
Method Article
Imagerie de la production gamma d'interféron in situ dans la rate de souris suite à l'infection à Listeria monocytogenes
Dans cet article
Résumé
Ici, nous décrivons une méthode simple de formation image confocale pour visualiser la localisation in situ des cellules sécrétant le gamma d'interféron de cytokine dans les organes lymphoïdes secondaires murines. Ce protocole peut être étendu pour la visualisation d'autres cytokines dans divers tissus.
Résumé
Les cytokines sont de petites protéines sécrétées par les cellules, qui sont des communications cellulaires qui sont cruciales pour des réponses immunitaires efficaces. Une caractéristique des cytokines est leur pléiotropisme, car ils sont produits par et peuvent affecter une multitude de types de cellules. En tant que tel, il est important de comprendre non seulement quelles cellules produisent des cytokines, mais aussi dans quel environnement ils le font, afin de définir des thérapies plus spécifiques. Ici, nous décrivons une méthode pour visualiser la production de cytokine in situ suivant l'infection bactérienne. Cette technique repose sur l'imagerie des cellules productrices de cytokine dans leur environnement naturel par microscopie confocale. Pour ce faire, les sections de tissu sont tachées pour les marqueurs de plusieurs types de cellules ainsi qu'une tache de cytokine. Clé de cette méthode, la sécrétion de cytokine est bloquée directement in vivo avant de récolter le tissu d'intérêt, permettant la détection de la cytokine qui s'est accumulée à l'intérieur des cellules productrices. Les avantages de cette méthode sont multiples. Tout d'abord, le microenvironnement dans lequel les cytokines sont produites est préservé, ce qui pourrait finalement informer sur les signaux nécessaires à la production de cytokine et les cellules touchées par ces cytokines. En outre, cette méthode donne une indication de l'emplacement de la production de cytokine in vivo, car elle ne repose pas sur la re-stimulation in vitro artificielle des cellules productrices. Cependant, il n'est pas possible d'analyser simultanément la signalisation en aval de cytokine dans les cellules qui reçoivent la cytokine. De même, les signaux cytokine observés ne correspondent qu'à la fenêtre temporelle au cours de laquelle la sécrétion de cytokine a été bloquée. Alors que nous décrivons la visualisation du gamma d'interféron de cytokine (IFN) dans la rate suivant l'infection de souris par les bactéries intracellulaires Listeria monocytogenes, cette méthode pourrait potentiellement être adaptée à la visualisation de n'importe quelle cytokine dans la plupart des organes.
Introduction
L'orchestration d'une réponse immunitaire efficace contre un agent pathogène nécessite une intégration complexe des signaux affichés par une variété de cellules immunitaires qui sont souvent dispersées entre l'organisme. Afin de communiquer, ces cellules produisent de petites protéines solubles avec de multiples fonctions biologiques qui agissent comme immunomodulateurs nommés cytokines. Cytokines contrôle le recrutement cellulaire, l'activation et la prolifération et sont donc connus pour être des acteurs clés dans la promotion des réponses immunitaires1. Les réponses immunitaires efficaces exigent que les cytokines soient libérés dans un modèle spatiotemporal très organisé reliant des cellules spécifiques pour induire des signaux spécifiques. Par conséquent, il est crucial d'étudier la production de cytokine et sa signalisation in situ, en tenant compte du microenvironnement dans lequel les cytokines sont produites.
Listeria monocytogenes (L. monocytogenes) est une bactérie intracellulaire Gram-positive utilisée comme modèle de choix pour étudier les réponses immunitaires aux agents pathogènes intracellulaires chez la souris. Une cytokine, IFN gamma (IFN) est produite rapidement, dans les 24 h suivant l'infection de L. monocytogenes. Il est nécessaire pour le dégagement d'agent pathogène, car les souris assommées pour IFN sont fortement sensibles à l'infection de L. monocytogenes 2. IFN est pléiotrope et produit par plusieurs cellules suite à l'infection3. Tandis que l'IFN produit par les cellules de tueurnaturel (NK) est exigé pour l'activité antibactérienne directe 4, IFNd d'autres sources ont été montrés pour avoir d'autres fonctions. En effet, nous et d'autres ont récemment constaté que l'IFN produit par les lymphocytes T CD8 A une fonction spécifique dans la régulation directe de la différenciation des lymphocytes T5,6,7. En tant que tel, la compréhension des cellules qui produisent IFN (et dans lequel le microenvironnement) est cruciale pour disséquer sa fonction.
La technique la plus courante pour étudier la production de cytokine repose sur la coloration cytokine intracellulaire analysée par cytométrie de flux. Cette méthode permet la détection simultanée de cytokines multiples combinés avec des marqueurs de surface cellulaire dans un seul échantillon, fournissant un outil extrêmement utile pour étudier la production de cytokine. Cependant, l'utilisation de la technique susmentionnée implique de perdre toute information spatiale. En outre, la détection de cytokine repose souvent sur la re-stimulation in vitro pour permettre la détection de cytokine. En tant que tel, la capacité d'une cellule donnée de produire une cytokine est analysée, et elle n'est pas nécessairement corrélée avec la sécrétion réelle de cytokine in situ. D'autres méthodes utilisent des souris reporter pour lesquelles l'expression des protéines fluorescentes est en corrélation avec la transcription cytokine et permet la visualisation sur un niveau unicellulaire8. Bien que cette méthode puisse suivre la transcription de cytokine in situ, il y a un nombre limité de souris cytokine-reporter disponibles. En outre, la transcription, la traduction et la sécrétion peuvent parfois être déconnectées, et les protéines fluorescentes ont une demi-vie différente de la cytokine qu'ils rapportent, ce qui rend cette méthode parfois inadéquate pour la visualisation cytokine in situ.
Ici, nous décrivons une méthode pour visualiser la production in situ de cytokine par microscopie confocale à la résolution simple de cellules. Cette technique permet la visualisation de la source cellulaire et de la niche environnante dans le tissu. Ce protocole décrit spécifiquement la visualisation de la production d'IFNMD dans la rate des souris infectées par L. monocytogenes, en se concentrant ici sur la production d'IFNMD par les cellules NK et les cellules CD8et T spécifiques à l'antigène. Cependant, il peut être étendu et adapté à la caractérisation de toute production de cytokine dans le contexte d'autres situations où les cytokines sont produites telles que l'infection, l'inflammation ou les maladies auto-immunes, tant que la cytokine ciblée peut être conservée dans les cellules par inhibiteur intracellulaire du transport des protéines.
Protocole
Toutes les expériences impliquant des souris étaient en accord avec la loi britannique sur les procédures scientifiques de 1986.
1. Transfert adoptif de CD8 antigénique-spécifique- cellules T chez les souris
- Isoler l'ovalbumine (OVA) spécifique CD8- lymphocytes T (OTI) exprimant la protéine fluorescente verte (OTI-GFP) ou la protéine fluorescente rouge (OTI-RFP) de la suspension des ganglions lymphatiques des souris transgéniques des récepteurs T-cell9,10 à l'aide d'une souris CD8- Kit d'isolation des lymphocytes T selon les instructions de fabrication. Préparer la suspension cellulaire en brisant les ganglions lymphatiques à l'aide d'un piston de seringue, comme décrit précédemment11.
- Transférer les cellules OTI-GFP ou OTI-RFP (3 x 106 cellules) dans les souris de type sauvage C57BL/6 par injection intraveineuse comme décrit par Cahalan, et al.12. Utilisez des souris qui sont généralement de 6 à 12 semaines.
REMARQUE: Cette étape est facultative et seulement nécessaire pour le suivi des cellules CD8et T spécifiques à l'antigène.
2. Infection de Listeria monocytogenes
- Élargir L. monocytogenes génétiquement modifié pour exprimer OVA (LM-OVA)13 à une phase exponentielle de croissance de l'infusion de cœur de bouillon à 37 oC sous une agitation douce jusqu'à ce que l'OD600 atteigne 0,08 à 0,1, comme décrit précédemment dans la référence 14.
- Injecter 100 L (volume maximum de 200 l) de 0,1 à 0,5 LD50 LM-OVA dilué dans des cellules saline tamponnées au phosphate (PBS) par injection intraveineuse à l'aide d'une seringue à insuline de 29 G, dans des souris de type sauvage C57BL/6 portant des cellules OTI-GFP ou OTI-RFP lorsqu'elles sont indiquées.
REMARQUE: Dans nos mains, 0,1 x LD50 LM-OVA correspond à 2 x 104 unités de formation de colonies (CFU). L. monocytogenes génétiquement modifié pour exprimer ova est utilisé pour activer les cellules PRÉCÉDEMMENT transférés OTI CD8 T, mais d'autres souches de L. monocytogenes peuvent être utilisés.
3. Traitement avec Brefeldin A (BFA) pour bloquer la sécrétion de cytokine
- Injecter 250 g de BFA dans 200 OL de PBS intrapéritonément 6 h avant le sacrifice de souris à l'aide d'une seringue à insuline de 29 G.
REMARQUE: Le BFA lyophilisé est d'abord resuspendu dans le sulfoxide de diméthyle (DMSO) pour préparer le stock de concentration de 25 mg/mL. Le BFA est ensuite dilué dans le PBS à température ambiante (RT) pour éviter la cristallisation avant l'injection. L'inhibition de la sécrétion de cytokine induit l'accumulation d'IFN Dans les cellules. Ceci est crucial pour la détection de cytokine.
4. Récolte de la rate
- Euthanasier les souris en utilisant une concentration croissante de CO2 suivie d'une luxation cervicale.
REMARQUE: Suivez les directives de l'institution locale pour l'euthanasie humaine des souris. - Nettoyer l'abdomen avec 70% d'éthanol, faire une incision avec des ciseaux pour faire une coupe de 1 à 2 cm à travers la peau sur le flanc gauche de la souris, où la rate est située. Faites soigneusement une incision dans le péritoine pour exposer la rate et retirez-la avec une pince à épiler. Récoltez la rate, en prenant soin de ne pas la presser avec des forceps ou de la couper pour éviter de perturber l'architecture de la rate.
5. Fixation de la rate avec paraformaldéhyde (PFA)
- Préparer la solution fixative en mélangeant 3,75 ml de PBS et 3,75 ml de 0,2 M L-lysine. Ajouter 21 mg de m-periodate de sodium et bien mélanger. Ajouter ensuite 2,5 ml de 4 % de PFA et 20 l de 12 NaOH.
REMARQUE: Utilisez la solution fixative le même jour et jetez l'excédent. Ne le stockez pas. Cette étape de fixation est importante si l'échantillon contient des protéines fluorescentes telles que le GFP. N'utilisez pas de PFA contenant des traces de méthanol, car il dénature les protéines fluorescentes.
MISE EN GARDE: La PFA est toxique et doit être traitée avec prudence. - Immerger la rate dans le fixatif et fixer pendant un minimum de 4 h, généralement 16 à 20 h à 4 oC sous une douce agitation.
- Jeter la solution fixative et ajouter 5 ml de PBS pendant 5 min à RT sous une douce agitation.
- Remplacer le PBS par 5 ml de PBS frais incuber pendant 1 h à 4 oC sous une douce agitation.
- Remplacer le PBS par 5 ml de saccharose à 30 %, incuber de 12 à 24 h.
REMARQUE: Cette méthode aide à maintenir la morphologie des tissus. Après l'incubation avec la solution de saccharose, l'organe devrait couler au fond du puits.
6. Congélation et sectionnement
- Mettre de la glace sèche dans un grand réceptacle et placer un plus petit réceptacle à l'intérieur contenant environ 50 ml de méthanol pur et quelques morceaux de glace sèche.
- Séchez délicatement la rate sur une lingette sans résineuse.
- Placez la rate à l'intérieur d'un moule de base contenant une goutte de température de coupe optimale (OCT) composé au fond. Veillez à ne pas produire de bulles. Ajouter OCT sur la rate.
- Avec les forceps, déposer le moule de base sur la surface du méthanol, en s'assurant qu'il ne touche pas l'OCT. Congeler le tissu aussi rapidement que possible pour minimiser les artefacts.
- Lorsqu'il est gelé, procéder à la section.
REMARQUE: La rate congelée peut être maintenue à -80 oC pendant plusieurs mois. -
Sectiondure le tissu à l'aide d'un cryomicrotome.
- Définir la température de la chambre sur le cryostat devrait être de -21 oC. Couper les sections de l'épaisseur désirée (généralement autour de 10 m). Ce protocole fonctionne avec des épaisseurs allant jusqu'à 30 m.
- Recueillir des sections sur des lames de microscope en verre (voir le tableau des matériaux)et inspecter visuellement.
REMARQUE: Les sections peuvent être conservées à -80 oC pendant plusieurs mois.
7. Coloration immunofluorescente
- Permettez à la section de venir à RT.
- Dessinez un cercle avec un bloqueur liquide (p. ex. stylo PAP) autour de la section tissulaire. Dessinez à l'extérieur de l'OCT ou il ne collera pas.
- Une fois qu'il a séché, réhydrater l'échantillon en plaçant PBS sur la section de tissu pendant 5 min.
REMARQUE: Le volume mis sur la section dépend de la taille de la section. Nous utilisons généralement 100 à 300 l. Ne laissez pas sécher les sections une fois qu'elles sont réhydratées. - Rincer avec PBS au moins deux fois pour s'assurer que la section est bien respectée à la diapositive.
-
Ajouter une solution de blocage à la section pour diminuer la liaison non spécifique des anticorps.
- Préparer la solution de blocage comme suit : PBS avec 0.1% Triton X100, 2% sérum foetal de veau (FCS), 2.5 -g/mL Fc bloqueur de récepteur (anti-souris CD16/32). Ajoutez ensuite 2 à 5 % de sérum normal de l'espèce de chaque anticorps secondaire du panneau de coloration.
REMARQUE: Si les anticorps sont directement conjugués/biotinylated, ajoutez 5% de sérum normal de l'espèce de chaque anticorps primaire. Si un anticorps primaire et l'un des anticorps secondaires proviennent de la même espèce (p. ex., anticorps primaires élevés chez le lapin et l'antirat de lapin secondaire), n'utilisez pas les espèces de sérum normales, car cela augmentera le signal de fond. - Retirez délicatement le PBS de la section par aspiration et ajoutez 100 l de la solution de blocage par section d'échantillon. Incuber dans une chambre humide couverte pour un minimum de 1 h à RT.
- Préparer la solution de blocage comme suit : PBS avec 0.1% Triton X100, 2% sérum foetal de veau (FCS), 2.5 -g/mL Fc bloqueur de récepteur (anti-souris CD16/32). Ajoutez ensuite 2 à 5 % de sérum normal de l'espèce de chaque anticorps secondaire du panneau de coloration.
-
Tache avec des anticorps primaires.
- Diluer les anticorps primaires à la concentration optimale dans la solution de blocage. La concentration générale d'anticorps de point de départ est de 5 g/mL, mais elle doit être optimisée pour chaque anticorps et tissu.
REMARQUE: Si les anticorps sont directement conjugués, centrifuger le mélange d'anticorps à 17,135 x g (13.500 tr/min) pendant 15 min à 4 oC avant de l'utiliser. Les fluorophores peuvent se précipiter. Cette étape permettra de granuler les précipités et ainsi empêcher le dépôt non spécifique des anticorps précipités sur la diapositive. - Remplacez la solution de blocage par le mélange d'anticorps primaire pour chaque échantillon.
- Incuber pendant 4 h à RT ou pendant la nuit (OVN) à 4 oC dans une chambre humide couverte.
- Diluer les anticorps primaires à la concentration optimale dans la solution de blocage. La concentration générale d'anticorps de point de départ est de 5 g/mL, mais elle doit être optimisée pour chaque anticorps et tissu.
-
Effectuer le lavage.
- Préparer le tampon de lavage en ajoutant 2 % de FCS à PBS.
- Laver 4 fois avec un tampon de lavage : un rapide (sans incubation), un pour 10 min et deux pour 5 min. Ensuite, effectuer un lavage final avec PBS pendant 5 min.
-
Coloration avec des anticorps secondaires.
- Diluer les anticorps secondaires d'intérêt à la concentration optimale dans la solution de blocage. Centrifuger le mélange, tel que décrit pour les anticorps primaires.
- Retirez la solution de lavage finale. Ajouter le mélange d'anticorps secondaires sur le dessus de la section et couver pendant 1 à 4 h à RT dans une chambre humide couverte.
- Laver 4 fois avec un tampon de lavage : un rapide (sans incubation), un pour 10 min et deux pour 5 min. Ensuite, effectuer un lavage final avec PBS pendant 5 min.
- Retirez la solution de lavage finale. Laisser le PBS s'évaporer, mais ne pas trop sécher la section. Placer une goutte du milieu de montage sur le dessus de l'échantillon et placer soigneusement le verre de couverture sur le dessus de celui-ci. Le support de montage doit récupérer toute la section. Laissez-le polymérase OVN à RT protégé de la lumière.
REMARQUE: Dessinez un cercle autour de la section sur le revers de la diapositive avant d'appliquer le support de montage. Une fois que le milieu de montage est appliqué, le tissu peut devenir difficile à voir. - Conserver les diapositives dans l'obscurité à 4 oC jusqu'à ce qu'elles soient prêtes à l'image.
8. Imagerie et analyse
- Effectuer l'imagerie de la coloration avec un microscope confocal.
REMARQUE: Dans ce protocole, un microscope à balayage laser spectral inversé a été utilisé (voir le Tableau des matériaux), ainsi que les objectifs 10x/NA 0,40 ou 60x/NA 1.4 (pour l'analyse de la localisation sous-cellulaire cytokine). Des longueurs d'onde d'excitation et d'émission sont affichées pour chaque fluorophore et protéine fluorescente dans le Tableau des matériaux. - Effectuer l'analyse et la quantification au besoin à l'aide d'un logiciel de traitement d'image (p. ex., Imaris ou Fidji).
Résultats
L'IFNMD produite dans les 24 premiers h après l'infection de Listeria monocytogenes est essentielle pour contrôler la propagation de cet agent pathogène. À l'aide de ce protocole, nous pouvons visualiser non seulement quelles cellules produisent des IFN, mais aussi si elles sont situées dans un microenvironnement spécifique. Pour nous aider à délimiter l'architecture de la rate, nous avons étiqueté les cellules connues pour avoir un emplacement particulier dans la rate. Le marqueur F4/80 étiquette tous les macrophages et met en évidence la pulpe rouge. Le marqueur B220 étiquette les cellules B et met en évidence les follicules cellulaires B entourant la zone des lymphocytes T. Le marqueur CD169 étiquette les macrophages de zone marginale, entourant la pulpe blanche (figure 1). La plupart des cellules OTI, qu'elles expriment OU non l'IFN, sont présentes dans la pulpe blanche et, en tant que telles, toutes les images sont celles de la pulpe blanche, à moins qu'elles ne soient indiquées.
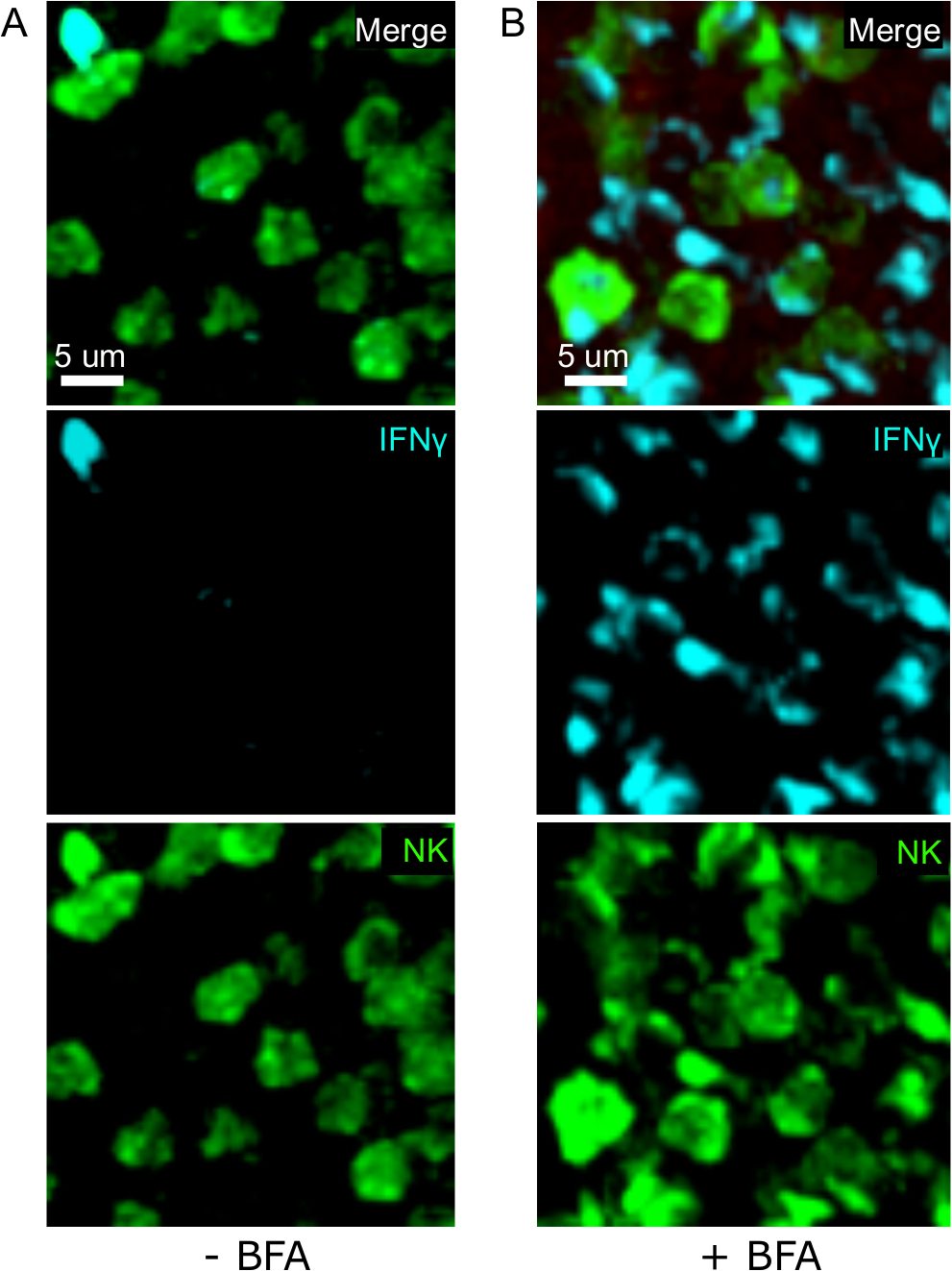
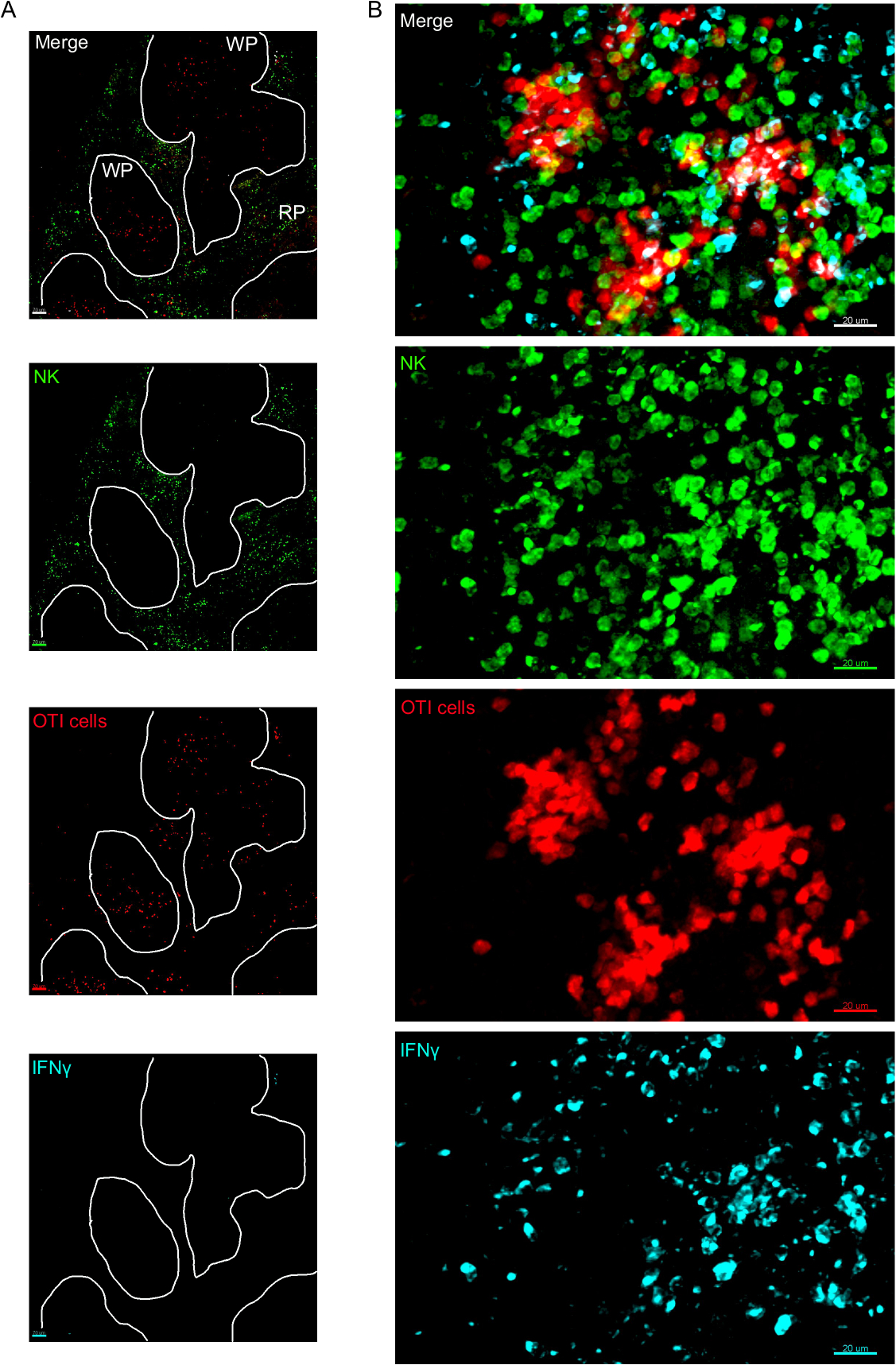
Une étape critique dans ce protocole est l'utilisation de BFA pour inhiber la sécrétion de cytokine. En effet, la détection de l'IFN MD par les cellules NK a été grandement altérée lorsque les souris n'ont pas été traitées par BFA (Figure 2). À l'aide de notre protocole, nous pourrions constater qu'au moins deux types de cellules produisent des IFN 24 h après l'infection — les cellules NK et les cellules CD8 et T spécifiques à l'antigène (figure 3) — de la même façon que ce qui a été trouvé précédemment par cytométrie d'écoulement3.
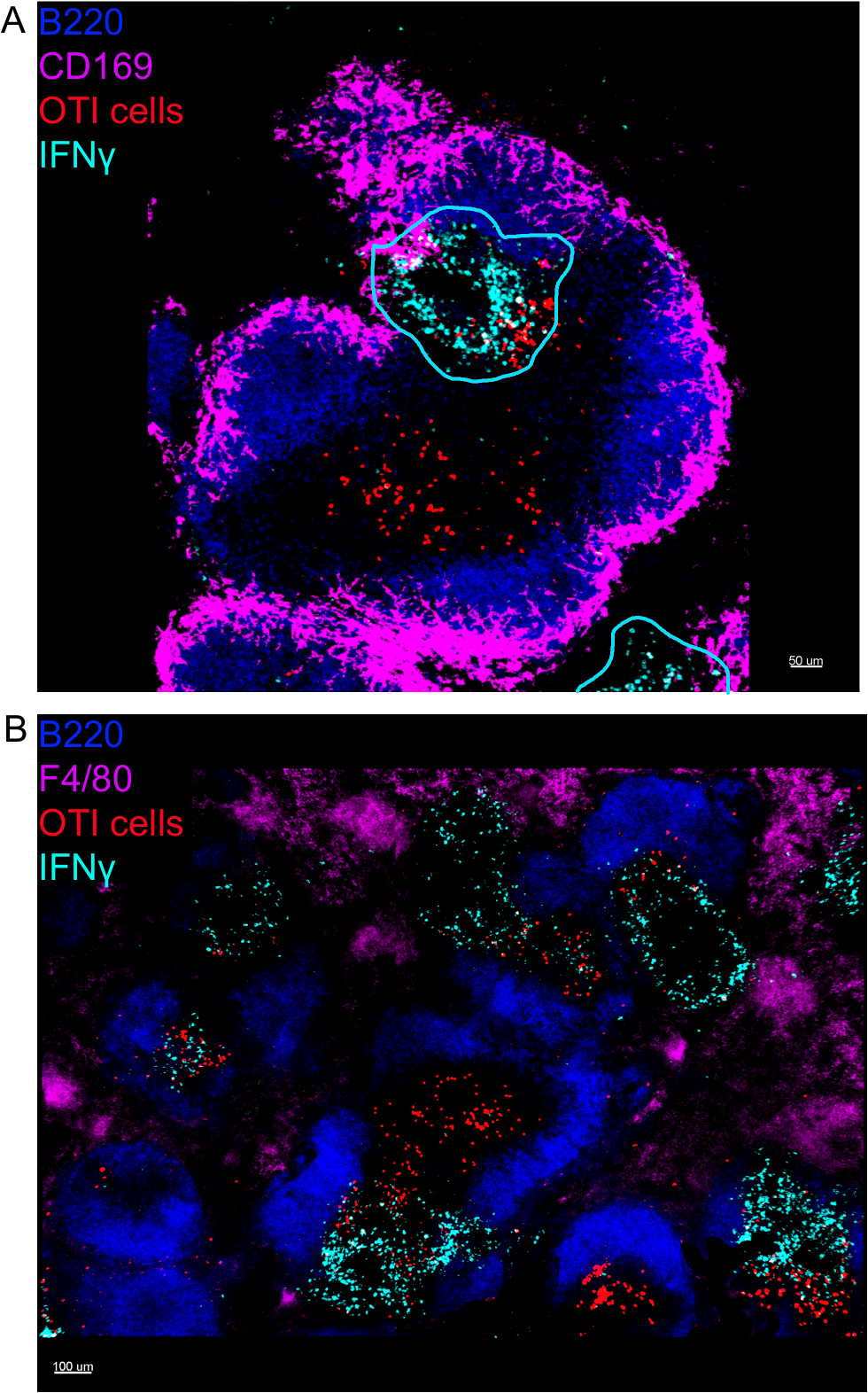
L'imagerie in situ des cellules productrices d'IFNmD a révélé que la production d'IFN n'est pas répartie dans toute la rate, mais concentrée dans des zones discrètes (figure 4). En effet, nous avons constaté que les lymphocytes T étaient activés dans toute la rate (mise en évidence par le clustering des lymphocytes T), ce qui n'était pas nécessairement corrélé avec la production d'IFN. Une explication probable est que la production d'IFN est limitée à l'emplacement des cellules infectées15,16, et l'activation des lymphocytes T, représentée par le regroupement, peut être soutenue à la fois par des personnes infectées (IFN) et non infectées (IFN négatif) des cellules présentant des antigènes. D'autres taches seront nécessaires pour identifier l'emplacement exact et obtenir une indication du mécanisme limitant la production d'IFN à cette zone et de sa relation avec le transfert d'antigène. Fait intéressant, nous avons constaté que les lymphocytes T activés, groupés et spécifiques à l'antigène sont situés dans toute la pulpe blanche de la rate, mais qu'ils ne produisent des IFN que dans les régions où les cellules NK coexistent avec elles (figure5). En tant que tel, la présence de cellules NK délimite un microenvironnement spécifique dans la pulpe blanche, dans laquelle les lymphocytes T groupés produisent des CELLULES IFNpar par opposition aux lymphocytes T groupés dans l'autre partie de la pulpe blanche. Cela suggère que l'activation des lymphocytes T n'est pas suffisante pour dicter la production d'IFN à ce moment-là.
Une autre caractéristique intéressante mise en évidence par notre protocole est la localisation sous-cellulaire différente de l'IFN en NK par rapport à CD8- lymphocytes T5. Comme le montre la figure 6, alors que la localisation de l'IFN MD dans les cellules NK est diffusée dans le cytosol, les cellules CD8et T recrutent souvent l'IFN vers une autre cellule T.

Figure 1 : Marqueurs mettant en valeur l'architecture de la rate. Les souris ont été infectées par 2 x 104 CFU LM-OVA et euthanasiées 24 h après l'infection. La rate a été explantée et traitée comme décrit dans le protocole. (A) Les sections étaient tachées pour les cellules NK (anti-NCR1 suivies de l'anti-chèvre IgG-FITC; vert), des cellules OTI-RFP (rouge) et des macrophages (anti-F4/80-APC; magenta). RP et Pulpe Rouge; WP et Pâte Blanche. Barre d'échelle de 200 m. (B) Les sections ont été tachées pour les cellules B (anti-B220-Pacific Blue; Bleu), cellules OTI-GFP (signal GFP montré en rouge) et macrophages de zone marginale (anti-CD169-Alexa647 ; magenta). RP et Pulpe Rouge; Follicule de cellules BF et B; TZ et zone de lymphocytes T. Barre d'échelle de 50 m. Il s'agit d'une image représentative de 3 expériences indépendantes (N ' 4). Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 2 : Le traitement bfA permet de détecter in situ l'IFN intracellulaire. La production de N est limitée à des zones spécifiques dans l'rate Les souris ont été infectées par 2 x 104 CFU LM-OVA et traitées par BFA (A) ou laissées non traitées (B) après 18 h. Les souris ont été euthanasiées 24 h après l'infection. La rate a été explantée et traitée comme décrit dans le protocole. Les sections ont été tachées pour les cellules NK (anti-NCR1 suivies de l'anti-chèvre IgG-FITC; vert), des cellules OTI-RFP (rouge) et IFNMD (anti-IFN-BV421; cyan). Barre d'échelle de 5 m. Il s'agit d'une image représentative des zones riches en cellules NK à partir de 3 expériences indépendantes (N - 3). Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 3 : Cellules productrices d'IFN MD dans la rate. Les souris ont été infectées par 2 x 104 CFU LM-OVA lorsqu'elles ont été indiquées et traitées par BFA après 18 h. Les souris ont été euthanasiées 24 h après l'infection. La rate a été explantée et traitée comme décrit dans le protocole. Les sections ont été tachées pour les cellules NK (anti-NCR1 suivies de l'anti-chèvre IgG-FITC; vert), des cellules OTI-RFP (rouge) et IFNMD (anti-IFN-BV421; cyan). (A) Image représentative d'une rate d'une souris naïve non infectée pour démontrer l'absence de coloration non spécifique DE l'IFN. Les lignes blanches délimitent la pulpe blanche. WP - Pulpe blanche; RP - Pulpe rouge. (B) Image représentative de la pulpe blanche de la rate d'une souris infectée par LM-OVA, montrant l'invasion des cellules NK à la pulpe blanche et la production de cellules IFNMD par les cellules NK, les cellules OTI et les cellules non étiquetées. Les images sont représentatives de 4 expériences indépendantes (N ' 4). Barres d'échelle de 70 m (A); et 20 m (B). Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 4 : La production d'IFN est limitée à des zones spécifiques de la rate à la suite de l'infection à LM-OVA. Les souris ont été infectées par 2 x 104 CFU LM-OVA et traitées avec BFA après 18 h. Les souris ont été euthanasiées 24 h après l'infection. La rate a été explantée et traitée comme décrit dans le protocole. Toutes les sections ont été tachées pour les cellules B (B220-Pacific Blue Ab, Blue) et IFNMD (anti-IFNMD-biotine suivie de streptavidin-PE; cyan). Cellules OTI-GFP (signal GFP indiqué en rouge). Les lignes cyannes correspondent à des zones de production élevée d'IFN. Il s'agit d'images représentatives de 4 expériences indépendantes (N et 4). (A) Des sections ont été tachées pour les macrophages de zone marginale (anti-CD169-Alexa 647, magenta). Barre d'échelle de 50 m. (B) Les sections ont été tachées pour tous les macrophages (F4/80). Barre d'échelle de 100 m. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 5 : La production d'IFNPar par des cellules OTI activées se produit dans un microenvironnement spécifique. Les souris ont été infectées par 2 x 104 CFU LM-OVA et traitées avec BFA après 18 h. Les souris ont été euthanasiées 24 h après l'infection. Les rates ont été explantées et traitées comme décrit dans le protocole. Les sections ont été tachées pour les cellules NK (anti-NCR1 suivies de l'anti-chèvre IgG-FITC; vert), des cellules OTI-RFP (rouge) et IFNMD (anti-IFN-BV421; cyan). Les lignes vertes et rouges mettent en évidence les zones cellulaires NK et OTI, respectivement. La flèche blanche indique des exemples de clusters de lymphocytes T ne produisant pas d'IFN. Flèches vertes exemples de clusters de lymphocytes T produisant IFN. Barre d'échelle de 100 m. Il s'agit d'une image représentative de quatre expériences indépendantes (N ' 4). Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 6 : Localisation sous-cellulaire de l'IFNMD dans les cellules NK et les lymphocytes T. Les souris ont été infectées par 2 x 104 CFU LM-OVA et traitées avec BFA après 18 h. Les souris ont été euthanasiées 24 h après l'infection. La rate a été explantée et traitée comme décrit dans le protocole. Toutes les sections ont été tachées pour IFNMD (anti-IFNMD-BV421; cyan). Les lignes blanches délimitent les bords cellulaires et les flèches blanches montrent la directionnalité de la sécrétion. Il s'agit d'une image représentative de deux expériences indépendantes (N ' 5). (A)- Les cellules OTI-RFP sont représentées en rouge. Barre d'échelle de 5 m. (B) Les sections ont été tachées pour les cellules NK (anti-NCR1 suivies de l'igG-FITC anti-chèvre; vert. Barre d'échelle de 2 m. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}
Discussion
Dans ce manuscrit, nous présentons une méthode pour visualiser la production d'IFNMD dans la rate suivant l'infection de L. monocytogenes chez la souris. Ce protocole est simple et peut être adapté à d'autres tissus et déclencheurs de cytokine, mais les aspects suivants doivent être pris en considération. Les cellules sécrètent souvent rapidement les cytokines qu'elles produisent, et les cytokines sont rapidement ramassées par les cellules voisines. Il est en tant que tel difficile de détecter les cytokines in situ. Une méthode commune pour relancer rapidement la production de cytokine est de re-stimuler les cellules ex vivo suivies de la détection de cytokine dans les médias par l'essai immunosorbent enzymatique-lié. Dans ce contexte, toute information sur la localisation spatiale des cellules productrices de cytokine est perdue. En outre, la production de cytokine après la re-stimulation ne reflète pas nécessairement si les cytokines sont effectivement produites et sécrétées in vivo, mais indique plutôt la capacité d'une population cellulaire donnée à produire des cytokines. Par conséquent, les deux méthodes fourniront des informations différentes et il faut considérer quelles informations sont les plus précieuses pour leur expérience.
Afin de détecter les cytokines intracellulaires, notre méthode utilise un inhibiteur intracellulaire de transport de protéine pour piéger des cytokines à l'intérieur des cellules et augmenter la détection de signal. Cependant, il est important de noter que ces inhibiteurs affectent le transport normal des protéines du réticulum endothélial (RE) à l'appareil Golgi et à la vésicule sécrétrice altérant leur libération, qui pourrait causer la toxicité. En conséquence, BFA, ou tout autre inhibiteur, devrait être utilisé pendant une courte période de temps, généralement pas plus de quelques heures. Par conséquent, il est important de trouver le bon équilibre entre la dose d'inhibiteur et le temps du traitement afin d'optimiser le niveau de cytokines emprisonnés à l'intérieur de la cellule sans causer d'effets cytotoxiques graves. Ces variables peuvent différer entre les cytokines et la voie d'administration pour le BFA. Dans notre modèle d'infection, le BFA a été administré intrapéritonement afin de fournir une dispersion systémique rapide, mais il peut également être livré par voie intraveineuse.
Les inhibiteurs intracellulaires les plus couramment utilisés de transport de protéine sont BFA, employé ici, et monensine (MN). Ces inhibiteurs sont souvent utilisés indistinctement pour accumuler et étudier la production de cytokine, mais ils ont de légères différences dans leurs mécanismes d'action. MN inhibe le transport des protéines dans l'appareil Golgi accumulant ainsi des protéines dans le Golgi17 tandis que BFA empêche le recrutement de protéines coatomères-I, inhibant le mouvement rétrograde des protéines vers le réticulum endoplasmique (ER) et favorisant ainsi l'accumulation de cytokines dans lesurgences 18. En tant que tel, le choix du meilleur inhibiteur intracellulaire de transport de protéine dépendra de différents facteurs, tels que la cytokine pour être détectée. Par exemple, il a été démontré dans la coloration intracellulaire induite par le lipopolysaccharide des monocytes que BFA est plus efficace pour mesurer les cytokines IL-1, IL-6 et TNF que MN19.
Ce protocole implique la visualisation de la cytokine in situ par microscopie confocale et donc il n'y a qu'un nombre limité de marqueurs que peut être utilisé pour étudier les cellules productrices de cytokine et leur microenvironnement. Il est également nécessaire de considérer que les inhibiteurs du transport de protéines tels que BFA ou MN perturbent l'expression normale de plusieurs protéines et donc leur utilisation lors de l'étude de l'expression simultanée de certains marqueurs de surface des cellules d'activation doit être approchée prudemment. Par exemple, BFA mais pas MN bloque l'expression de CD69 dans les lymphocytes murines20. Malgré cette limitation, la formation image confocale permet la localisation sous-cellulaire des cytokines, ainsi que la direction de la sécrétion de cytokine dans la cellule. Les données générées à l'aide de ce protocole suggèrent que les cellules NK ont tendance à sécrétiser IFN-y dans un modèle diffus tandis que les cellules CD8T semblent diriger la sécrétion IFN vers d'autres cellules CD8T qui sont en interaction directe avec eux5.
Pour conclure, ce protocole est approprié pour visualiser une variété de cytokines in situ et identifier les cellules productrices et leur microenvironnement suivant de nombreux déclencheurs tels que l'infection ou l'auto-immunité. L'information obtenue est instrumentale pour comprendre l'importance de l'orchestration spatiale in vivo de différents types de cellules et de la cytokine qu'ils produisent, nécessaire pour une réponse immunitaire efficace.
Déclarations de divulgation
Les auteurs n'ont rien à révéler.
Remerciements
Nous remercions le personnel de l'installation d'imagerie de l'Institut Kennedy pour son aide technique en imagerie. Ce travail a été soutenu par des subventions du Kennedy Trust (à A.G.) et du Biotechnology and Biological Sciences Research Council (BB/R015651/1 à A.G.).
matériels
| Name | Company | Catalog Number | Comments |
| Brefeldin A | Cambridge bioscience | CAY11861 | |
| Paraformaldehyde | Agar scientific | R1018 | |
| L-Lysin dihydrochloride | Sigma lifescience | L5751 | |
| Sodium meta-periodate | Thermo Scientific | 20504 | |
| D(+)-saccharose | VWR Chemicals | 27480.294 | |
| Precision wipes paper Kimtech science | Kimberly-Clark Professional | 75512 | |
| O.C.T. compound, mounting medium for cryotomy | VWR Chemicals | 361603E | |
| Fc block, purified anti-mouse CD16/32, clone 93 | Biolegend | 101302 | Antibody clone and Concentration used: 2.5 mg/ml |
| Microscope slides - Superfrost Plus | VWR Chemicals | 631-0108 | |
| anti-CD169 - AF647 | Biolegend | 142407 | Antibody clone and Concentration used: clone 3D6.112 1.6 mg/ml Excitation wavelength: 650 Emission wavelength: 65 |
| anti-F4/80 - APC | Biolegend | 123115 | Antibody clone and Concentration used: clone BM8 2.5 mg/ml Excitation wavelength: 650 Emission wavelength: 660 |
| anti-B220 - PB | Biolegend | 103230 | Antibody clone and Concentration used: clone RA3-6B2 1.6 mg/mL Excitation wavelength: 410 Emission wavelength: 455 |
| anti-IFNg - biotin | Biolegend | 505804 | Antibody clone and Concentration used: clone XMG1.2 5 mg/mL |
| anti-IFNg - BV421 | Biolegend | 505829 | Antibody clone and Concentration used: clone XMG1.2 5 mg/mL Excitation wavelength: 405 Emission wavelength: 436 |
| anti-Nkp46/NCRI | R&D Systems | AF2225 | Antibody clone and Concentration used: goat 2.5 mg/mL |
| anti-goat IgG-FITC | Novusbio | NPp 1-74814 | Antibody clone and Concentration used: 1 mg/mL Excitation wavelength: 490 Emission wavelength: 525 |
| Streptavidin - PE | Biolegend | 405203 | Antibody clone and Concentration used: 2.5 mg/mL Excitation wavelength: 565 Emission wavelength: 578 |
| Streptavidin - FITC | Biolegend | 405201 | Antibody clone and Concentration used: 2.5 mg/mlL Excitation wavelength: 490 Emission wavelength: 525 |
| Fluoromount G | SouthernBiotech | 0100-01 | |
| Cover glasses 22 mm x 40 mm | Menzel-Glazer | 12352128 | |
| Liquid blocker super PAP PEN mini | Axxora | CAC-DAI-PAP-S-M | |
| Imaris - Microscopy Image Analysis Software | Bitplane | ||
| Confocal microscope - Olympus FV1200 Laser scanning microscope | Olympus | ||
| Cryostat - CM 1900 UV | Leica | ||
| Base mould disposable | Fisher Scientific UK Ltd | 11670990 | |
| PBS 1x | Life Technologies Ltd | 20012068 | |
| BHI Broth | VWR Brand | 303415ZA | |
| GFP | Excitation wavelength: 484 Emission wavelength: 507 | ||
| RFP | Excitation wavelength: 558 Emission wavelength: 583 | ||
| Insulin syringe, with needle, 29 G | VWR International | BDAM324824 | |
| C57BL/6 wild type mice | Charles River |
Références
- Iwasaki, A., Medzhitov, R. Control of adaptive immunity by the innate immune system. Nature Immunology. 16 (4), 343-353 (2015).
- Harty, J. T., Bevan, M. J. Specific immunity to Listeria monocytogenes in the absence of IFN gamma. Immunity. 3 (1), 109-117 (1995).
- Kubota, K., Kadoya, Y. Innate IFN-gamma-producing cells in the spleen of mice early after Listeria monocytogenes infection: importance of microenvironment of the cells involved in the production of innate IFN-gamma. Frontiers in Immunology. 2 (26), (2011).
- Dunn, P. L., North, R. J. Early gamma interferon production by natural killer cells is important in defense against murine listeriosis. Infection and Immunity. 59 (9), 2892-2900 (1991).
- Krummel, M. F., et al. Paracrine costimulation of IFN-gamma signaling by integrins modulates CD8+ T cell differentiation. Proceedings of the National Academy of Sciences of the United States of America. 115 (45), 11585-11590 (2018).
- Curtsinger, J. M., Agarwal, P., Lins, D. C., Mescher, M. F. Autocrine IFN-gamma promotes naive CD8+ T cell differentiation and synergizes with IFN-alpha to stimulate strong function. Journal of Immunology. 189 (2), 659-668 (2012).
- Hosking, M. P., Flynn, C. T., Whitton, J. L. Antigen-specific naive CD8++ T cells produce a single pulse of IFN-gamma in vivo within hours of infection, but without antiviral effect. Journal of Immunology. 193 (4), 1873-1885 (2014).
- Croxford, A. L., Buch, T. Cytokine reporter mice in immunological research: perspectives and lessons learned. Immunology. 132 (1), 1-8 (2011).
- Gerard, A., et al. Secondary T cell-T cell synaptic interactions drive the differentiation of protective CD8++ T cells. Nature Immunology. 14 (4), 356-363 (2013).
- Engelhardt, J. J., et al. Marginating dendritic cells of the tumor microenvironment cross-present tumor antigens and stably engage tumor-specific T cells. Cancer Cell. 21 (3), 402-417 (2012).
- Matheu, M. P., Cahalan, M. D. Isolation of CD4+ T cells from mouse lymph nodes using Miltenyi MACS purification. Journal of Visualized Experiments. (9), 409 (2007).
- Matheu, M. P., Parker, I., Cahalan, M. D. Dissection and 2-photon imaging of peripheral lymph nodes in mice. Journal of Visualized Experiments. (7), 265 (2007).
- Pope, C., et al. Organ-specific regulation of the CD8+ T cell response to Listeria monocytogenes infection. Journal of Immunology. 166 (5), 3402-3409 (2001).
- Jones, G. S., D'Orazio, S. E. Listeria monocytogenes: cultivation and laboratory maintenance. Current Protocols in Microbiology. 31, 1-7 (2013).
- Kang, S. J., Liang, H. E., Reizis, B., Locksley, R. M. Regulation of hierarchical clustering and activation of innate immune cells by dendritic cells. Immunity. 29 (5), 819-833 (2008).
- Chang, S. R., et al. Characterization of early gamma interferon (IFN-gamma) expression during murine listeriosis: identification of NK1.1+ CD11c+ cells as the primary IFN-gamma-expressing cells. Infection and Immunity. 75 (3), 1167-1176 (2007).
- Mollenhauer, H. H., Morre, D. J., Rowe, L. D. Alteration of intracellular traffic by monensin; mechanism, specificity and relationship to toxicity. Biochimica et Biophysica Acta. 1031 (2), 225-246 (1990).
- Helms, J. B., Rothman, J. E. Inhibition by brefeldin A of a Golgi membrane enzyme that catalyses exchange of guanine nucleotide bound to ARF. Nature. 360 (6402), 352-354 (1992).
- Schuerwegh, A. J., Stevens, W. J., Bridts, C. H., De Clerck, L. S. Evaluation of monensin and brefeldin A for flow cytometric determination of interleukin-1 beta, interleukin-6, and tumor necrosis factor-alpha in monocytes. Cytometry. 46 (3), 172-176 (2001).
- Nylander, S., Kalies, I. Brefeldin A, but not monensin, completely blocks CD69 expression on mouse lymphocytes: efficacy of inhibitors of protein secretion in protocols for intracellular cytokine staining by flow cytometry. Journal of Immunology Methods. 224 (1-2), 69-76 (1999).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.