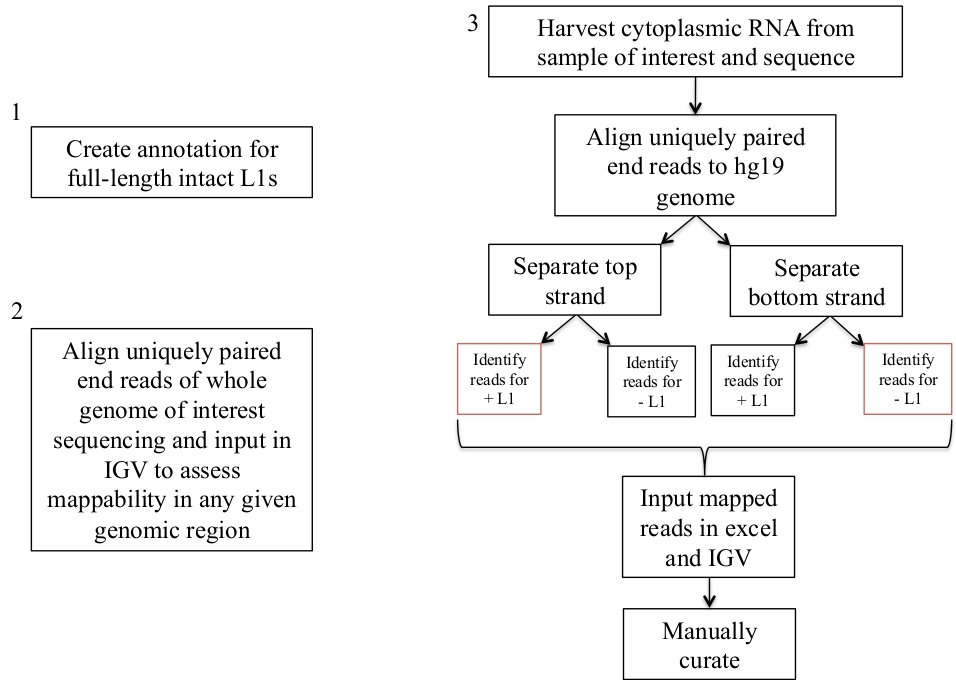
Figure 1: Workflow schematic.
Graphically described are the steps to identify expressed L1s in a human sample. Note that steps 1 and 2 do not need to be repeated if the appropriate files are already available. These appropriate files may be downloaded from Supplement File 1a-b and Supplement File 2. The boxes in red indicate the steps where bedtools coverage program is used to count the number of reads mapping to L1s in the same sense direction. These loci with sense oriented mapping reads are the L1s that should be manually curated. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Examples of curated L1 loci in DU145.
Loaded into IGV are the reference genome, the full-length L1 gff annotation file matching the reference genome version (Supplement File 1), the DU145 bam file, and lastly the genomic HeLa bam file to assess mappability, which are all available upon author request. Arrows have been added to aid in the visualization of direction of the annotated L1. Arrows and reads in red are oriented in sequence from right to left. Arrows and reads in blue are oriented in sequence from left to right. a) In IGV, this L1 locus appears to be expressed off its own promoter as there are no reads upstream of the L1 in the sense orientation for over 5 kb. This L1 has low mappability, it is not in a gene, and has evidence of expected antisense promoter activity26. b) In IGV, this L1 locus appears to be expressed off its own promoter as there are no reads upstream the L1 in the sense orientation for over 5 kb. This L1 has low mappability and is within a gene of opposite direction. c) In IGV, this L1 locus was rejected as an expressed L1 as there are upstream reads in the same orientation within 5 kb. This L1 is within a gene of the same direction so the transcript reads are most likely originating from the promoter of the expressed gene. d) In IGV, this L1 locus was rejected as an expressed L1 as there are upstream reads in the same orientation within 5 kb. This L1 is downstream of a highly expressed gene in the same direction so the transcript reads are most likely originating from the promoter of that expressed gene and extending beyond the normal gene terminator. e) In IGV, this L1 locus was rejected as an expressed L1 as there are upstream reads in the same orientation within 5 kb. This L1 is not within or near an annotated gene in the reference gene so the origin of these transcripts within and upstream of the L1 element suggest an un-annotated promoter. Please click here to view a larger version of this figure.
{kind=link}

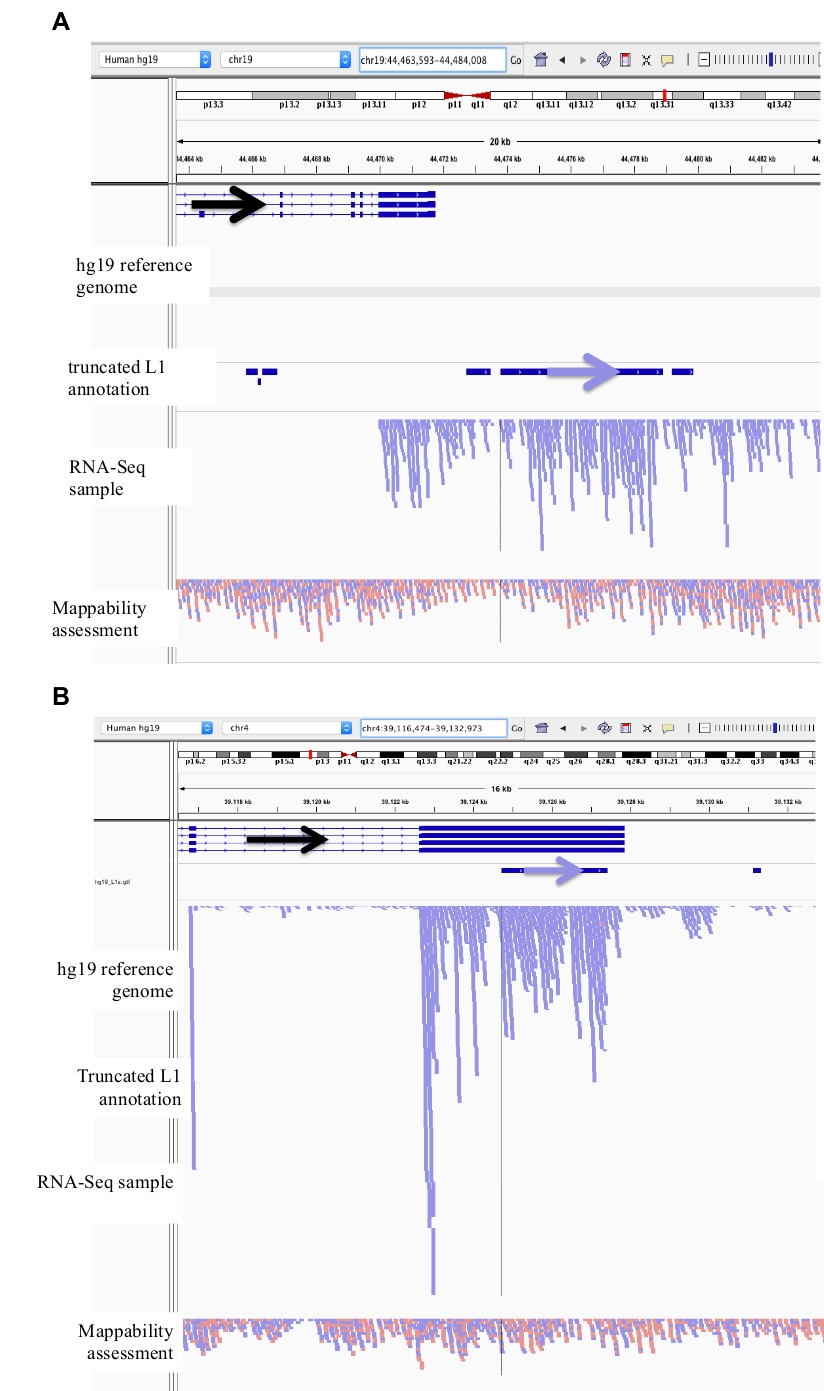
Figure 3: Background noise originates from truncated L1s as well.
Our L1 annotation does not include truncated L1s as they are a major source of background noise. Arrows have been added to aid in the visualization of direction of the annotated L1. Arrows and reads in blue are oriented in sequence from left to right. a) Demonstrated is an example of a truncated L1 in the L1MB5 sufamily that is 2706 bps. In IGV it is apparent that the reads originate from downstream extension of an expressed gene. b) Shown is another example of a truncated L1. This L1 is an L1PA11 that is 4767 bps long. In IGV it is apparent that the reads mapping uniquely to the L1 originate from the expressed exon, which the L1 is within. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Transcript reads that map uniquely to all full-length intact L1s in the human genome expressed in DU145 prostate tumor cell line.
In black are the specific loci to be identified as authentically expressed after manual curation and in red are the specific loci to be rejected as authentically expressed reads after manual curation. In grey are loci with less than ten reads mapping to each. As these loci represent a small fraction of transcript reads, they were not manually curate. The x-axis tick marks denote every 100 full-length, intact L1s. Approximately 4,500 loci are not graphically shown as they had zero mapped reads. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Transcript reads that map uniquely to authentically expressed full-length intact L1s in DU145 prostate tumor cell line.
Shown are the numbers of transcript reads that map to specific loci in DU145 cells after manual curation. Please click here to view a larger version of this figure.
{kind=link}

Figure 6: Reads mapping to authentically expressed L1 when adjusted by mappability.
Shown are the numbers of transcript reads adjusted by loci-specific mappability scores that map to manually curated L1 loci in DU145 cells. Please click here to view a larger version of this figure.
{kind=link}
Supplemental File 1: Annotations for full-length, intact human L1s according to orientation. a) FL-L1-BLAST_RM_minus.gff. b) FL-L1-BLAST_RM_plus.gff. Please click here to download this file.
Supplemental File 2: Supercomputer scripts used to automate the bioinformatics pipeline detailed in section 4. Please click here to download this file.
Supplemental Figure 1: Genomic DNA sample used to determine L1 mappability.
Shown are the number of genomic transcript reads from HeLa cell line sample that map uniquely to all 5,000 full-length L1 loci in the genome. It was designated that an L1 has full coverage mappability when 400 reads map to the L1. Please click here to download this figure.
Supplemental Table 1: Manual Curation of L1s in DU145. Please click here to download this table.
Supplemental Table 2: Curated L1s in DU145 with mappability adjustment. Please click here to download this table.
Discussion
L1 activity has been shown to cause genetic damage and instability contributing to disease27,28,29. Of the approximately 5,000 full-length L1 copies, only a few dozen evolutionarily young L1s account for the majority of retrotransposition activity2. However, there is evidence that even some older, retrotranspositionally-incompentent L1s are still able to produce DNA damaging proteins30. To fully appreciate the role of L1s in genomic instability and disease, L1 expression at the locus-specific level must be understood. However, the high background of L1-related sequences incorporated into other RNAs unrelated to L1 retrotransposition poses a significant challenge in interpreting authentic L1 expression. Another challenge in identifying and therefore understanding expression patterns of individual L1 loci occurs because of their repetitive nature that does not allow many short read sequences to map to a single unique locus. To overcome these challenges, we developed the above-described approach in identifying expression of individual L1 loci using RNA-Seq data.
Our approach filters the high level (over 99%) of transcriptional noise generated from L1 sequences that are unrelated to L1 retrotransposition by taking a number of steps. The first step involves the preparation of cytoplasmic RNA. By selecting for cytoplasmic RNA, L1-related reads found within expressed intronic mRNA in the nucleus are significantly depleted. In the sequencing library preparation, another step taken to reduce transcriptional noise unrelated to L1s include the selection of polyadenylated transcripts. This removes L1-related transcript noise found in non-mRNA species. Another step includes strand-specific sequencing in order to identify and eliminate antisense L1-related transcripts. The use of an annotation for full-length L1s with functional promoter regions when identifying the number of RNA-Seq transcripts that map to L1s also eliminates background noise that otherwise originate from truncated L1s. Finally, the last critical step in eliminating transcriptional noise of L1 sequences unrelated to L1 retrotransposition is the manual curation of full-length L1s identified to have mapped RNA-Seq transcripts. The manual curation involves the visualization of each bioinformatically identified-to-be-expressed L1 locus in the context of its surrounding genomic environment to confirm that expression originates from the L1 promoter. This approach was applied to DU145, a prostate tumor cell line. Even with all the preparation-related steps taken to reduce background noise, approximately 50% of L1 loci identified bioinformatically in DU145 were rejected as L1 background noise originating from other transcriptional sources (Figure 4), emphasizing the rigor required to produce reliable results. This approach using manual curation is labor intensive, but necessary in the development of this pipeline to evaluate and understand the genomic environment surrounding a full-length L1. The next steps include reducing the amount of necessary manual curation by automating some of the curation rules, though due to the still not completely known nature of genomic expression, un-annotated sources of expression in the reference genome, regions of low mappability, and even complicating factors involved with the construction of a reference genome it is not be possible to fully automate L1 curation at this time.
The second challenge in identifying expression of individual L1 loci with sequencing relates to the mapping of repetitive L1 transcripts. In this alignment strategy, it is required that a transcript must align uniquely and co-linearly to the reference genome in order to be mapped. By selecting for paired-end sequences that map concordantly, the amount of transcripts that uniquely align to L1 loci found in the reference genome increases. This unique-mapping strategy provides confidence in the calling of reads mapping specifically to a single L1 locus, though it potentially underestimates the expression quantity of each identified-to-be-authentically expressed, repetitive L1. To approximately correct for this underestimation, a “mappability” score for each L1 locus based on its mappability was developed and applied to the number of uniquely mapped transcript reads (Figure 6). It is of note that ideally, mappability should be scored to full coverage reads across the full-length L1 according to the matched WGS sample. Here, we use WGS of HeLa cells to determine mappability scores of each L1 loci in order to inflate or deflate reads mapping to L1 loci in DU145 prostate tumor cell lines. This mappability calculation is a crude correction score, but the chosen ‘complete coverage mappability’ of 400 reads was determined with the dynamic nature of tumor cell lines in mind. It can be observed in Supplemental Figure 1, that there are a few L1 loci with HeLa WGS with extremely high number of mapped reads. These likely come from duplicated chromosome sequences within HeLa that are not within the reference genome, which is why those loci were not chosen to be representative of complete mappability coverage. Instead it was determined that the average of 100% read coverage occurs around 400 reads according to Supplemental Figure 1 and was then assumed that this average applies to the DU145 tumor prostate cell line as well.
This alignment strategy with 100-200 bp reads from RNA-Seq technology also preferentially selects for evolutionarily older L1s within the reference genome as older L1s have accumulated over time unique mutations that make them more mappable. This approach, therefore, has limited sensitivity when it comes to identifying the youngest of L1s as well as non-reference, polymorphic L1s. To identify the youngest of L1s, we suggest using 5’ RACE selection of L1 transcripts and sequencing technology like PacBio that make use of longer reads21. This permits more unique mapping and therefore confident identification of the expressed, young L1s. Using RNA-Seq and PacBio approaches together can lead to a more comprehensive list of authentically expressed L1s. To identify authentically expressed polymorphic L1s, the first next steps include construction and insertion of polymorphic sequences into the reference genome.
The biological and technical challenges in studying repeat sequences are great, though with the above rigorous procedure to remove transcriptional noise of L1 sequences un-related to retrotransposition using RNA-sequencing technology, we begin to sift through the large levels of transcriptional background noise and being to confidently and stringently identify L1 expression patterns and quantity at the individual locus level.
Disclosures
The authors have nothing to disclose.
Acknowledgements
We would like to thank Dr. Yan Dong for the DU145 prostate tumor cells. We would like to thank Dr. Nathan Ungerleider for his guidance and advice in creating supercomputer scripts. Some of this work was funded by NIH grants R01 GM121812 to PD, R01 AG057597 to VPB, and 5TL1TR001418 to TK. We would also like to acknowledge support from the Cancer Crusaders and the Tulane Cancer Center Bioinformatics Core.
Materials
| Name | Company | Catalog Number | Comments |
| 1 M HEPES | Affymetrix | AAJ16924AE | |
| 5 M NaCl | Invitrogen | AM9760G | |
| Agilent bioanalyzer 2100 | Agilent technologies | ||
| Agilent RNA 6000 Nano Kit | Agilent technologies | 5067-1511 | |
| bedtools.26.0 | https://bedtools.readthedocs.io/en/latest/content/installation.html | ||
| bowtie-0.12.8 | https://sourceforge.net/projects/bowtie-bio/files/bowtie/0.12.8/ | ||
| Cell scraper | Olympus plastics | 25-270 | |
| Chloroform | Fisher | C298-500 | |
| Digitonin | Research Products International Corp | 50-488-644 | |
| Ethanol | Fisher | A4094 | |
| Gibco (Phosphate Buffered Saline) | Invitrogen | 10-010-049 | |
| Homogenizer | Thomas Scientific | BBI-8541906 | |
| IGV 2.4 | https://software.broadinstitute.org/software/igv/download | ||
| Isopropanol | Fisher | A416-500 | |
| mac2unix | https://sourceforge.net/projects/cs-cmdtools/files/mac2unix/ | ||
| Q-tips | Fisher | 23-400-122 | |
| RNAse later solution | Invitrogen | AM7022 | |
| RNaseZap RNase Decontamination Solution | Invitrogen | AM9780 | |
| samtools-1.3 | https://sourceforge.net/projects/samtools/files/ | ||
| sratoolkit.2.9.2 | https://github.com/ncbi/sra-tools/wiki/Downloads | ||
| SUPERase·In RNase Inhibitor | Invitrogen | AM2694 | |
| Trizol | Invitrogen | 15-596-018 | |
| Water (DNASE, RNASE free) | Fisher | BP2484100 |
References
- International Human Genome Sequencing. Initial sequencing and analysis of the human genome. Nature. 409, 860 (2001).
- Brouha, B., et al. Hot L1s account for the bulk of retrotransposition in the human population. Proceedings of the National Academy of Sciences of the United States of America. 100 (9), 5280-5285 (2003).
- Dombroski, B. A., Mathias, S. L., Nanthakumar, E., Scott, A. F., Kazazian, H. H. Isolation of an active human transposable element. Science. 254 (5039), 1805 (1991).
- Swergold, G. D. Identification, characterization, and cell specificity of a human LINE-1 promoter. Molecular and Cellular Biology. 10 (12), 6718-6729 (1990).
- Speek, M. Antisense promoter of human L1 retrotransposon drives transcription of adjacent cellular genes. Molecular and Cellular Biology. 21 (6), 1973-1985 (2001).
- Deininger, L., Batzer, M. A., Hutchison, C. A., Edgell, M. H. Master genes in mammalian repetitive DNA amplification. Trends in Genetics. 8 (9), 307-311 (1992).
- Boissinot, S., Chevret, P., Furano, A. L1 (LINE-1) Retrotransposon Evolution and Amplification in Recent Human History. Molecular Biology and Evolution. 17 (6), 915-918 (2000).
- Khazina, E., Weichenrieder, O. Non-LTR retrotransposons encode noncanonical RRM domains in their first open reading frame. Proceedings of the National Academy of Sciences of the United States of America. 106 (3), 731-736 (2009).
- Martin, S. L., Bushman, F. D. Nucleic acid chaperone activity of the ORF1 protein from the mouse LINE-1 retrotransposon. Molecular and Cellular Biology. 21 (2), 467-475 (2001).
- Feng, Q., Moran, M. H., Kazazian, H. H., Boeke, J. D. Human L1 Retrotransposon Encodes a Conserved Endonuclease Required for Retrotransposition. Cell. 87 (5), 905-916 (1996).
- Mathias, S. L., Scott, A. F., Kazazian, H. H., Boeke, J. D., Gabriel, A. Reverse transcriptase encoded by a human transposable element. Science. 254 (5039), 1808 (1991).
- Luan, D. D., Korman, M. H., Jakubczak, J. L., Eickbush, T. H. Reverse transcription of R2Bm RNA is primed by a nick at the chromosomal target site: A mechanism for non-LTR retrotransposition. Cell. 72 (4), 595-605 (1993).
- van den Hurk, J. A. J. M., et al. Novel types of mutation in the choroideremia (CHM) gene: a full-length L1 insertion and an intronic mutation activating a cryptic exon. Human Genetics. 113 (3), 268-275 (2003).
- Miné, M., et al. A large genomic deletion in the PDHX gene caused by the retrotranspositional insertion of a full-length LINE-1 element. Human Mutation. 28 (2), 137-142 (2007).
- Solyom, S., et al. Pathogenic orphan transduction created by a nonreference LINE-1 retrotransposon. Human Mutation. 33 (2), 369-371 (2012).
- Hancks, D. C., Kazazian, H. H. Roles for retrotransposon insertions in human disease. Mobile DNA. Mobile DNA. 7, 9-9 (2016).
- Tubio, J. M. C., et al. Mobile DNA in cancer. Extensive transduction of nonrepetitive DNA mediated by L1 retrotransposition in cancer genomes. Science. 345 (6196), 1251343-1251343 (2014).
- Ewing, A. D., et al. Widespread somatic L1 retrotransposition occurs early during gastrointestinal cancer evolution. Genome Research. 25 (10), 1536-1545 (2015).
- Beck, C. R., Garcia-Perez, J. L., Badge, R. M., Moran, J. V. LINE-1 elements in structural variation and disease. Annual Review of Genomics and Human Genetics. 12, 187-215 (2011).
- Philippe, C., et al. Activation of individual L1 retrotransposon instances is restricted to cell-type dependent permissive loci. eLife. 5, e13926 (2016).
- Deininger, P., et al. A comprehensive approach to expression of L1 loci. Nucleic Acids Research. 45 (5), e31-e31 (2017).
- Jin, Y., Tam, O. H., Paniagua, E., Hammell, M. TEtranscripts: a package for including transposable elements in differential expression analysis of RNA-seq datasets. Bioinformatics. 31 (22), 3593-3599 (2015).
- . . Agilent RNA 6000 Nano Kit Guide. , (2017).
- Mueller, O. L., Schroeder, A. . RNA Integrity Number (RIN) –Standardization of RNA Quality Control. , (2016).
- Robinson, J. T., et al. Integrative genomics viewer. Nature Biotechnology. 29, 24 (2011).
- Speek, M. Antisense promoter of human L1 retrotransposon drives transcription of adjacent cellular genes. Molecular Cellular Biology. 21 (6), 1973-1985 (2001).
- Belancio, V. P., Deininger, L., Roy-Engel, A. M. LINE dancing in the human genome: transposable elements and disease. Genome Medicine. 1 (10), 97-97 (2009).
- Iskow, R. C., et al. Natural Mutagenesis of Human Genomes by Endogenous Retrotransposons. Cell. 141 (7), 1253-1261 (2010).
- Scott, E. C., et al. A hot L1 retrotransposon evades somatic repression and initiates human colorectal cancer. Genome Research. 26 (6), 745-755 (2016).
- Kines, K. J., Sokolowski, M., deHaro, D. L., Christian, C. M., Belancio, V. P. Potential for genomic instability associated with retrotranspositionally-incompetent L1 loci. Nucleic Acids Research. 42 (16), 10488-10502 (2014).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved