
Method Article
Ottimizzazione di proteine sintetiche: identificazione di interposizione Dipendenze Indicazione Strutturalmente e / o residui funzionalmente collegate
In questo articolo
Riepilogo
Synthetic protein sequences based on consensus motifs typically ignore co-evolving residues, that imply interpositional dependencies (IPDs). IPDs can be essential to activity, and designs that disregard them may result in suboptimal results. This protocol uses StickWRLD to identify IPDs and help inform rational protein design, resulting in more efficient results.
Abstract
Allineamenti proteine sono comunemente utilizzati per valutare la somiglianza dei residui proteici, e la sequenza di consenso derivata utilizzati per identificare le unità funzionali (ad esempio domini). Modelli di costruzione del consenso tradizionali non riescono a spiegare le dipendenze interposizione - funzionalmente necessaria covarianza dei residui che tendono ad apparire contemporaneamente in tutta l'evoluzione e attraverso l'albero phylogentic. Queste relazioni possono rivelare importanti indizi circa i processi di ripiegamento delle proteine, termostabilità, e la formazione di siti funzionali, che a loro volta possono essere utilizzati per informare l'ingegneria delle proteine sintetiche. Purtroppo, queste relazioni essenzialmente formano sub-motivi che non può essere previsto da semplice "regola di maggioranza" o modelli di consenso anche a base di HMM, e il risultato può essere un "consenso" biologicamente non valido, che non è solo mai visto in natura, ma è meno praticabile di qualsiasi proteina esistente. Abbiamo sviluppato un un visivostrumento alytics, StickWRLD, che crea una rappresentazione interattiva 3D di un allineamento di proteine e chiaramente mostra covarying residui. L'utente ha la possibilità di pan e zoom, nonché dinamicamente modificare la soglia statistica alla base della individuazione di covarianti. StickWRLD è già stata utilizzata con successo per identificare i residui covarying funzionalmente necessarie in proteine come adenilato chinasi e in sequenze di DNA, come siti di destinazione endonuclease.
Introduzione
Allineamenti proteine sono da tempo utilizzati per valutare la somiglianza dei residui in una famiglia di proteine. Frequentemente le caratteristiche più interessanti di una proteina (per esempio, siti di legame catalitici o altri) sono il risultato di ripiegamento delle proteine portando regioni distali del sequenza lineare a contatto, e di conseguenza queste regioni apparentemente indipendenti l'allineamento tendono ad evolversi e cambiare modo coordinato. In altri casi, la funzione di una proteina può essere dipendente dalla sua firma elettrostatica, e mutazioni che influenzano il dipolo elettronica sono compensate da modifiche residui carichi lontane. Effetti allosterici possono anche indurre a lungo raggio dipendenze sequenziali e spaziali tra le identità di residui. Indipendentemente dalla loro origine, questi covariations funzionalmente richieste di residui - dipendenze tra posizionali (IPDS) - potrebbe non essere evidente con esame visivo del tracciato (Figura 1). Identificazione DPI - nonchéche i residui specifici all'interno di tali posizioni tendono ad covary come unità - può rivelare importanti indizi sui processi di folding delle proteine e la formazione di siti funzionali. Queste informazioni possono poi essere utilizzate per ottimizzare (ingegnerizzati) proteine sintetiche in termini di stabilità termica e di attività. E 'noto da tempo che non tutte le mutazioni puntiformi verso consenso forniscono una migliore stabilità o attività. Più recentemente, proteine progettati per sfruttare DPI noti loro sequenza hanno dimostrato di causare un aumento dell'attività della stessa proteina progettata rigorosamente da consensus 1,2 (manoscritto in preparazione), simile al concetto di stabilizzazione mutazioni puntiformi 3.
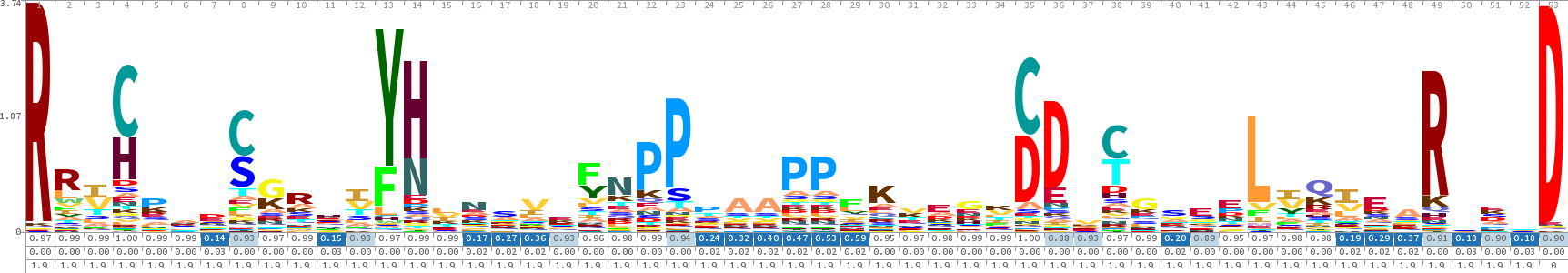
Purtroppo, i modelli di costruzione del consenso tradizionali (ad esempio, la regola di maggioranza) solo catturano DPI per caso. Metodi di consenso e posizione di punteggio specifico Matrix sono ignoranti di DPI e solo 'correttamente' li includono nei modelli, quando i residui dipendentisono anche i residui più popolari per quelle posizioni in famiglia. Modelli catena di Markov possono catturare DPI quando sono in sequenza prossimale, ma la loro tipica implementazione ignora tutto tranne vicini sequenziali immediati, e anche al loro meglio, calcoli Hidden Markov Model (vedi figura 2) diventano intrattabile quando le dipendenze sono separati nella sequenza di oltre una dozzina di posizioni 4. Dal momento che questi DPI essenzialmente formano "sub-motivi", che non può essere previsto da semplice "regola di maggioranza" o modelli consenso addirittura basati su HMM 5,6 il risultato può essere un "consenso" biologicamente non valido, che non è solo mai visto in natura, ma è meno praticabile di qualsiasi proteina esistente. I sistemi basati su Markov casuale Fields, quali GREMLIN 7, tentano di superare questi problemi. Inoltre mentre sofisticate tecniche biologiche / biochimici come noncontiguous ricombinazione 3,8 possono essere utilizzate per identify elementi essenziali di proteine per regione, richiedono molto tempo e banco di lavoro per singolo-base-pair di precisione da raggiungere.
StickWRLD 9 è un programma basato su Python che crea una rappresentazione interattiva 3D di un allineamento proteina che rende DPI chiaro e facile da capire. Ogni posizione nell'allineamento è rappresentata come una colonna nella visualizzazione, in cui ogni colonna è costituita da una pila di sfere, una per ciascuno dei 20 aminoacidi che potrebbero essere presenti in quella posizione entro l'allineamento. La dimensione della sfera dipende dalla frequenza di occorrenza degli aminoacidi, in modo tale che l'utente può raccogliere immediatamente il residuo consenso o la relativa distribuzione di amminoacidi all'interno di tale posizione semplicemente guardando le dimensioni delle sfere. Le colonne rappresentano ciascuna posizione sono avvolti attorno ad un cilindro. Questo dà ogni sfera che rappresenta una possibile amminoacido in ciascuna posizione l'allineamento, una chiara 'linea di vista'ad ogni altra possibilità ammino acido in ogni altra posizione. Prima della visualizzazione, StickWRLD calcola la forza della correlazione tra tutte le possibili combinazioni di residui per identificare il DPI 9. Per rappresentare DPI, linee sono disegnate tra i residui che sono coevolving ad una maggiore o minore di quanto ci si aspetterebbe se i residui presenti nelle posizioni erano indipendenti (DPI).
Non solo questa visualizzazione spettacolo che le posizioni di sequenza interagiscono evolutivamente, ma come le strisce di margine IPD sono disegnate tra le sfere di aminoacidi in ogni colonna, l'utente può determinare rapidamente che gli amminoacidi specifici tendono ad essere coevolving in ciascuna posizione. L'utente ha la possibilità di ruotare e di esplorare la struttura IPD visualizzato, nonché dinamicamente modificare le soglie statistiche che controllano la visualizzazione delle correlazioni, rendendo StickWRLD un potente strumento di scoperta per DPI.
Applicazioni come GREMLIN 7 similarly visualizzare le informazioni relazionali complesse tra i residui - ma queste relazioni sono calcolati con più modelli tradizionali di Markov, che non sono progettati per determinare eventuali relazioni condizionate. Come tali, questi sono in grado di essere visualizzato come proiezioni 2D. Per contro, StickWRLD può calcolare e visualizzare le dipendenze condizionali multi-nodo, che può essere offuscati se reso come un grafico 2D (un fenomeno noto come occlusione bordo).
Vista 3D di StickWRLD ha anche diversi altri vantaggi. Consentendo agli utenti di manipolare le visuali - caratteristiche che possono essere offuscati o poco intuitivo in una rappresentazione 2D può essere più facilmente visibile nel cilindro 3D di StickWRLD - panning, rotazione e zoom. StickWRLD è essenzialmente uno strumento di analisi visiva, sfruttando la potenza di pattern recognition capacità del cervello umano di vedere i modelli e le tendenze, e la possibilità di esplorare i dati da diversi punti di vista si presta a questo.
Protocollo
1. Software Download e installazione
- Utilizzare un computer ha un processore Intel i5 o meglio processore con almeno 4 GB di RAM, e sia in esecuzione Mac OS X o GNU / Linux (ad esempio, Ubuntu) OS. Inoltre, sono necessari Python 2.7.6 10 e il wxPython 2.8 11, SciPy 12, e PyOpenGL librerie 13 pitone - scaricare e installare ogni dalle rispettive repository.
- Scarica StickWRLD come un archivio zip contenente tutti i relativi script Python. Scaricare lo script "fasta2stick.sh" per la conversione di allineamenti di sequenze / proteine DNA FASTA standard per formato StickWRLD.
- Estrarre l'archivio e mettere la cartella StickWRLD risultante sul tuo desktop. Posizionare lo script "fasta2stick.sh" sul desktop pure.
2. Preparare il Allineamento
- Creare un allineamento delle sequenze proteiche utilizzando qualsiasi stansoftware allineamento Dard (ad esempio, ClustalX 14). Salvare l'allineamento sul desktop in formato FASTA.
- Aprire l'applicazione terminale sul computer Mac o GNU / Linux e passare al desktop (la posizione del "fasta2stick.sh" script di shell) digitando cd ~ / Desktop e premendo il ritorno. Eseguire lo script "fasta2stick.sh" digitando ./fasta2stick.sh nel terminale. Se lo script non viene eseguito, assicurarsi che sia eseguibile - nel tipo chmod + x terminale fasta2stick.sh per rendere lo script eseguibile.
- Seguire le istruzioni visualizzate fornite dallo script per specificare il nome del file di input (il file creato in 1.2 sopra) e il nome di output desiderato. Salvare il file di output (che ora è nel formato corretto per StickWRLD) sul desktop.
3. Avvio StickWRLD
- Navigare nelle eseguibili StickWRLD cartella utilizzando la applic terminalezione del computer Mac o GNU / Linux. Ad esempio, se la cartella StickWRLD si trova sul desktop, digitare cd ~ / Desktop / StickWRLD / exec nel terminale.
- Avviare StickWRLD digitando python-32 stickwrld_demo.py nel terminale.
- Verificare che il pannello StickWRLD Data Loader è visibile sullo schermo (Figura 3).
4. Caricamento dei dati
- Caricare l'allineamento sequenza proteica convertito premendo il pulsante "Load Protein ..." pulsante.
- Selezionare il file creato nel passaggio 3 sopra e premere "Apri". StickWRLD aprirà numerose finestre, tra cui "StickWRLD Control" (Figura 4) e "StickWRLD - OpenGL" (Figura 5).
- Selezionare la - finestra "StickWRLD OpenGL". Scegliere "Ripristina vista" dal menu "OpenGL" per visualizzare la visualizzazione StickWRLD default in un "top-downVista "attraverso il cilindro che rappresenta i dati nelle finestre ridimensionabili OpenGL ..
5. Opzioni di visualizzazione
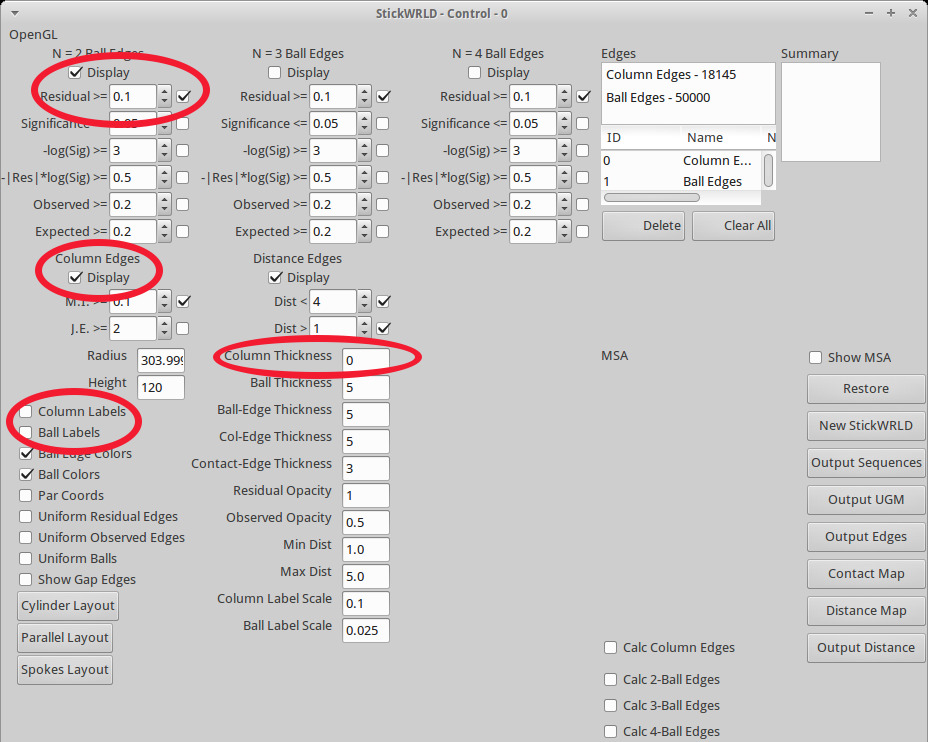
- Selezionare le caselle per "etichette di colonna" e "Etichette Ball" nel riquadro "StickWRLD Control" (Figura 4) per visualizzare i valori per le colonne e le palle.
- Deselezionare la casella per "Bordi colonna" nel riquadro "StickWRLD Control" per nascondere le strisce di margine di colonna.
- Impostare il "spessore Colonna" per 0,1 nel riquadro "StickWRLD Control" per disegnare una linea sottile attraverso le colonne, rendendo più facile la navigazione nella vista 3D. Premere Invio per accettare la modifica.
- Ripristina la vista nella "StickWRLD - OpenGL" finestra al precedente punto 5.3, quindi premere il pulsante "schermo intero" per massimizzare la vista.
6. Navigation
- Ruotare il display 3D StickWRLD tenendo premuto il tasto sinistro del mouse WHIle spostando il mouse in qualsiasi direzione.
- Zoom il display 3D StickWRLD tenendo premuto il pulsante destro del mouse mentre si sposta il mouse verso l'alto o verso il basso.
7. Individuazione interposizionale Dipendenze (DPI)
- Sfoglia la vista panoramica e lo zoom come descritto al punto 6. residui Coevolving superano i requisiti di soglia sia p e residuale sono collegati attraverso strisce di margine come si vede in figura 6. Se ci sono troppi o troppo pochi bordi di collegamento residui, cambiare il residuo soglia (nel riquadro "StickWRLD Control") per visualizzare un numero inferiore, o più, i bordi.
- Aumentare la soglia residua sul riquadro di controllo fino a quando non StickWRLD strisce di margine IPD sono mostrati e lentamente rampa fino a quando appaiono le relazioni. Continuare ad aumentare il residuo fino ad avere un numero sufficiente di relazioni da esaminare.
- Identificare le relazioni che coinvolgono sia i residui di interesse noto (ad esempio, all'interno di un motivo o di legame / divertimentosito finzionale) o residui che sono distale fra loro entro l'allineamento (suggerendo che sono prossimale nella proteina ripiegata)
8. Selezione e risultati di risparmio
- Utilizzando il comando + click sinistro su tutti i bordi di interesse. Il riquadro StickWRLD controllo indicherà le colonne e collegare specifici residui, ad esempio, "(124 | G) (136 | H)" (Figura 7). Le linee continue rappresentano associazioni positive; linee tratteggiate rappresentano associazioni negative.
- Premere il pulsante "Edges uscita" sul pannello "StickWRLD di controllo" per salvare un file in formato solo testo (edge_residual.csv) di tutti i bordi visibili, tra cui i residui uniti ei loro valori residui effettivi, in / StickWRLD / exec directory /.
Risultati
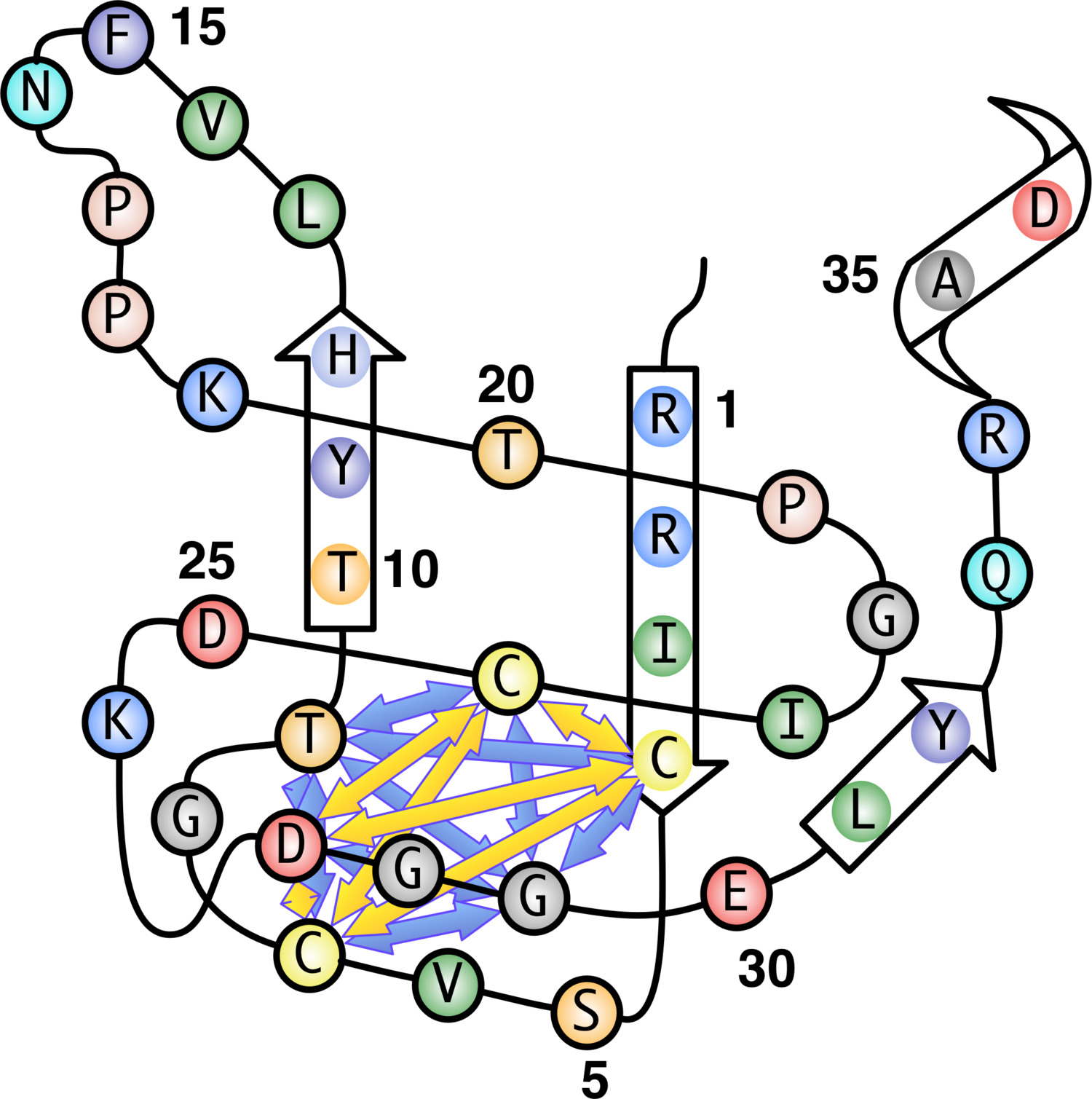
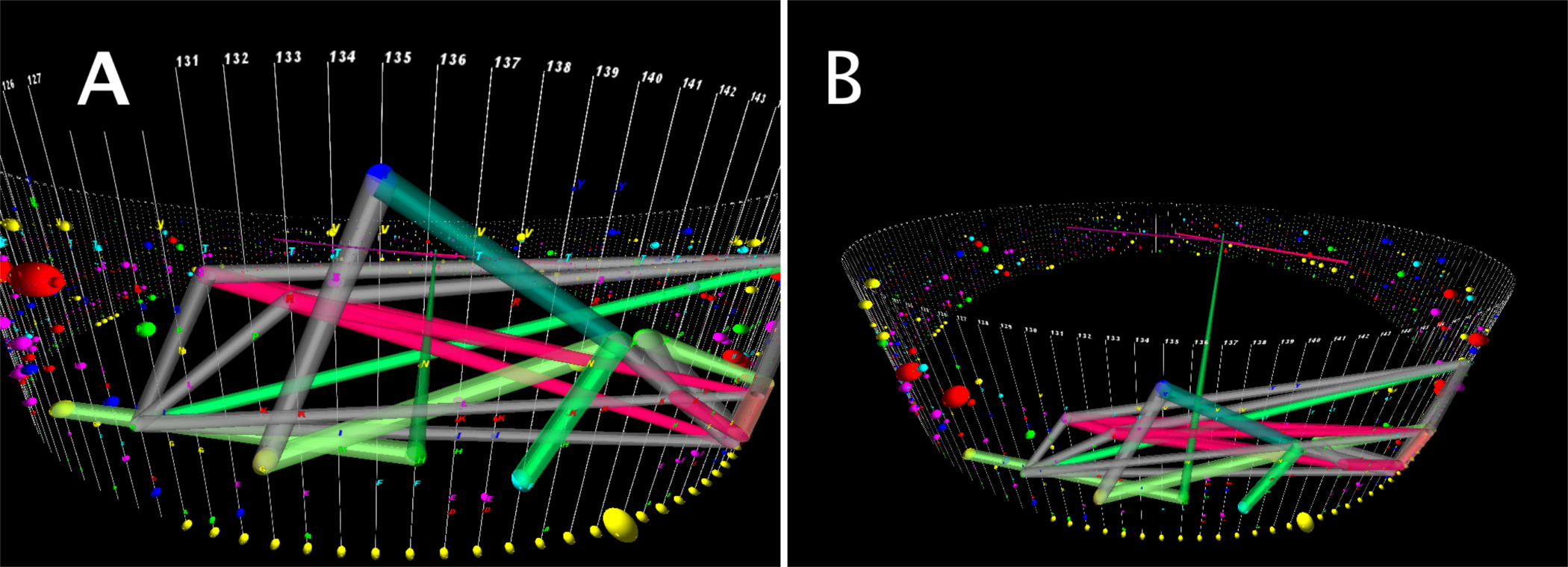
StickWRLD è stato utilizzato in precedenza per rilevare le dipendenze interposizione (IPDS) tra residui sia DNA 3 e proteine 15-17 allineamenti. Questi residui co-evoluzione, mentre spesso distale uno dall'altro in allineamento di sequenze, sono spesso prossimale tra loro nella proteina ripiegata. StickWRLD permette una rapida scoperta di specifici residui di co-occorrenza di tali siti, ad esempio., Una alanina in posizione "X" è fortemente correlato a una treonina alla posizione "y". Tali correlazioni possono essere indicativi di relazioni strutturali dimostrabili, e in genere sono siti che, per necessità, co-evolvono. StickWRLD è in grado di rilevare questi rapporti anche se più "tradizionali" Approcci usando HMM per descrivere motivi sicuro. Ad esempio, l'analisi dell'allineamento PFAM del dominio coperchio ADK usando StickWRLD rivela una forte correlazione positiva tra cisteine (C) nelle posizioni 4 e 8 e una coordinatacoppia di C nelle posizioni 35 e 38. Allo stesso tempo, StickWRLD mostrato una simile forte relazione positiva tra istidina (H) e serina (S) a 4 e 8, con forti relazioni negative tra questi e il quartetto C a 4, 8, 35, e 38, e una forte relazione positiva con acido aspartico (D) e treonina (T) in posizioni 35 e 38 rispettivamente. Esistono DPI aggiuntivi tra la H, S, D, T motivo e una T e G alla posizione **** 10 e 29 in b subtilis **** evidenziando la natura condizionale di questi DPI - il motivo tetracysteine non 'cura' sulle identità a queste due posizioni, mentre l'H idrofila, S, D, T triade richiede specifici residui in queste posizioni quasi assolutamente. Questi due completamente diversi motivi di residui in funzione della posizione possono svolgere lo stesso ruolo del coperchio ADK. Come si vede in figura 6, un grande cluster di DPI, compresa un'associazione 3-nodo fra G (glicina) alla posizione 132, Y (tirosina) alla posizione 135, e un P (Proline) alla posizione 141, è visibile in primo piano (Figura 6A). In figura 6B, la vista è distorta per posizionare l'utente leggermente sopra il cilindro, rivelando una IPD tra un H (istidina) alla posizione 136 e una M (metionina) alla posizione 29, 107 residui lontana. Un motivo PFAM HMM di derivazione dello stesso dominio (figura 2), nel frattempo, non solo non rileva questi come in particolare le varianti di co-occuring motivi, ma definisce anche i raggruppamenti complessivi in uno schema biologicamente non supportato 16.

Figura 1. "Subway Map" rappresentazione del B. subtilis adenosina chinasi struttura di dominio (ADK) coperchio. Le frecce indicano DPI individuati nell'allineamento PFAM di ADK dominio coperchio StickWRLD. StickWRLD è in grado di identificare correttamente DPI all'interno di un cluster of residui che si trovano in prossimità della proteina ripiegata. Di particolare interesse sono la coppia T e G alla posizione 9 e 29, che costituiscono solo IPD quando tetrade di residui a 4, 7, 24, e 27 non è C, C, C, C). Numeri residui visualizzato rappresenta B. subtilis posizione e non Pfam posizioni di allineamento. Cliccate qui per vedere una versione più grande di questa figura.
{kind=link}

Figura 2. Skylign 18 Hidden Markov Model (HMM) Sequenza Logo per il dominio coperchio ADK. Mentre HMM sono strumenti potenti per determinare le probabilità in ogni posizione, così come il contributo di ciascun sito per il modello generale, l'indipendenza posizionale di HMM li rende inadatto per la rilevazione di DPI. Questo modello non suggeriscono alcunadipendenze visto nelle rappresentazioni StickWRLD (Figura 6). Cliccate qui per vedere una versione più grande di questa figura.
{kind=link}

Figura 3. Il StickWRLD Data Loader. Gli utenti possono scegliere tra i dati demo esistenti oppure caricare i propri dati in forma di DNA o sequenze proteiche allineamenti.

Figura 4. La finestra StickWRLD di controllo. Il pannello di controllo consente all'utente di modificare varie proprietà della vista, nonché regolare le soglie che controllano la visualizzazione delle strisce di margine che indicano le relazioni tra residui (DPI). Cerchiati in rosso sono i valori predefiniti che in genere necessitano di t o essere regolata per una migliore visualizzazione di qualsiasi set di dati. Il valore residuo imposta la soglia di (osservato del previsto) per il quale le linee connettore / associazione sono disegnati. I comandi per colonna e palle etichette controllano se la posizione della colonna e dei valori residui (per esempio, "A" per arginina) vengono visualizzati. La colonna Bordo cavicchi di controllo di linea e fuori la visualizzazione delle linee di bordo che collegano le colonne - per i set di dati densi questo è meglio spento. I controlli Colonna Spessore se la colonna stessa viene visualizzato -. Impostazione a un valore molto piccolo (ad esempio, 0,1) disegnerà una linea attraverso le sfere della colonna, rendendo più semplice per distinguere le colonne le une dalle altre Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
ghres.jpg "width =" 600 "/>
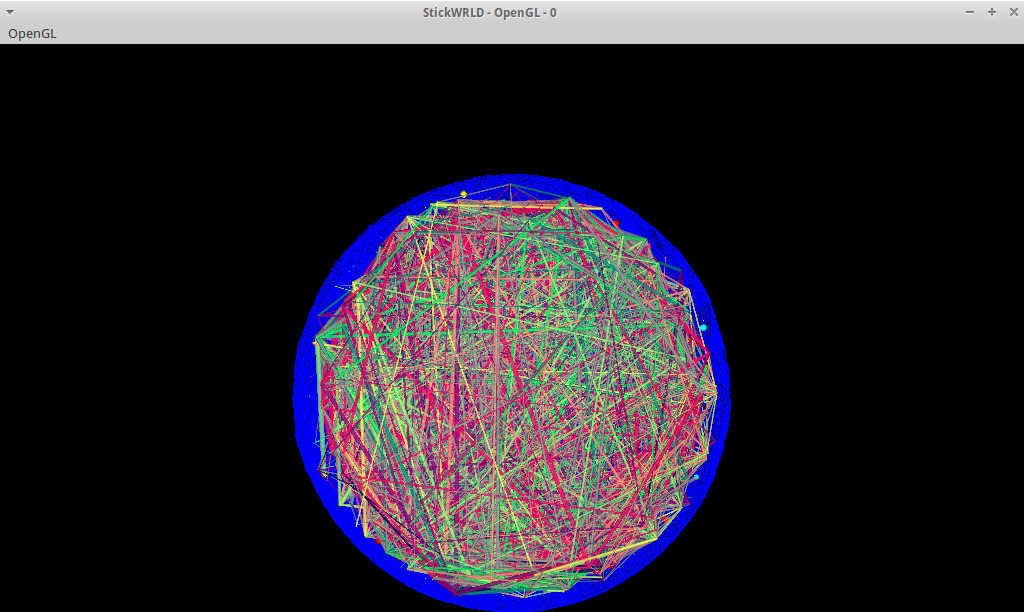
Figura 5. vista iniziale della finestra StickWRLD OpenGL con l'adenilato chinasi dominio coperchio set di dati proteina caricato. La prospettiva iniziale sembra "down" attraverso il cilindro costituito delle posizioni di allineamento sequenza. L'utente può ruotare il cilindro con sinistro del mouse fare clic e trascinare, e lo zoom in / out con il pulsante destro del mouse fare clic e trascinare. La vista iniziale è abbastanza denso, perché la visualizzazione predefinita mostra anche piccole percentuali di co-evoluzione. Per molte proteine, in questa impostazione, moduli distinti possono essere rilevati, ma anche in aree densamente co-evoluzione proteine il display può essere rapidamente e in modo interattivo semplificato per trovare le IPD più importanti utilizzando l'interfaccia StickWRLD. Cliccate qui per vedere una versione più grande questa cifra.
{kind=link}
ghres.jpg "width =" 700 "/>
Figura 6. Vista del primo piano di una visualizzazione StickWRLD della proteina dominio coperchio adenilato chinasi. Qui abbiamo cambiato il default residuo di 0,2. Questo aumenta la soglia per la visualizzazione dei bordi tra residui, mostrando meno bordi. I bordi che rimangono indicano DPI fortemente associati. Inoltre la vista è stata ruotata e ingrandita per consentire per facilitare la visualizzazione dei bordi. (A) Un grande cluster di DPI è visibile in primo piano, tra cui un'associazione 3-nodo fra G (glicina) alla posizione 132, Y (tirosina) alla posizione 135, e P (prolina) nella posizione 141. (B) Il punto di vista è stata distorta per posizionare l'utente leggermente al di sopra del cilindro, svelando un IPD tra un H (istidina) alla posizione 136 e una M (metionina) alla posizione 29, 107 residui distante. Clicca qui per vedere una versione più grande di questo figura.
{kind=link}

Figura 7. Finestra StickWRLD Controllo visualizzare informazioni in basso a destra. CTRL + clic sinistro su un oggetto (ad esempio, sfera o bordo) nella finestra OpenGL visualizza le informazioni per l'oggetto in basso a destra della finestra StickWLRD controllo. Qui vediamo le informazioni per un bordo IPD tra una metionina nella posizione 29 e una istidina in posizione 136.
Discussione
StickWRLD è stato utilizzato con successo per identificare tali DPI nel dominio coperchio adenilato chinasi 16, così come le basi del DNA associate in terminatori rho-dipendente 9, e un romanzo splice-site specificità archeali tRNA endonucleasi introni 6 siti di destinazione. Questi DPI non erano rilevabili attraverso un esame diretto degli allineamenti.
StickWRLD visualizza ogni posizione di allineamento come una colonna di 20 "sfere", dove ogni sfera rappresenta uno dei residui 20 aminoacidi e la dimensione della sfera indica la frequenza di occorrenza di quel particolare residuo dentro tale colonna (Figura 4). Le colonne sono disposte in un cilindro, con strisce di margine collegamento residui in colonne diverse (che indica un IPD). Queste strisce di margine sono disegnate solo se i residui corrispondenti sono covarying con una frequenza che supera sia il valore p (significato) e (attesi - osservate) residue soglie.
Il rilevamento di residui concomitanti interdipendenti, o DPI, nelle regioni distali di un allineamento di DNA o sequenze proteiche è difficile utilizzare la sequenza standard di strumenti di allineamento 6. Mentre tali strumenti generano un consenso, o motivo, la sequenza, questo consenso è in molti casi una semplice maggioranza regola media e non trasmettere relazioni covariazione che si possono formare uno o più sub-motivi - gruppi di residui che tendono a co-evolvere. Anche i modelli HMM, che sono in grado di rilevare le dipendenze vicini, non possono accuratamente modello motivi di sequenza con DPI distali 5. Il risultato è che il consenso calcolato può infatti essere una sequenza "sintetico" non trovato in natura - e proteine ingegnerizzate basate su tale consenso computazionale non può, infatti, essere ottimale. Infatti, la Pfam HMM per ADK suggerirebbe che una proteina chimerica contenente la metà del motivo tetracysteine, e la metà della H, S, D, T motivo, è funzionalmente altrettanto accettabilicome ogni ADK realmente esistente. Questo non è il caso, in quanto tali chimere (e molti altri blendings di questi motivi) sono cataliticamente morti 4,19.
Quando cerchi correlazioni, è fondamentale che la soglia residua essere regolata per consentire la scoperta di correlazioni pertinenti impostando la soglia al di sopra del livello al quale eventuali spigoli sono visti e poi gradualmente rampa soglia indietro. In questo modo solo i bordi più significativi sono considerati inizialmente.
Un approccio alternativo è quello di iniziare con la soglia impostata residua estremamente bassa. Ciò provoca la visualizzazione di tutti i bordi significativi. Da qui la soglia residuo può essere aumentata lentamente, permettendo bordi di drop out fino a modelli di emergere. Anche se questo approccio è meno utile quando si cerca per l'inserimento di nodi specifici (ad esempio, l'applicazione della conoscenza di dominio), permette la scoperta di relazioni inaspettate usando StickWRLD come a visUAL strumento analitico per scoprire modelli emergenti nella visualizzazione dei dati.
StickWRLD è limitata principalmente dalla memoria disponibile del sistema in cui viene eseguito e la risoluzione del dispositivo di visualizzazione. Mentre non vi è alcun limite teorico al numero di punti dati StickWRLD può esaminare, e sequenze fino a 20.000 posizioni sono stati testati, in pratica StickWRLD funziona meglio con le sequenze fino a circa 1.000 posizioni.
Il vantaggio principale di StickWRLD risiede nella sua capacità di identificare gruppi di residui che covary uno con l'altro. Questo è un vantaggio significativo rispetto all'approccio tradizionale della sequenza consenso statistica, che è una semplice media statistica e non tiene conto coevolution. Mentre in alcuni casi i residui covarying possono semplicemente essere un artefatto della filogenesi, anche questi residui hanno resistito alla "test di selezione", e come tale è improbabile che sminuire l'functionalità di ogni proteina progettata per includerli.
Durante l'utilizzo StickWRLD identificare DPI in una sequenza di DNA o proteine canonica consenso / motif prima varianti ingegneria sintetici ridurrà il potenziale di errore e sostenere ottimizzazione rapida della funzione, va notato che StickWRLD può essere usato come strumento di identificazione di correlazione generalizzato e non si limita esclusivamente ai dati di proteine. StickWRLD può essere utilizzato per scoprire visivamente la co-occorrenza di eventuali variabili in ogni insieme di dati codificati correttamente.
Divulgazioni
The authors declare that they have no competing financial interests.
Riconoscimenti
StickWRLD was made possible in part through funding provided to Dr. Ray by the Research Institute at Nationwide Children's Hospital, and by NSF grant DBI-1262457.
Materiali
| Name | Company | Catalog Number | Comments |
| Mac or Ubuntu OS computer | Various | Any Mac or GNU/Linux (e.g., Ubuntu) computer capable of running Python & associated shell scripts | |
| Python programming language | python.org | Python version 2.7.6 or greater recommended | |
| wxPython library | wxpython.org | Latest version recommended | |
| SciPy library | scipy.org | Latest version recommended | |
| PyOpenGL library | pyopengl.sourceforge.net | Latest version recommended | |
| StickWRLD Python scripts | NCH BCCM | Available from http://www.stickwrld.org | |
| fasta2stick.sh file converter | NCH BCCM | Available from http://www.stickwrld.org | |
| Protein and/or DNA sequence data | Samples available at http://www.stickwrld.org |
Riferimenti
- Ray, W. C. Addressing the unmet need for visualizing conditional random fields in biological data. BMC. 15, 202 (2014).
- Sullivan, B. J., Durani, V., Magliery, T. J. Triosephosphate isomerase by consensus design: dramatic differences in physical properties and activity of related variants. Journal of molecular biology. 413, 195-208 (2011).
- Smith, M. A., Bedbrook, C. N., Wu, T., Arnold, F. H. Hypocrea jecorina cellobiohydrolase I stabilizing mutations identified using noncontiguous recombination. ACS synthetic biology. 2, 690-696 (2013).
- Ray, W. C. Understanding the sequence requirements of protein families: insights from the BioVis 2013 contests. BMC proceedings. 8, S1 (2014).
- Eddy, S. R. What is a hidden Markov model?. Nature biotechnology. 22, 1315-1316 (2004).
- Ray, W. C., Ozer, H. G., Armbruster, D. W., Daniels, C. J. Beyond identity - when classical homology searching fails, why, and what you can do about it. Proceedings of the 4th Ohio Collaborative Conference on Bioinformatics. , 51-56 (2009).
- Ovchinnikov, S., Kamisetty, H., Baker, D. Robust and accurate prediction of residue-residue interactions across protein interfaces using evolutionary information. eLife. 3, e02030 (2014).
- Trudeau, D. L., Lee, T. M., Arnold, F. H. Engineered thermostable fungal cellulases exhibit efficient synergistic cellulose hydrolysis at elevated temperatures. Biotechnology and bioengineering. 111, 2390-2397 (2014).
- Ray, W. C. MAVL and StickWRLD: visually exploring relationships in nucleic acid sequence alignments. Nucleic acids research. 32, W59-W63 (2004).
- . Python Language Reference v.2.7.6 Available from: https://www.python.org/download/releases/2.7.6/ (2014)
- . . PyOpenGL The Python OpenGL Binding. , (2014).
- Larkin, M. A. Clustal W and Clustal X version 2.0. Bioinformatics. 23, 2947-2948 (2007).
- Ozer, H. G., Ray, W. C. MAVL/StickWRLD: analyzing structural constraints using interpositional dependencies in biomolecular sequence alignments. Nucleic acids research. 34, W133-W136 (2006).
- Ray, W. C. MAVL/StickWRLD for protein: visualizing protein sequence families to detect non-consensus features. Nucleic acids research. 33, W315-W319 (2005).
- Ray, W. C. A Visual Analytics approach to identifying protein structural constraints. IEEE. , 249-250 (2010).
- Wheeler, T. J., Clements, J., Finn, R. D. Skylign: a tool for creating informative, interactive logos representing sequence alignments and profile hidden Markov models. BMC bioinformatics. 15, 7 (2014).
- Perrier, V., Burlacu-Miron, S., Bourgeois, S., Surewicz, W. K., Gilles, A. M. Genetically engineered zinc-chelating adenylate kinase from Escherichia coli with enhanced thermal stability. The Journal of biological chemistry. 273, 19097-19101 (1998).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneEsplora altri articoli
This article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati