
Method Article
Analyse des facteurs d'expression des gènes tumoraux avec le portail Web CorExplorer
Dans cet article
Résumé
Nous introduisons le portail Web CorExplorer, une ressource pour l'exploration des facteurs de séquençage de l'ARN tumoral trouvés par l'algorithme d'apprentissage automatique CorEx (Correlation Explication), et montrons comment les facteurs peuvent être analysés par rapport à la survie, les annotations de base de données, interactions protéines-protéines, et les uns les autres pour obtenir un aperçu de la biologie tumorale et des interventions thérapeutiques.
Résumé
L'analyse différentielle d'expression de gène est une technique importante pour comprendre des états de la maladie. L'algorithme d'apprentissage automatique CorEx a montré l'utilité dans l'analyse de l'expression différentielle des groupes de gènes dans la tumeur RNA-seq d'une manière qui peut être utile pour faire progresser l'oncologie de précision. Cependant, CorEx produit de nombreux facteurs qui peuvent être difficiles à analyser et à se connecter à la compréhension existante. Pour faciliter de telles connexions, nous avons construit un site Web, CorExplorer, qui permet aux utilisateurs d'explorer les données de manière interactive et de répondre à des questions communes liées à son analyse. Nous avons formé CorEx sur les données d'expression de gène d'ARN-seq pour quatre types de tumeur : ovaire, poumon, mélanome, et côlorectal. Nous avons ensuite incorporé les éléments de survie correspondants, les interactions protéines-protéines, l'entologie génique (GO) et l'encyclopédie des gènes et des génomes de Kyoto (KEGG) et les cartes thermiques dans le site Web pour association avec la visualisation du graphique de facteur. Ici nous employons des protocoles d'exemple pour illustrer l'utilisation de la base de données pour comprendre l'importance des facteurs de tumeur appriss dans le contexte de ces données externes.
Introduction
Depuis son introduction il y a un peu plus d'une décennie, l'ARN-seq est devenu un outil omniprésent pour mesurer l'expression des gènes1. C'est parce qu'il permet un profilage rapide et bon marché de novo de l'ensemble transcriptome d'un échantillon. Cependant, les données de tumeur d'ARN-seq reflètent une biologie fondamentale qui est intrinsèquement complexe et souvent sous-échantillonnée, alors que les données elles-mêmes sont de haute dimension et bruyantes. Cela présente un défi important pour l'extraction de signaux fiables. L'algorithme CorEx tire parti de l'information mutuelle multivariée pour trouver des modèles subtils dans de telles situations2,3 . Cette technique a été précédemment adaptée pour analyser des échantillons d'ARN-seq de tumeur ovarienne de l'Atlas de génome de cancer (TCGA) et dans ce contexte elle a semblé avoir des avantages significatifs au-dessus des méthodes plus couramment employées d'analyse4.
Bien que l'utilisation de l'ARN-seq soit extrêmement répandue dans les applications de recherche, y compris en oncologie, ces efforts n'ont pas conduit à une large utilisation aux fins des interventions cliniques5. Cela s'explique en partie par le manque d'algorithmes et de logiciels conviviaux ciblés sur ces problèmes spécifiques. Pour aider à combler cette lacune, nous avons conçu le portail Web CorExplorer pour permettre aux chercheurs de divers milieux d'étudier les facteurs d'expression génique des échantillons d'ARN-seq tumoraux tels que trouvés par l'algorithme d'apprentissage automatique CorEx. Le portail CorExplorer prend en charge la visualisation interactive et l'interrogation des facteurs de plusieurs types de tumeurs différentes, y compris le poumon, le côlon, le mélanome, et l'ovaire6,7,8,9, 10, dans le but d'aider les chercheurs à passer au crible les corrélations de données et à identifier les voies des candidats pour stratifier les patients à des fins thérapeutiques.
Nous nous attendons à ce que le portail CorExplorer soit utile à plusieurs types d'utilisateurs. Le portail a été conçu avec l'utilisateur à l'esprit qui souhaite comprendre les grands facteurs qui conduisent les différences d'expression des gènes tumoraux dans les bases de données publiques et peut-être aussi placer des profils individuels d'expression des gènes dans le contexte de tumeurs avec des Caractéristiques. En plus des protocoles représentatifs décrits ici, les enquêtes CorExplorer peuvent servir de point de départ pour suggérer des hypothèses pour d'autres tests, pour comparer et contraster les résultats de CorEx sur les ensembles de données à l'extérieur du CorExplorer, et pour se connecter signatures d'expression pathologique d'un ou quelques gènes dans une tumeur individuelle à de plus grands groupes qui peuvent être coordonnés affectés. Enfin, il peut servir d'introduction conviviale à l'application de l'apprentissage automatique à l'ARN-seq pour ceux qui commencent dans le domaine.
Protocole
1. Explorer les facteurs contenant un gène d'intérêt
- Ouvrez un navigateur Web et rendez-vous sur http://corex.isi.edu, la page d'accueil de CorExplorer.
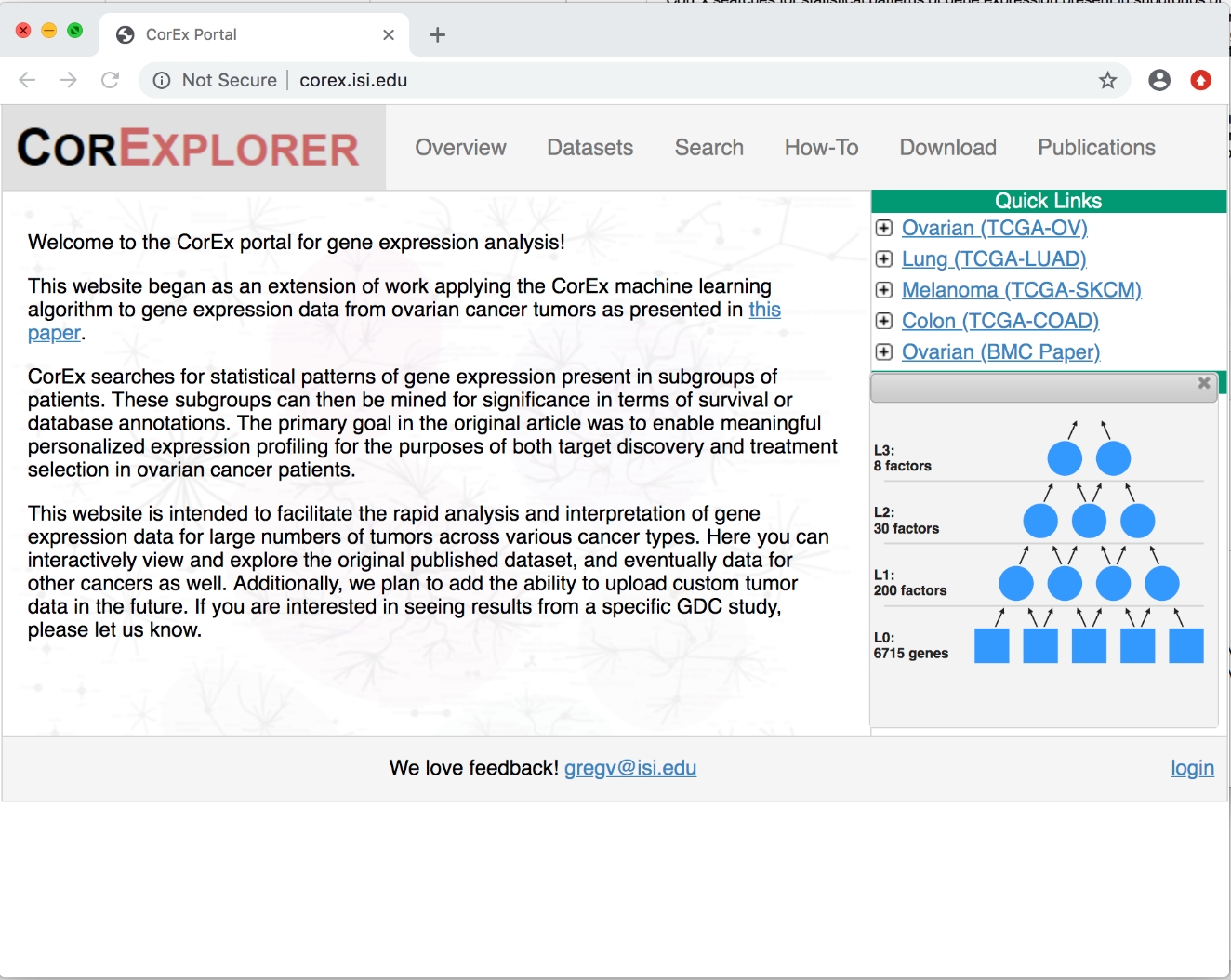
- Sur le côté droit sous Quick Links, cliquez sur le bouton d'extension à côté de l'ovaire (TCGA-OV) pour voir un résumé du graphique du facteur CorEx qui a été formé sur les données sur le cancer de l'ovaire TCGA (voir dans la figure 1). En option, cliquez sur les autres pour comparer.
- Une fois terminé l'inspection des graphiques de facteur, cliquez surPoumon (TCGA-LUAD)pour accéder à la page CorExplorer pour le cancer du poumon RNA-seq.
- Explorez le graphique du facteur CorEx pour trouver un gène d'intérêt à l'aide de la fenêtre CorExplorer 'Factor Graph'.
- Déplacez le curseur de la souris sur la fenêtre d'affichage graphique facteur. Zoom sur le graphique de facteur à l'aide de la roue de défilement de la souris ou trackpad pour voir les détails du graphique tels que les gènes les plus importants dans chaque facteur et les connexions entre les nœuds à différentes couches. Vous pouvez également cliquer et faire glisser pour déplacer la zone de vue ou tout nœud.
- Pour trouver un gène cible (ici, nous utiliserons BRCA1), cliquez sur le menu Dedropdown Gene en haut de la fenêtre graphique de facteur. Tapez 'BRCA1' pour le sélectionner dans la liste de déclassement et appuyez sur Retour pour faire le zoom de vue au facteur 26, le facteur avec lequel BRCA1 est le plus fortement corrélé.
- Repositionnez la souris sur l'écran graphique et faites défiler pour effectuer un zoom arrière pour voir le nœud de niveau 2, L2-8, et ses facteurs associés qui sont voisins au facteur 26. Notez que seuls les gènes dont le poids est supérieur au seuil indiqué sur le curseur de poids du maillon Min sont indiqués.
- Pour voir tous les gènes associés au facteur, cliquez sur le nœud L1-26 et sélectionnez Charger des gènes supplémentaires dans la fenêtre contextuelle. Lorsque le mot «Fait» apparaît, fermez la fenêtre contexty.
- Maintenant, retournez à la section en-tête au-dessus de la fenêtre graphique facteur et saisir et faire glisser le modificateur de poids lien Min. Maintenant, comme le curseur de poids de lien est déplacé vers le bas à 0,05, d'autres gènes dans le facteur L1-26, y compris BRCA2, apparaîtra dans l'ordre de poids. En option, repositionner les nœuds en saisissant et en faisant glisser pour améliorer la mise en page.
- Déterminer comment la stratification des patients par rapport au facteur affecte la survie en interrogeant dans la fenêtre de survie.
- Dans la fenêtre de survie, décocher Sort by p-val, puis sélectionnez le facteur 26 dans le menu de déclassement single Factor afin d'afficher les courbes de survie pour le facteur 26.
- Faites défiler le graphique de survie pour montrer le nombre de patients à risque le long de l'axe X.
- Trouvez des associations avec la fonction biologique en interrogeant dans la fenêtre Annotation.
- Dans la fenêtre d'annotation, pour trier le menu de baisse de facteur par nombre de facteur plutôt que le taux de découverte faux (FDR), uncheck FDR trier.
- Faites défiler et cliquez pour sélectionner le facteur 26 dans la fenêtre d'annotation pour afficher les annotations d'enrichissement pour le facteur.
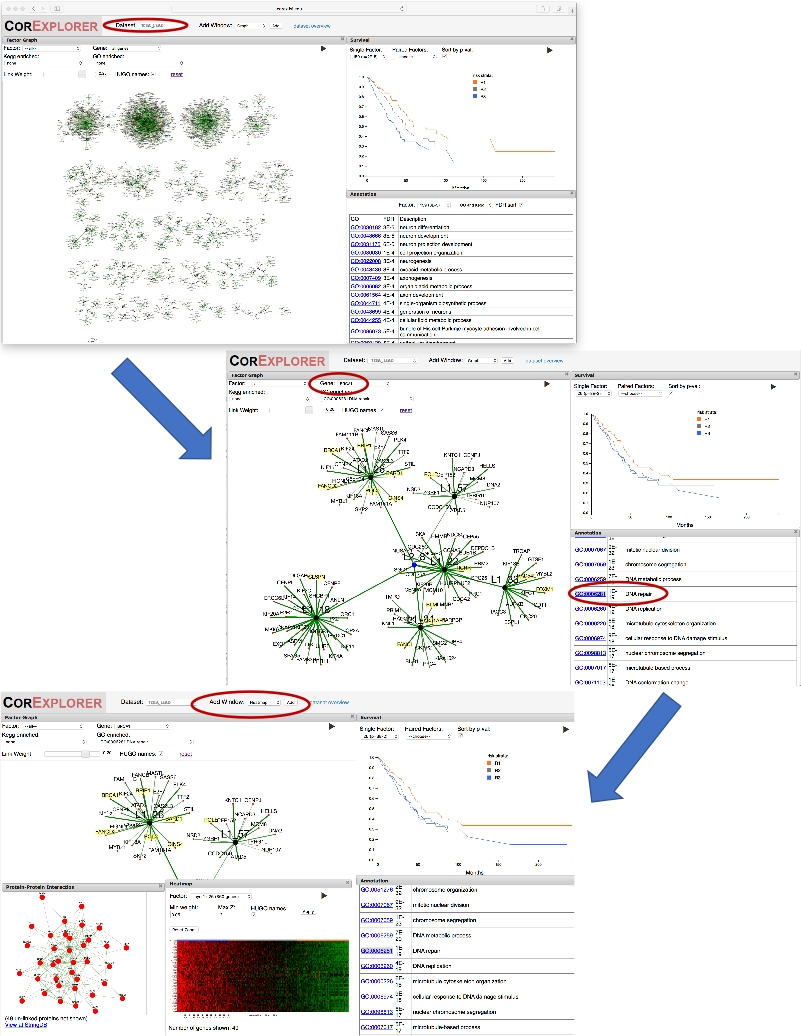
- Faites défiler la liste d'annotation jusqu'à ce que la réparation de l'ADN soit visible et cliquez dessus pour voir immédiatement les gènes associés mis en évidence en jaune sur l'écran graphique. Voir le panneau du milieu de la figure 2.
- Notez que les facteurs disparaissent ou apparaissent sous forme de termes GO différents, selon qu'ils sont enrichis ou non pour des gènes avec l'annotation sélectionnée, par exemple « voie de signalisation apoptotique intrinsèque en réponse à des dommages à l'ADN ».
- Explorez les facteurs plus loin en ajoutant des fenêtres avec des fonctionnalités différentes.
- À partir de la barre de menu supérieure, ajoutez une fenêtre de réseau d'interaction protéines-protéines (PPI) en sélectionnant PPI à partir du dropdown Add Window, puis cliquez sur le bouton Ajouter pour ajouter une fenêtre graphique PPI à la zone d'affichage. Dans la fenêtre graphique PPI, choisissez le facteur 'Layer1: 26' pour montrer les interactions protéines-protéines. Notez la densité des connexions.
- À partir de la barre de menu supérieure, au lieu de PPI, sélectionnez Heatmap à partir de la fenêtre Add dropdown, puis cliquez sur le bouton Ajouter pour ajouter une fenêtre de carte thermique à la zone d'affichage. Dans la fenêtre de la carte thermique, choisissez le facteur 'Layer1: 26' pour afficher les modèles d'expression génique.
- Saisissez et repositionnez la fenêtre de la carte chauffre afin que la fenêtre de survie soit également visible. Le long du haut de la carte thermique, observez comment la barre de couleur orange/bleu/gris correspond aux strates de risque des patients sur le graphique de survie. Les résultats sont affichés en bas de la figure 2.
- Explorez le graphique du facteur CorEx pour trouver un gène d'intérêt à l'aide de la fenêtre CorExplorer 'Factor Graph'.
2. Filtrer et interpréter les facteurs CorEx à l'aide de données sur le poids, la survie et l'annotation des gènes
- Filtrer les facteurs d'intérêt en utilisant la survie et la qualité des grappes.
- À partir du menu de déclassement de Dataset en haut, sélectionnez TCGA-OVCA pour aller à la page CorExplorer pour l'ARN-seq du cancer de l'ovaire TCGA.
- Une fois que la page a chargé, notez de la fenêtre de survie que le facteur avec le plus grand différentiel de survie pour différentes strates est 114.
- En haut de la fenêtre graphique de facteur, sélectionnez 'Layer1: 114' à partir du déclassement du facteur.
- Prenez le curseur de poids de lien avec la souris et déplacez-le jusqu'à 0,5. Notez que le grand nombre de gènes dans le facteur 114 (1609), aucun n'ayant de poids -0,35, indique un regroupement relativement faible.
- Ensuite, élargissez la liste des facteurs dans la fenêtre de survie et sélectionnez le meilleur facteur suivant dans la fenêtre de survie, facteur 39, pour montrer ses courbes de survie associées.
- Sélectionnez le facteur 39 dans la fenêtre d'annotation en cliquant dessus. Les annotations GO et KEGG importantes sont montrées.
- Pour mieux comprendre le rôle biologique des gènes dans le facteur 39, interpréter les facteurs en utilisant les informations d'annotation de voisinage comme suit.
- En haut de la fenêtre graphique de facteur, sélectionnez le facteur 'Layer1: 39' dans le facteur de descente. Ensuite, déplacez la souris sur la fenêtre graphique de facteur et effectuez un zoom arrière pour révéler l'ensemble du cluster L2-14 avec 6 facteurs : 14, 32, 39, 42, 52 et 82 (voir la figure 3).
- Pour comprendre l'importance relative des facteurs liés au nœud L2-14, commencez par visualiser les écarts de survie pour chacun des facteurs L2-14. Décochez trier par p-val dans la fenêtre de survie, puis cliquez sur chacun des nombres de facteurs successifs. Pour ce faire, notez que seuls les facteurs 14, 32 et 39 présentent une association de survie.
- Maintenant, à partir de la barre de menu supérieure, sélectionnez PPI à partir de la fenêtre Add dropdown une fois de plus. Appuyez sur Ajouter pour ajouter une fenêtre graphique PPI à la zone d'affichage. Dans la fenêtre graphique PPI, sélectionnez le facteur 'Layer1: 52' pour montrer les interactions protéines-protéines qui sont significatives. Un exemple de disposition des fenêtres à ce stade est indiqué dans la figure 3.
- Cliquez sur le lien Afficher à StringDB en bas de la fenêtre PPI pour vous connecter à la base de données en ligne De StringDB. Cliquez sur Continuer à partir du premier écran, puis sélectionnez l'onglet Analyse ci-dessous le graphique réseau comme avant pour obtenir une analyse GO en ligne pour les gènes du réseau PPI. Le composant cellulaire supérieur est le complexe protéique de classe II du MHC.
- Retournez à l'onglet CorExplorer et à la fenêtre PPI et sélectionnez le facteur 32, cette fois à partir de la baisse du facteur. Cliquez sur le lien Afficher à StringDB à l'analyse StringDB. Le composant cellulaire supérieur est «complexe protéique de classe I du MHC», contrairement à la classe II pour le facteur 52 dans l'étape précédente!
- Enfin, retournez à la fenêtre PPI et sélectionnez 'Layer1: 39' à partir du menu de baisse des facteurs en haut. Cliquez sur le lien Afficher à StringDB pour un lien vers l'analyse StringDB.
- Cliquez sur Continuer à partir du premier écran, puis sélectionnez l'onglet Analyse ci-dessous le graphique réseau pour obtenir une analyse GO en ligne pour les gènes du réseau PPI. Observez que la fonction moléculaire supérieure est ' CXCR3 chemokine récepteur liant.'
3. Utilisation des annotations de survie et de base de données pour rechercher des combinaisons thérapeutiques prometteuses
- Passez au TCGA mélanome CorExplorer en sélectionnant TCGA-SKCM à partir du menu de déclassement Dataset.
- Notez que le facteur ayant le plus grand différentiel de survie est le facteur 171. Examinez le facteur 171 annotations en faisant défiler et notez que la « réponse immunitaire » et la « voie de signalisation à médiation cytokine » sont près du sommet (comme c'était le cas pour le facteur ovarien supérieur).
- Pour trouver un facteur complémentaire, examinez les principaux facteurs associés à la survie ainsi que leurs termes d'annotation supérieurs. Pour ce faire, cliquez sur le lien d'aperçu de Dataset dans la barre de menu supérieure pour ouvrir un onglet séparé contenant un tableau avec des détails de traitement de jeu de données ainsi qu'un résumé des principaux facteurs en fonction de la valeur p du différentiel de survie. Notez que le premier facteur non immunitaire est de 88.
- Retournez à l'onglet navigateur TCGA-SKCM.
- Sélectionnez le facteur 88 dans les fenêtres de survie, d'annotation et de graphique. Les termes GO supérieurs sont liés au « traitement de rRNA » et à « organisation de mitochondndrion», confirmant qu'il est distinct des facteurs liés au système immunitaire.
- Dans la fenêtre de survie, sur les facteurs appariés dropdown, sélectionnez '88-171' pour voir comment la survie est améliorée pour les patients dans la strate moyenne pour les facteurs d'expression combinés 171 et 88. Les comparaisons d'annotation et de survie sont illustrées à la figure 4.
4. Trouver des points communs et des différences de variation d'expression génique entre les types de tumeurs à l'aide de la page de recherche
- Cliquez sur la rubrique CorExplorer pour revenir à la première page.
- Cliquez sur Rechercher sur la barre de menu supérieure pour aller à une page permettant la recherche sur tous les jeux de données sur le site CorExplorer.
- Dans la zone de recherche Gene, entrez 'FLT1' (VEGFR1) et appuyez sur Return ou appuyez sur Search. FLT1 se trouve avec un poids relativement élevé dans les facteurs suivants: OVCA - 76, LUAD - 162, SKCM - 195 et SKCM - 184, ainsi que COAD - 112 et COAD - 74.
- Vous pouvez également rechercher un terme GO connexe dans tous les ensembles de données. Essayez ceci dans la case 'GO Search' en tapant 'angiogenèse' et en appuyant sur Return ou en appuyant sur Search. Tous les facteurs FLT1, à l'exception de SKCM-195, sont répertoriés comme statistiquement enrichis pour les gènes de l'angiogenèse - le facteur 195 a, en fait, l'annotation, mais en dessous du seuil par défaut de 10-8. Les résultats de la recherche et l'étape précédente sont indiqués à la figure 5.
- À titre d'exemples supplémentaires, dans la boîte de recherche GO, premier type de « récepteur du facteur de croissance épidermique ». Seul LUAD est enrichi pour ce terme, un facteur de stratification bien connu pour le cancer du poumon. Ensuite, tapez 'mesenchymal' dans la zone de recherche. Ce terme est enrichi dans les groupes d'expression génique pour OVCA, où il est un facteur de stratification bien étudié.
Résultats
La recherche du gène 'BRCA1' dans l'ensemble de données sur le cancer du poumon révèle qu'il est le plus fortement associé au facteur CorEx 26 (figure 2). L'enrichissement à terme GO pour ce facteur est considéré comme extrêmement élevé, avec la réparation d'ADN présentant un FDR de seulement 1 x 10-19. La sélection attire également l'attention sur le groupe de deuxième niveau L2-8 qui a six facteurs étroitement liés comme les enfants. La sélection de la « réparation de l'ADN » dans les annotations du terme GO ou dans le graphique de facteur GO enrichi déroulant met en évidence les gènes associés dans chacun des facteurs, le facteur 26 ayant de loin le plus, comme prévu11. Le réseau d'interaction protéine-protéine est fortement relié, soutenant davantage la fonctionnalité étroitement liée des gènes dans le facteur 26. Le graphique de survie associé suggère une association possible avec la survie du patient, mais ceci devrait être confirmé dans un ensemble de données plus large.
Commencer par la survie peut permettre la dissection des raisons de l'amélioration de la survie associée à des groupes d'expression génétique particuliers. À titre d'exemple, le principal facteur influençant la survie du cancer de l'ovaire est le numéro 39, qui est fortement enrichi pour les gènes associés au système immunitaire (figure 3). Cinq autres facteurs associés au même nœud de niveau 2 sont également indiqués comme étant liés au système immunitaire, mais l'impact sur la survie semble être fortement variable parmi eux, 39 étant le plus élevé et 52 étant le plus faible. L'ajout d'une fenêtre d'interaction protéine-protéine pour un facteur montre le réseau d'interaction immédiat et permet de se connecter au site Web StringDB12 pour interroger divers enrichissements pour les gènes du réseau PPI. En faisant cela pour chacun des facteurs L2-14 à son tour, on constate que les enrichissements de StringDB pour les gènes du réseau PPI suggèrent l'explication possible suivante pour les associations avec la survie. Le facteur 32 contient des gènes qui constituent le complexe protéique majeur de classe I du complexe d'histocompatibilité (MHC), qui est reconnu par les lymphocytes T cytotoxiques. Le facteur 39 correspond à la signalisation cytokine et à la liaison des récepteurs CXCR3, liées aux lymphocytes T CD8MD. Ces deux facteurs semblent conférer un avantage significatif de survie pour les patients présentant l'expression relativement élevée des gènes correspondants. Les lymphocytes Cytotoxic CD8MD T sont principalement responsables de l'immunité antitumorale. Le facteur 52, d'autre part, est composé de gènes codant pour les protéines dans le complexe de classe II de MHC qui sont reconnus principalement par les cellules d'aide de CD4 MD plutôt que directement par les lymphocytes T cytotoxiques. Les autres facteurs De L2-14 reflètent l'activation généralisée du système immunitaire qui ne différencie pas les deux types de populations de lymphocytes. Une association de survie spécifique à la reconnaissance cytotoxique de lymphocyte de T des antigènes cellulaires de classe I de MCH est compatible avec notre compréhension de l'immunité antitumorale en général et d'autres cancers tels que le mélanome13,14.
Le portail Web soutient la découverte de paires de facteurs avec des fonctions complémentaires qui peuvent suggérer des thérapies combinées tumeur-spécifiques efficaces. La vue d'ensemble de données peut être numérisée pour les facteurs qui montrent une corrélation avec la survie tout en ayant des enrichissements GO distincts. Pour le mélanome (TCGA-SKCM; Figure 4), on voit que le principal facteur de survie 171 est lié au système immunitaire, tandis que le facteur 88 en bas de la liste montre l'enrichissement des gènes liés à l'organisation de la mitochondrie. En effet, cela a été suggéré comme une cible dans le mélanome15. L'ajout de fenêtres de survie à la page CorExplorer permet de comparer la stratification en utilisant la paire de facteurs à celle de chaque facteur individuellement, montrant que les modèles d'expression génique favorables des deux groupes présentent une tendance de survie meilleure que celle de l'un ou l'autre facteur seul. La strate supérieure ne semble pas être améliorée cependant, suggérant l'immunothérapie seulement peut être la meilleure option pour quelques patients.
Les points communs et les différences entre les tumeurs peuvent être observés en cherchant des gènes ou des termes GO dans tous les ensembles de données(figure 5). À titre d'exemple, FLT1 (alias VEGFR1) est un marqueur pro-angiogénique bien étudié16,17. Quand il est mis dans la barre de recherche, toutes les tumeurs ont des facteurs dans lesquels FLT1 joue un rôle majeur. Inversement, lorsque le terme GO «angiogenèse» est entré sur la page de recherche, 5 des 6 groupes FLT1 apparaissent avec cet enrichissement. Tous les facteurs FLT1, à l'exception de SKCM-195, sont répertoriés comme statistiquement enrichis pour les gènes d'angiogenèse. Le sixième facteur a, en fait, l'annotation, mais en dessous du seuil par défaut de 10-8. Lorsque la pondération dans la liste des facteurs est utilisée dans une calculatrice d'enrichissement alternative, par exemple, Gene Set Enrichment Analysis (GSEA)18, le sixième facteur s'est avéré être considérablement enrichi pour les gènes d'angiogenèse ainsi.
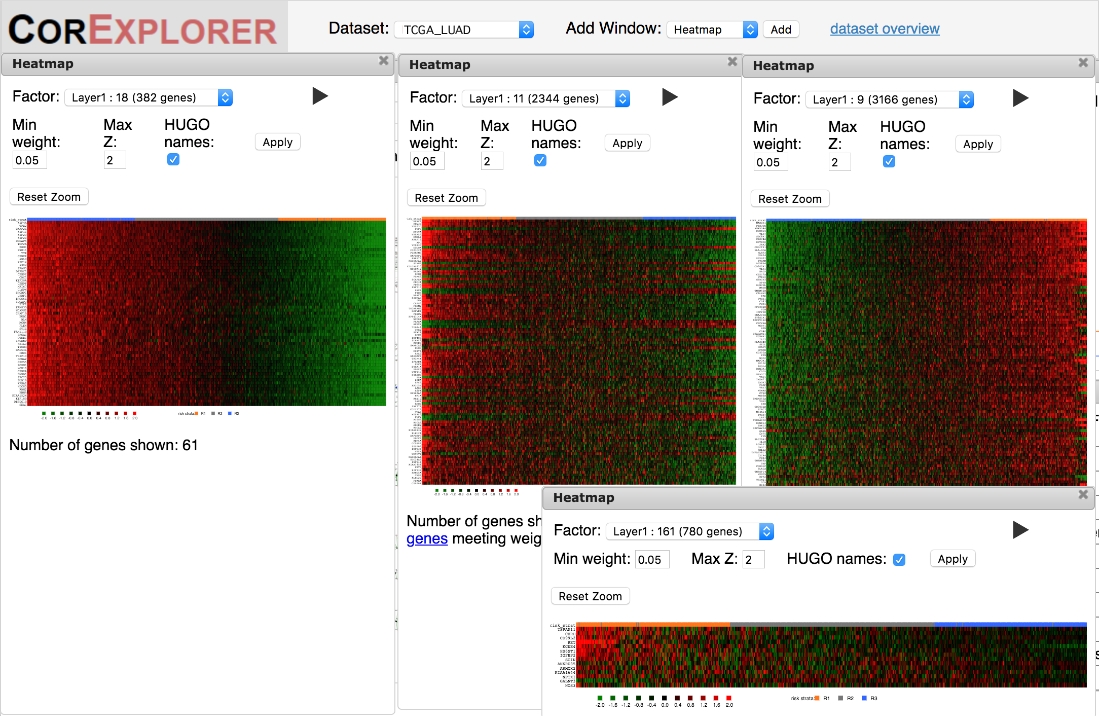
Il est important de vérifier les cartes thermiques pour s'assurer que le modèle d'expression génique est de qualité suffisante pour soutenir les interprétations biologiques. Les cartes thermiques qui présentent une forte variation claire peuvent présenter une expression coordonnée des gènes du facteur allant de modèles faibles à élevés ou plus complexes, certains gènes ayant une faible expression étant corrélés avec d'autres ayant des facteurs élevés(figure 6). Un marqueur clé d'un groupement de haute qualité est la présence de plusieurs gènes avec une variation lisse de l'expression en fonction du score de facteur. Les cartes thermiques de facteur montrent des échantillons commandés selon le score de facteur, ainsi il devrait y avoir un gradient lisse se déplaçant de gauche à droite. Cependant, cela peut ne pas se produire d'au moins deux façons différentes. Le plus souvent, les corrélations peuvent être extrêmement bruyantes (Figure 5C), remettant en question la robustesse et l'utilité de toute inférence concernant la survie et/ou la fonction biologique. En outre, les modèles qui ne se produisent que dans une petite minorité d'échantillons peuvent ne pas se conformer au modèle de trois états d'expression supposés par l'algorithme CorEx, ce qui entraîne une classification trompeuse des échantillons (côté droit de la figure 5D).

Figure 1 : Première page CorExplorer. Après avoir cliqué sur le cancer de l'ovaire sous Quick Links,les détails du graphique de facteur sont affichés. Le modèle hiérarchique CorEx est composé de variables d'entrée (expression génique dans ce cas) sur la couche inférieure et de facteurs latents inférés dans les couches supérieures. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 2 : Utilisation d'un nom de gène pour guider l'exploration. La figure montre une série de captures d'écran illustrant l'exploration des facteurs de cancer du poumon CorEx fortement liés à BRCA1. Tout d'abord, la sélection de «BRCA1» dans la boîte de dépôt de gènes pour le graphique facteur provoque la vue graphique de zoomer sur le facteur pour lequel BRCA1 a le plus de poids. Zoom sur un peu encadre la couche deux nœuds L2-8 reliant ce facteur à d'autres connexes. La survie et les annotations peuvent être comparées : en cliquant sur le terme GO, la réparation de l'ADN met en évidence les gènes annotés. Une fenêtre PPI est ajoutée pour afficher les interactions réseau pour les gènes dans le facteur. L'utilisation du bouton Add Window pour ajouter une carte thermique montre l'association des modèles d'expression avec la survie, suggérant une expression accrue des gènes de réparation de l'ADN peut être associée à une diminution de la survie. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 3 : Utilisation de données cliniques (survie) pour guider l'exploration. L'exploration du facteur associé à la survie (39) pour le cancer de l'ovaire révèle des relations intéressantes entre les facteurs voisins. Après avoir sélectionné le facteur 39 dans le graphique de facteur et zoom sur un peu, le facteur de la couche deux lié au facteur 39 est considéré comme ayant cinq autres facteurs associés. Une fenêtre de survie supplémentaire permet une comparaison directe des différentiels de survie associés. Les facteurs 39 et 32 montrent tous deux une corrélation de survie positive, contrairement au facteur 52, ce qui n'est pas le cas. Les réseaux d'interaction protéines-protéines sont tous bien définis. Lier à StringDB permet de comparer les annotations GO (non montrées) : le facteur 39 est associé à un réseau de signalisation cytokine lié à l'activation cytotoxique des lymphocytes T CD8MD et le facteur 32 est dominé par les protéines présentant des protéines de classe I du MHC qui présentent des protéines qui déclencher la reconnaissance par ces lymphocytes; les facteurs voisins, cependant, sont dominés par d'autres composants du système immunitaire tels que les lymphocytes T d'aide CD4 MD et ne montrent aucune corrélation de survie. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 4 : L'exploration des principaux facteurs de survie suggère des combinaisons thérapeutiques potentielles. Le lien 'Datasets' sur la barre de menu de la page d'accueil conduit à une table concise des facteurs de survie classés par p-valeur, avec l'annotation GO haut (non montré). Utilisant cette information pour le mélanome, la combinaison du facteur 171 pour la fonction immunisée avec le facteur 88 pour l'organisation de mitochondndrion semble complémentaire. La figure montre des fenêtres d'annotation pour chacun des facteurs côte à côte pour les contraster. Les courbes de survie pour les patients stratifiés par les deux facteurs individuellement ou ensemble indiquent que la combinaison augmente le différentiel de survie par rapport à l'un ou l'autre facteur seul. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 5 : La page de recherche facilite l'analyse pancancéreuse. Les gènes ou les termes de processus biologique GO peuvent être recherchés dans tous les ensembles de données à l'aide du lien de recherche à partir de la page d'accueil. La figure montre les résultats de recherche pour le gène FLT1 et le terme GO 'angiogenesis'. Les résultats montrent la présence de FLT1 dans les facteurs annotés avec le terme «angiogenèse» à travers les cancers. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 6 : Les cartes thermiques peuvent être utilisées pour évaluer qualitativement les corrélations entre les gènes et les échantillons en fonction du score des facteurs. Les relations d'expression génique de haute qualité sont montrées par gradation lisse quand les patients sont commandés par le score de facteur dans les cartes thermiques. La carte thermique la plus à gauche pour le facteur 18 en est un exemple. Les modèles peuvent également englober des signatures complexes d'expression de haut en bas comme dans la carte thermique moyenne grande pour le facteur 11. Des modèles de qualité inférieure montrent parfois des changements brusques dans l'expression pour un sous-groupe de patients comme dans le facteur 9 heatmap sur les corrélations droites ou simples très bruyantes comme dans le facteur 161 heatmap en bas à droite. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}
Discussion
Nous avons présenté le site CorExplorer, un serveur Web accessible au public pour l'exploration interactive des facteurs d'expression génique au maximum corrélés appris de l'ARN-seq tumoral par l'algorithme CorEx. Nous avons montré comment le site Web peut être employé pour stratifier des patients selon l'expression de gène de tumeur, et comment une telle stratification correspond à la fonction et à la survie biologiques.
D'autres serveurs Web pour l'analyse RNA-seq ont été construits. L'analyse différentielle et de co-expression pour des tumeurs peut être examinée et intégrée avec d'autres types de données dans cbioPortal19,20. Les serveurs GenePattern21, Mev22, et Morpheus23, intègrent des techniques de clustering établies telles que l'analyse des composants principaux (PCA), kmeans, ou des cartes auto-organisées (SOMs). Les efforts plus novateurs incluent CamurWeb24, basé sur un classificateur automatisé génératrice de règles, et TACCO25, qui met en œuvre des classificateurs et des lassos aléatoires de forêt. L'algorithme CorEx utilisé ici optimise les informations multivariées afin de trouver une hiérarchie de facteurs qui expliquent les modèles dans les données. L'apprentissage des facteurs non linéaires et hiérarchiques semble permettre d'améliorer l'interprétabilité par rapport aux facteurs globaux linéaires trouvés par l'intermédiaire de PCA4. En outre, l'analyse fine de la technique des signaux d'échantillon permet des comparaisons précises de tumeur vis-à-vis des sous-types larges plus couramment utilisés. Cette combinaison d'analyse des chevauchements et des facteurs hiérarchiques distingue le CorExplorer de la plupart des autres approches et nécessite de nouveaux outils de visualisation et de synthèse.
Une partie essentielle de l'analyse des facteurs CorExplorer est la capacité d'explorer non seulement plusieurs, mais plus de 100 facteurs avec des modèles de gènes informatifs qui sont placés dans une hiérarchie qui se chevauche. Le CorExplorer facilite l'extraction de ces innombrables facteurs pour les associations biologiques et cliniques et permet une caractérisation exceptionnellement détaillée des tumeurs individuelles. L'apprentissage non supervisé d'un si grand nombre de facteurs signifie que tous ne seront pas pertinents pour la biologie des maladies. Dans un tel cas, il est essentiel d'utiliser des annotations ou des gènes connus pour éliminer les facteurs d'intérêt ou rechercher des facteurs associés à des données cliniques telles que la survie. Ainsi, le CorExplorer permet aux utilisateurs d'implémenter cette étape de filtrage très importante. La présence des modèles de gène de facteur dans une tumeur peut même suggérer une approche au traitement personnalisé d'oncologie. En outre, la multiplicité des scores de facteur pour chaque tumeur qui permet la découverte des combinaisons thérapeutiques potentiellement utiles.
Il arrive parfois qu'aucune annotation GO significative n'apparaisse pour des facteurs fortement corrélés avec la survie. Bien que cela puisse se produire en raison de données bruyantes ou sous échantillonnées, il existe d'autres causes possibles telles qu'une taille de grappe qui est trop petite pour enregistrer des scores d'enrichissement significatifs ou le groupe étant un « panier » de gènes uniques provenant de diverses voies sans association. En outre, une catégorie d'annotation différente du processus biologique KEGG et GO, par exemple compartiment cellulaire, peut être appropriée. Ceux-ci peuvent être consultés en liant à StringDB comme démontré dans le protocole. L'analyse de l'enrichissement en généologie sur le site de CorExplorer ne tient pas compte de la pondération génétique dans un facteur, bien que cela sera probablement corrigé dans un proche avenir. Notez qu'une option de liste de gènes est disponible sous 'Add Window' qui permet le téléchargement de la liste complète des gènes facteurs pour une analyse plus approfondie avec des outils externes.
Aux fins du site Web, CorEx a été exécuté sur chacun des jeux de données cinq fois et l'exécution qui a abouti à la plus grande corrélation totale totale a été conservée. Avoir une représentation statistique des résultats de plusieurs séries peut être plus instructif et est un objectif pour les travaux futurs. En outre, l'ensemble des types de tumeurs disponibles sur le serveur est plutôt faible, mais nous nous attendons à ce que cela se développe au fil du temps en fonction de l'intérêt des utilisateurs.
Comme indiqué ci-dessus, le CorExplorer visualise les relations corEx ARN-seq facteur avec des informations cliniques et de base de données, permettant ainsi une variété de différents modes d'interrogatoire. Nous espérons que cet outil mènera à d'autres travaux pour utiliser la puissance de l'analyse ARN-seq pour la découverte et l'application clinique en oncologie.
Déclarations de divulgation
Les auteurs déclarent qu'ils n'ont pas d'intérêts financiers concurrents.
Remerciements
GV a été soutenu par DARPA prix W911NF-16-0575.
matériels
| Name | Company | Catalog Number | Comments |
| Public server for CorExplorer website | USC | http://corex.isi.edu | Intel Xeon E5-2690 4-core 2.6 GHz, 8GB RAM. Backend architecture is LAMP: Linux, Apache, MySQL, PHP. |
| Web browser | Google/Apple | Chrome/Safari | Verified web browsers. |
Références
- Petryszak, R., et al. The RNASeq-er API-a gateway to systematically updated analysis of public RNA-seq data. Bioinformatics. 33, 2218-2220 (2017).
- Steeg, G. V., Galstyan, A. Maximally Informative Hierarchical Representations of High-Dimensional Data. Proceedings of the Eighteenth International Conference on Artificial Intelligence and Statistics (AISTATS). , (2015).
- Ver Steeg, G., Galstyan, A. Discovering structure in high-dimensional data through correlation explanation. Advances in Neural Information Processing Systems. , (2014).
- Pepke, S., Ver Steeg, G. Comprehensive discovery of subsample gene expression components by information explanation: therapeutic implications in cancer. BMC medical Genomics. 10, 12 (2017).
- Byron, S. A., Van Keuren-Jensen, K. R., Engelthaler, D. M., Carpten, J. D., Craig, D. W. Translating RNA sequencing into clinical diagnostics: opportunities and challenges. Nature Reviews Genetics. 17, 257 (2016).
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 511, 543 (2014).
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 487, 330 (2012).
- Akbani, R., et al. Genomic classification of cutaneous melanoma. Cell. 161, 1681-1696 (2015).
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 474, 609 (2011).
- Grossman, R. L., et al. Toward a shared vision for cancer genomic data. New England Journal of Medicine. 375, 1109-1112 (2016).
- Moynahan, M. E., Chiu, J. W., Koller, B. H., Jasin, M. Brca1 controls homology-directed DNA repair. Molecular Cell. 4, 511-518 (1999).
- Szklarczyk, D., et al. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Research. 47, 607-613 (2018).
- Durgeau, A., Virk, Y., Corgnac, S., Mami-Chouaib, F. Recent advances in targeting CD8 T-cell immunity for more effective cancer immunotherapy. Frontiers in Immunology. 9, 14 (2018).
- Sato, E., et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proceedings of the National Academy of Sciences of the United States of America. 102, 18538-18543 (2005).
- De Moura, M. B., et al. Mitochondrial respiration-an important therapeutic target in melanoma. PLoS One. 7, 40690 (2012).
- Folkman, J., Merler, E., Abernathy, C., Williams, G. Isolation of a tumor factor responsible for angiogenesis. Journal of Experimental Medicine. 133, 275-288 (1971).
- Takahashi, S. Vascular endothelial growth factor (VEGF), VEGF receptors and their inhibitors for antiangiogenic tumor therapy. Biological and Pharmaceutical Bulletin. 34, 1785-1788 (2011).
- Subramanian, A., et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 102, 15545-15550 (2005).
- Cerami, E., et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discovery. 2, 401-404 (2012).
- Gao, J., et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Science Signalling. 6, 1 (2013).
- Reich, M., et al. GenePattern 2.0. Nature Genetics. 38, 500 (2006).
- Wang, Y. E., Kutnetsov, L., Partensky, A., Farid, J., Quackenbush, J. WebMeV: A Cloud Platform for Analyzing and Visualizing Cancer Genomic Data. Cancer Research. 77, 11-14 (2017).
- . Morpheus Available from: https://software.broadinstitute.org/morpheus (2019)
- Weitschek, E., Lauro, S. D., Cappelli, E., Bertolazzi, P., Felici, G. CamurWeb: a classification software and a large knowledge base for gene expression data of cancer. BMC Bioinformatics. 19, 354 (2018).
- Chou, P. -. H., et al. tACCo, a Database Connecting transcriptome Alterations, pathway Alterations and Clinical outcomes in Cancers. Scientific Reports. 9, 3877 (2019).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.