Method Article
Utilizzando tecnica LUCE microscopia a fluorescenza all'Immagine sviluppo Zebrafish Eye
* Questi autori hanno contribuito in egual misura
In questo articolo
Riepilogo
Light sheet fluorescence microscopy is an excellent tool for imaging embryonic development. It allows recording of long time-lapse movies of live embryos in near physiological conditions. We demonstrate its application for imaging zebrafish eye development across wide spatio-temporal scales and present a pipeline for fusion and deconvolution of multiview datasets.
Abstract
Light sheet fluorescence microscopy (LSFM) is gaining more and more popularity as a method to image embryonic development. The main advantages of LSFM compared to confocal systems are its low phototoxicity, gentle mounting strategies, fast acquisition with high signal to noise ratio and the possibility of imaging samples from various angles (views) for long periods of time. Imaging from multiple views unleashes the full potential of LSFM, but at the same time it can create terabyte-sized datasets. Processing such datasets is the biggest challenge of using LSFM. In this protocol we outline some solutions to this problem. Until recently, LSFM was mostly performed in laboratories that had the expertise to build and operate their own light sheet microscopes. However, in the last three years several commercial implementations of LSFM became available, which are multipurpose and easy to use for any developmental biologist. This article is primarily directed to those researchers, who are not LSFM technology developers, but want to employ LSFM as a tool to answer specific developmental biology questions.
Here, we use imaging of zebrafish eye development as an example to introduce the reader to LSFM technology and we demonstrate applications of LSFM across multiple spatial and temporal scales. This article describes a complete experimental protocol starting with the mounting of zebrafish embryos for LSFM. We then outline the options for imaging using the commercially available light sheet microscope. Importantly, we also explain a pipeline for subsequent registration and fusion of multiview datasets using an open source solution implemented as a Fiji plugin. While this protocol focuses on imaging the developing zebrafish eye and processing data from a particular imaging setup, most of the insights and troubleshooting suggestions presented here are of general use and the protocol can be adapted to a variety of light sheet microscopy experiments.
Introduzione
Morfogenesi è il processo che dà forma l'embrione e, insieme con la crescita e la differenziazione guida la ontogenesi da un ovulo fecondato in un organismo multicellulare maturo. I processi morfogenetici durante lo sviluppo degli animali possono essere analizzati meglio da immagini di esemplari viventi intatti 1-3. Ciò perché tale l'imaging intero embrione conserva tutti i componenti che guidano e regolano lo sviluppo compreso gradienti di molecole di segnalazione, matrice extracellulare, vascolarizzazione, innervazione così come le proprietà meccaniche del tessuto circostante. Per colmare le scale, in cui si verifica la morfogenesi, gli eventi subcellulare veloci devono essere catturato su una scala temporale minuto nel contesto dello sviluppo di tutto il tessuto sopra ore o giorni. Per soddisfare tutte queste esigenze, una moderna implementazione 4 del ortogonali illuminazione piano microscopio 5 è stato sviluppato. In origine, è stato chiamato il piano di illuminazione selettiva Microscopia (SPIM) 4; ora un Term Sheet chiaro che tutto abbraccia microscopia a fluorescenza (LSFM) viene utilizzato in genere. LSFM permette l'imaging ad elevata risoluzione temporale, mentre induce meno fototossicità di scansione laser o disco rotante microscopi confocale 6,7. Al giorno d'oggi, ci sono già molte implementazioni del principio di base foglio di luce di illuminazione ed è stato utilizzato per l'immagine di una grande varietà di esemplari e dei processi precedentemente inaccessibili ai ricercatori 8-11.
Per prima cosa vorremmo sottolineare alcuni vantaggi chiave di LSFM rispetto agli approcci confocale microscopia convenzionale:
Per acquisire risultati significativi Dall'imaging vivo esperimenti microscopiche, è importante che l'osservazione influisce solo minimamente il campione. Tuttavia, molti organismi, tra cui zebrafish sono molto sensibili alla esposizione alla luce laser, il che rende difficile per l'immagine di loro in un microscopio confocale con elevata risoluzione temporale senza fototossicità efdifetti come in fase di stallo o ritardato 6,7 sviluppo. LSFM è attualmente la tecnica di imaging di fluorescenza con minimi effetti dirompenti sul campione 7. Poiché il sottile foglio leggero laser illumina solo la parte del campione che viene esposta in un particolare punto di tempo, il foglio microscopio ottico è usando fotoni molto efficiente. Di conseguenza, la bassa esposizione alla luce permette più a lungo le osservazioni time-lapse di esemplari sani, ad esempio 12-17. Inoltre, grazie alla minima invasività della LSFM, il numero di immagini acquisite non è più dettata dalla quantità di luce che il campione può tollerare, ma piuttosto da quanti dati possono essere elaborati e memorizzati.
Sulla stessa linea di mantenere il campione in condizioni fisiologiche vicino, LSFM viene fornito con il montaggio del campione strategie alternative ben si adatta per gli embrioni dal vivo. Nelle tecniche LSFM, gli embrioni sono tipicamente inseriti all'interno di una colonna sottile di bassa agarosio percentuale. il mounting in cilindri agarosio permette la completa libertà di rotazione, in modo che il campione può essere osservato dalla angolo perfetto (in LSFM denominato vista) e da diverse viste simultaneamente. Imaging MultiView e la successiva fusione MultiView è utile in particolare per i grandi campioni di scattering e permette loro cattura ad alta risoluzione isotropo. Una sintesi di altre possibili strategie di montaggio LSFM può essere trovato nel manuale operativo del microscopio ufficiale, nel capitolo dedicato alla preparazione del campione scritto dal laboratorio di E. Reynaud. È una lettura consigliata, soprattutto se l'obiettivo è quello di diversi campioni dell'immagine quanto descritto qui.
L'acquisizione delle immagini in LSFM è grande campo, a base di fotocamera, al contrario di scansione laser microscopio confocale. Ciò si traduce in un rapporto segnale rumore (SNR) per le immagini acquisite superiore e può essere estremamente veloce (decine a centinaia di fotogrammi al secondo). L'elevata sensibilità del LSFM permette inoltre di imaging di samp debolmente fluorescenteles, come fattori di trascrizione espressi a livelli endogeni 18 o, in un prossimo futuro, le proteine endogene marcati usando CRISPR / Cas9. L'elevato SNR è anche importante per il successo analisi dell'immagine valle. L'alta velocità è necessario non solo per catturare processi intracellulari rapidi, ma anche per l'immagine dell'intero embrione da viste multiple abbastanza veloce. Una fusione armoniosa delle visualizzazioni multiple può essere raggiunto solo se il fenomeno osservato non cambia durante l'acquisizione di queste diverse pile z provenienti da viste separate.
I vantaggi di LSFM tipicamente non va a scapito della qualità dell'immagine. La risoluzione laterale di LSFM è leggermente peggiore rispetto alla risoluzione di un microscopio confocale. Questo perché gli obiettivi di rilevazione utilizzati in LSFM hanno una minore apertura numerica (di solito 1,0 o inferiore) rispetto a 1.2-1.3 di acqua o immersione silicio obiettivi su configurazioni confocale standard. Inoltre, grazie al rilevamento ampio campo in LSFM (Absence di un foro), c'è più luce fuori fuoco rispetto ad un microscopio confocale. La quantità di out-of-focus luce è determinato dallo spessore foglio leggero. Tuttavia, questi svantaggi sono compensati dal SNR superiore in LSFM. In pratica, questo si traduce in immagini di qualità simili rispetto a, per esempio filatura disco acquisizione confocale 15. Di conseguenza, ciò consente l'estrazione affidabile di caratteristiche come membrane cellulari o nuclei, per esempio, per cell lineage tracing 15,19.
La risoluzione assiale LSFM è determinata, oltre all'obiettivo rilevamento, dallo spessore foglio leggero. La risoluzione assiale LSFM può in alcuni casi superare la risoluzione dei microscopi confocale. In primo luogo, il miglioramento della risoluzione viene quando il foglio leggero è più sottile rispetto alla risoluzione assiale dell'obiettivo rilevamento, che tipicamente si verifica per grandi campioni viste con obiettivo a basso ingrandimento. Il secondo modo, come il LSFM può achIEVE migliore risoluzione assiale, è la fusione MultiView, in cui le informazioni ad alta risoluzione XY Da diversi punti di vista si combina in un'unica serie di immagini. Lo stack fusa risultante ha una risoluzione isotropa avvicina i valori di risoluzione nella direzione laterale 20,21. La strategia per la registrazione delle molteplici vista sulla vicenda descritta in questo articolo si basa sull'utilizzo di perle di polistirene fluorescenti come marcatori fiduciari incorporati nel agarosio intorno al campione 20,21.
Come risultato della commercializzazione LSFM, questa tecnica è ora disponibile per una vasta comunità di scienziati 22. Pertanto, la motivazione per la scrittura di questo protocollo è quello di rendere questa tecnologia accessibile a biologi dello sviluppo che mancano di esperienza pratica in LSFM e per ottenere questi scienziati iniziato a utilizzare questa tecnologia con i loro campioni. Il nostro protocollo utilizza il microscopio foglio di luce commerciale, che costituisce un concettualmente semplice microscopio tcappello è facile da usare. Vorremmo inoltre segnaliamo altri protocolli più recenti per l'imaging zebrafish con configurazioni LSFM casa costruita, che potrebbero essere adatti per rispondere a particolari domande 23-25. Un'altra opzione ingresso al LSFM sono le piattaforme aperte 26,27, che utilizzano i principi di accesso aperto per portare la microscopia foglio di luce per una comunità più ampia. La documentazione sia l'hardware e gli aspetti del software può essere trovato alla http://openspim.org e https://sites.google.com/site/openspinmicroscopy/.
In questo protocollo, si usa il pesce zebra teleosteo come sistema modello per studiare i processi di sviluppo con LSFM. Morfogenesi dell'occhio zebrafish è un esempio che sottolinea molti dei vantaggi di LSFM. LSFM è già stato utilizzato in passato per indagare lo sviluppo degli occhi in Medaka 28 e in zebrafish 29,30. Alla fase iniziale di sviluppo dell'occhio è complicato per orientare l'embrione in modo corretto per la microscopia convenzionale,come il tuorlo ingombranti non consente l'embrione a giacere sul lato con il suo occhio rivolto verso l'obiettivo. Tuttavia, con il montaggio LSFM in una colonna di agarosio, il campione può essere riproducibile posizionato. Inoltre, durante la transizione da vescicola ottica allo stadio tazza ottica, l'occhio eseguono importanti riarrangiamenti morfogenetiche accompagnati dalla crescita, che richiede catturando una grande z stack e un grande campo di vista. Anche per queste sfide LSFM è superiore a confocale convenzionale. Il processo di formazione tazza ottica è tridimensionale, quindi è difficile da comprendere e visualizzare esclusivamente da immagini da una visualizzazione. Questo rende l'imaging MultiView con risoluzione isotropo vantaggioso. Dopo la formazione tazza ottica, la retina diventa sempre più sensibile all'esposizione laser. Così, la fototossicità bassa associata a LSFM è un grande vantaggio per l'imaging a lungo termine.
Qui vi presentiamo un protocollo ottimizzato per l'imaging di uno a tre giorni embrioni di zebrafish unlarve ND con focus sullo sviluppo dell'occhio. Il nostro metodo permette la registrazione di filmati time-lapse che copre fino a 12-14 ore con alta risoluzione spaziale e temporale. È importante sottolineare che, mostriamo anche una pipeline per l'elaborazione dei dati, che è un passo essenziale per LSFM, come questa tecnica genera invariabilmente grandi insiemi di dati, spesso nella gamma terabyte.
Protocollo
NOTA: Tutto il lavoro animale è stato eseguito in conformità con l'Unione Europea (UE) la direttiva 2011/63 / UE e la legge sul benessere degli animali tedesca. Il protocollo è destinata ad essere seguita senza interruzione, dal montaggio di imaging del campione. A seconda della esperienza pratica, ci vorranno 2-3 ore per avviare un esperimento di time-lapse. Il trattamento dei dati non è incluso in questo calcolo del tempo. Tutto il materiale necessario per l'esperimento può essere trovato nella lista di materiale necessario prima di iniziare che viene fornito come documento complementare. Indossare guanti senza polvere durante le fasi 1, 2, 3 e 4. Per i passaggi 2, 3, 4 e 5 del protocollo si riferisce anche il manuale operativo del microscopio ufficiale.
1. I lavori preparatori prima dell'esperimento Imaging
- Fluorescente tallone stock Soluzione
- Per questo protocollo, utilizzare i 500 o 1.000 nm perle di polistirene del diametro (marcati con colorante fluorescente che emettono rosso). La diluizione di lavoro delle perle è 1: 4,000. In primo luogo, vortex la soluzione tallone stock per 1 min. Diluire 10 ml di perline in 990 ml di DDH 2 O. Conservare la soluzione al buio a 4 ° C e utilizzare questa diluizione 1: 100 come soluzione di riserva per l'ulteriore diluizione 1:40.
- Preparazione Soluzione per la camera del campione
- In un becher da 100 ml mix 38.2 ml di terreno E3 colpo blu di metilene, 0,8 ml di 10 mM N -phenylthiourea e 1 ml di 0,4% MS-222. E 'utile usare medio E3 filtrata per evitare piccole particelle di polvere o cristalli insoluti che galleggiano nel pozzetto di misurazione in seguito.
- Pesce fluorescente la selezione degli embrioni
- Nei giorni precedenti l'esperimento, preparare gli embrioni che esprimono proteine fluorescenti. A destra prima dell'esperimento, ordinare gli embrioni sotto lo stereoscopio fluorescente per forza desiderabile del segnale fluorescente. Prendere 5-10 embrioni sani e dechorionate utilizzando delle pinzette.
NOTA: Questo protocollo è ottimizzato per 16-72 ore vecchioembrioni.
- Nei giorni precedenti l'esperimento, preparare gli embrioni che esprimono proteine fluorescenti. A destra prima dell'esperimento, ordinare gli embrioni sotto lo stereoscopio fluorescente per forza desiderabile del segnale fluorescente. Prendere 5-10 embrioni sani e dechorionate utilizzando delle pinzette.
2. Configurazione del Campione Camera
- Montaggio del Tre Camera di Windows
- Ci sono 4 finestre nella camera del campione, una per l'obiettivo e tre per essere sigillato con il coprioggetto (18 mm di diametro, selezionati spessore 0,17 millimetri). Conservare queste coprioggetto in etanolo al 70%. Pulirli prima dell'uso con etere: etanolo (1: 4).
- Inserire il vetrino nella finestra utilizzando una pinza sottile e assicurarsi che si inserisce nel più piccolo solco. Coprire con l'O-ring in gomma di diametro 17 mm e avvitare l'anello adattatore di illuminazione utilizzando lo strumento della finestra della camera. Ripetere la procedura per gli altri due finestre.
- Montaggio delle parti rimanenti della Camera
- Avvitare l'adattatore per un obiettivo appropriato nel quarto lato rimanente della camera. Inserire l'O-ring 15 mm di diametro nel centro dell'adattatore.
- Avvitare il bianco adattatore Luer-Lock nell'apertura in basso a destra nella chamber e il connettore di scarico grigia nell'apertura superiore sinistro. Bloccare tutte le tre aperture rimanenti con i tappi ciechi neri.
- Fissare il blocco Peltier e il metallo coda di rondine diapositiva fondo della camera utilizzando una chiave a brugola. Collegare il tubo con la siringa da 50 ml per l'adattatore Luer-Lock. Inserire la sonda di temperatura nella camera.
- Inserimento degli obiettivi e la Camera nel microscopio
- Controllare sotto lo stereoscopio che tutti gli obiettivi siano puliti. Utilizzare gli obiettivi di illuminazione 10X / 0.2 e l'obiettivo di rilevazione Plan-Apochromat 20X / 1.0 W e fare in modo che il suo collare di correzione indice di rifrazione è impostato su 1,33 per l'acqua. Avvitare l'obiettivo rilevazione nel microscopio, mantenendo gli obiettivi illuminanti coperti.
- Togliere i tappi di plastica protettive che coprono gli obiettivi illuminanti. far scorrere con attenzione la camera nel microscopio e fissarlo con la vite di fissaggio.
- Collegare il pro di temperaturaessere e il blocco Peltier con il microscopio.
NOTA: I due tubi che circolano il liquido di raffreddamento del blocco Peltier sono compatibili con entrambi i connettori. Essi formano un circuito funzionante indipendentemente dall'orientamento della connessione. - Riempire la camera attraverso la siringa con soluzione preparata al punto 1.2 fino al bordo superiore delle finestre della camera. Controllare che la camera non perda.
- Avviare il microscopio, incubazione ei computer di controllo e di stoccaggio. Avviare il software microscopio operativo e impostare la temperatura di incubazione di 28,5 ° C.
NOTA: Ci vorrà 1 ora per equilibrare completamente. Preparare il campione nel frattempo.
Preparazione del campione 3.
- Preparazione del agarosio Mix
- 15 min prima di fare del mix agarosio, sciogliere una 1 ml un'aliquota di 1% agarosio punto basso punto di fusione (disciolto in media E3) in un blocco di riscaldamento impostata a 70 ° C. Una volta che l'agarosio è completamente moldieci, trasferire 600 ul in una nuova provetta da 1,5 ml, aggiungere 250 microlitri di media E3, 50 ml di 0,4% MS-222 e 25 ml di soluzione madre in agitazione tallone.
NOTA: Questo rende 925 microlitri della miscela, mentre altri 75 microlitri è calcolato per il liquido aggiunto successivamente insieme alle embrioni. - Mantenere il tubo in un secondo blocco di riscaldamento a 38-40 ° C o verificare che l'agarosio è molto vicino al punto di gelificazione prima di mettere embrioni campione in esso.
- 15 min prima di fare del mix agarosio, sciogliere una 1 ml un'aliquota di 1% agarosio punto basso punto di fusione (disciolto in media E3) in un blocco di riscaldamento impostata a 70 ° C. Una volta che l'agarosio è completamente moldieci, trasferire 600 ul in una nuova provetta da 1,5 ml, aggiungere 250 microlitri di media E3, 50 ml di 0,4% MS-222 e 25 ml di soluzione madre in agitazione tallone.
- Montaggio del Embrioni
- Prendere cinque capillari di vetro di 20 volumi microlitri (con un segno nero, ~ 1 mm di diametro interno) e inserire i pistoni punta corrispondenza Teflon in loro. Spingere lo stantuffo attraverso il capillare in modo che la punta Teflon è al fondo del capillare.
- Trasferimento cinque embrioni (un numero che può essere montato in una sola volta), con un bicchiere o pipetta di plastica nel tubo di 37 ° C in agitazione caldo stile agarosio.
NOTA: provare a riportare un volume minimo di spirito insieme liquidoh gli embrioni. - Inserire il capillare nel mix e succhiare un embrione all'interno tirando lo stantuffo fino. Assicurarsi che il capo dell'embrione entra nel capillare prima della coda. Evitare eventuali bolle d'aria tra il pistone e il campione. Ci dovrebbe essere ± 2 cm di agarosio sopra l'embrione e ± 1 cm al di sotto di esso. Ripetere l'operazione per i rimanenti embrioni.
- Attendere l'agarosio solidifica completamente, che avviene in pochi minuti e quindi memorizzare i campioni in terreno E3, per attaccarle a parete di un bicchiere con plastilina o nastro. L'apertura inferiore del capillare deve essere appeso, nella soluzione, consentendo lo scambio di gas al campione.
NOTA: Un protocollo simile alle sezioni 3.1 e 3.2 si possono trovare anche sulla pagina wiki OpenSPIM http://openspim.org/Zebrafish_embryo_sample_preparation.
Posizionamento 4. Campione
- Montaggio Campione
- Inserire 2 buste di plastica della misura giusta (nero)contro l'altro nel gambo portacampioni. I loro lati fessura devono affrontare verso l'esterno. Fissare la vite di serraggio liberamente ruotandola in 2-3 turni. Inserire il capillare attraverso la vite di bloccaggio e spingerlo attraverso il supporto fino alla striscia di colore nero diventa visibile sull'altro lato. Evitare di toccare lo stantuffo.
- Serrare la vite di fissaggio. Inserire l'eccesso 1 cm di agarosio sotto l'embrione dal capillare e tagliarlo. Inserire lo stelo nel disco porta-campioni.
- Conferma nel software che la fase microscopio è in posizione di carico. Utilizzare rotaie di guida per scivolare tutta supporto con il campione verticalmente verso il basso nel microscopio. Ruotare, in modo che i magnetici blocca disco supporto nella posizione.
- Individuazione del capillare
- D'ora in poi, controllare il posizionamento del campione dal software. Nella scheda Individuare scegliere l'opzione capillare Individuare e posizionare il capillare in x, yez a fuoco appena sopra l'oggetto di rilevazionelente ive. Utilizzare la rappresentazione grafica nel navigatore del campione per l'orientamento.
- Inserire l'embrione delicatamente del capillare finché è di fronte alla pupilla dell'obiettivo rilevamento.
NOTA: Il 'Individuare capillare' è l'unico passo nel protocollo rimanente, durante la quale il coperchio superiore del microscopio deve essere aperto e il campione spinto fuori.
- Individuazione del campione
- Passare all'opzione 'Individuare campione' e al 0,5 zoom portare l'occhio zebrafish al centro del campo visivo. Ruotare l'embrione, in modo che il foglio leggero non passerà attraverso parti altamente rifrazione o assorbimento del campione prima che raggiunga l'occhio. Analogamente, la fluorescenza emessa bisogno di un percorso chiaro dal campione. Clicca su 'Imposta posizione iniziale'.
- Aprire lo sportello anteriore del microscopio e mettere il coperchio di plastica con una apertura 3 mm superiore della camera per evitare l'evaporazione.
NOTA: Se il livello del liquido scende al di sottoil livello di imaging, l'esperimento sarà compromessa. - Controllare il battito cardiaco dell'embrione come proxy per la salute globale. Se è troppo lento, utilizzare un altro campione (confronto ai controlli non-montato; valori specifici dipendono dalla fase di sviluppo). Passare a un'impostazione di zoom finale e regolare la posizione di un embrione.
5. La creazione di un multidimensionale acquisizione
- I parametri di acquisizione
- Passare alla scheda 'acquisizione'. Definire il percorso della luce comprese le linee laser, obiettivo di rilevazione, filtro laser blocco, divisore di fascio e le telecamere.
- Attivare la casella di controllo scansione perno. Definire le altre impostazioni di acquisizione, come la profondità di bit, formato immagine, spessore della lamiera leggera e scegliere l'illuminazione solo lato.
- Premere 'continuo' e seconda dell'intensità dell'immagine ottenuta modificare il tempo di potenza del laser e l'esposizione della fotocamera.
NOTA: Per regolare tutte le impostazioni di imaging utilizzano lesalimentazione s laser (0,5% di 100 mW laser, 30 tempo di esposizione msec), che per l'esperimento vero per evitare danni photo inutili al campione.
- Regolazione tecnica LUCE
- Passare alla 'Illumination Sided doppio' e attivare il 'casella linea laterale doppia Fusio'n. Avviare la 'Lightsheet guidata Auto-adjust'. Seguire le istruzioni passo per passo.
NOTA: La procedura guidata sposta il foglio leggero nel piano focale dell'obiettivo rilevamento, e assicura che non è inclinato e la sua vita è al centro del campo visivo. Dopo aver terminato la regolazione automatica, le posizioni dei fogli luce sinistro e destro vengono automaticamente aggiornati nel software. Un miglioramento nella qualità dell'immagine ora evidente. Attivare la casella di controllo 'Z-stack'. - Controllare la regolazione foglio leggero controllando la simmetria della funzione di diffusione di punto (PSF) in perline fluorescenti nella xz e yz vista orto. Se non èt simmetrica, regolare manualmente la posizione parametro foglio di luce su e giù fino ad ottenere una PSF forma simmetrica clessidra (Figura 1A).
- Passare alla 'Illumination Sided doppio' e attivare il 'casella linea laterale doppia Fusio'n. Avviare la 'Lightsheet guidata Auto-adjust'. Seguire le istruzioni passo per passo.
- Impostazioni di acquisizione multidimensionali
- Definire lo z-stack con il 'First Slice' e le opzioni 'ultima fetta' e impostare il passo z a 1 micron.
NOTA: Il foglio leggero in questo microscopio è statica e la z sezionamento viene ottenuto muovendo il campione attraverso. Usare sempre l'opzione 'Drive continuo' per l'acquisizione veloce di Z-stack. - Attivare la casella di controllo 'Time Series'. Definire il numero totale di punti di tempo e l'intervallo tra di loro.
- Attivare la casella di controllo 'Multiview'. Aggiungere la vista corrente nella lista MultiView, in cui è memorizzato l'x, y, z e l'angolo informazioni. Utilizzare il controller fase di ruotare il capillare e definire le altre viste desiderati. Impostare un z-stack in ogni vista e aggiungerli alla lista multivista.
NOTA: Ilsoftware ordina le viste in modo seriale, in modo che il capillare viene acceso unidirezionalmente, mentre l'immagine viene acquisita.
- Definire lo z-stack con il 'First Slice' e le opzioni 'ultima fetta' e impostare il passo z a 1 micron.
- Drift Correzione e di iniziare l'esperimento
- Una volta che l'acquisizione set up è completa, attendere 15-30 minuti prima di iniziare l'esperimento vero e proprio.
NOTA: Il campione derive inizialmente pochi micrometri in x, y, z, ma deve fermarsi in 30 min. Se il campione continua alla deriva per più tempo, utilizzare un altro campione o rimontare. - Passare alla scheda 'Mantenere' e l'opzione 'streaming' definiscono come i dati devono essere salvati, ad esempio, un file per ogni punto di tempo o file separato per ciascuna vista e il canale. Torna alla scheda 'acquisizione', premere 'Inizia Experiment' e definire il nome del file, posizione in cui deve essere salvato e il formato del file (uso .czi).
- Osservare l'acquisizione del primo punto di tempo per confermare che tutto funziona alla perfezione. Immediatamente procedere con la registrazionee fusione del primo punto temporale come descritto ai punti 6 e 7, per confermare che sarà possibile elaborare l'intera serie di dati.
- Una volta che l'acquisizione set up è completa, attendere 15-30 minuti prima di iniziare l'esperimento vero e proprio.
6. Registrazione Multiview
- Multiview ricostruzione Application
- Alla fine della sessione di imaging, trasferire i dati dal computer memorizzazione dei dati al microscopio a un computer di elaborazione dati. Utilizzare il 'MultiView ReconstructionApplication '20,21,31 implementato in Fiji 32 per l'elaborazione dei dati (Figura 1B).
- definire Dataset
- Aggiornare Figi: Fiji> Aiuto> Aggiorna ImageJ e Fiji> Aiuto> Aggiorna Fiji. Utilizzare il ImageJ principale e siti di aggiornamento Fiji.
- Trasferire l'intero insieme di dati in una cartella. I risultati e le file intermedi del trattamento saranno salvati in questa cartella. Avviare il MultiView ricostruzione di applicazione: Fiji> Plugin> Multiview Ricostruzione> Multiview Ricostruzione applicazione.
- Seleziona 'definire un nuovo set di dati'. Come tipo di set di dati selezionare l'opzione meu "Zeiss Lightsheet Z.1 set di dati (LOCI Bioformats)" e creare un nome per il file XML. Quindi selezionare il primo file .czi del set di dati (ad esempio file senza indice). Esso contiene i dati di immagine, nonché i metadati della registrazione.
NOTA: Una volta che il programma si apre il primo file .czi, i metadati vengono caricati nel programma. - Verificare che il numero degli angoli, canali, illuminazioni e osservare la dimensione voxel dai metadati. Premendo OK, osservare tre finestre separate aperte (Figura 1C): una finestra di log, che mostra lo stato di avanzamento del processo e dei suoi risultati, il 'ViewSetup Explorer' e una console, che mostra i messaggi di errore di Fiji.
NOTA: Il 'ViewSetup Explorer' è un interfaccia user-friendly che mostra ogni vista, canale e illuminazione e consente la selezione di i file di interesse. Inoltre, il 'ViewSetup Explorer' permette di sterzatura di tutte le fasi di lavorazione. - Selezionare i file che devono essere elaborati e premere il tasto destro del mouse l'esploratore. Osservare una finestra aperta con diverse fasi di lavorazione (Figura 1C).
- Al momento di definire il set di dati, osserviamo che viene creato un file XML nella cartella con i dati.
NOTA: Questo file contiene i metadati che sono stati confermata prima. - Nell'angolo in alto a destra osservare 'Info' due bottoni e 'salva'. Premendo 'info' mostra una sintesi del contenuto del file XML. Premendo 'salvare' salverà i risultati di elaborazione.
NOTA: Durante l'elaborazione del 'MultiView ricostruzione Application' le varie fasi di lavorazione devono essere salvati nella .xml prima di chiudere Fiji.
OAD / 53966 / 53966fig1.jpg "/>
Figura 1: Multiview la ricostruzione del flusso di lavoro e punto di interesse di rilevamento (A) allineamento foglio di luce sulla base di imaging tallone fluorescente.. Il sistema è allineato (centro), quando l'immagine tallone ha una forma a clessidra simmetrici proiezioni XZ e YZ. Gli esempi di foglio leggero disallineato in entrambe le direzioni sono mostrati a sinistra ea destra. Le proiezioni di massima intensità di un cordone 500 nm in xz, sono mostrati assi YZ e xy. Si noti che il rapporto di intensità del picco centrale del disco di Airy (in xy) ai lobi laterali è maggiore, quando il lightsheet è allineata correttamente rispetto alla situazione disallineato. Le immagini sono state scattate con 20X / 1.0 obiettivo W a 0,7 zoom. barra della scala rappresenta il 5 micron. (B) Il set di dati è definito e poi salvato in formato HDF5. Le perle sono segmentati e quindi registrati. Per una serie di tempo ogni punto di tempo viene registrato su un punto di tempo di riferimento. I dati sono infine fusi in ununico volume isotropo. (C, in alto) Il ViewSetup Explorer mostra i diversi punti di tempo, gli angoli, i canali e le parti di illuminazione del set di dati. (C, a sinistra in basso) La finestra BigDataViewer mostra la vista che viene selezionato nella ViewSetup Explorer. (C, al centro a destra) Fare clic destro nella ViewSetup Explorer apre le opzioni di elaborazione. (C, nell'angolo in basso a destra) Lo stato di avanzamento ei risultati del trattamento vengono visualizzati nel file di registro. (D ed E) L'obiettivo della rilevazione è di segmentare più punti di interesse (grani) con il minor rilevamento nel campione come possibile qui indicato come schermate dalla segmentazione tallone interattivo. (D ed E, in alto a sinistra) La segmentazione è definito da due parametri, i valori di differenza-di-gaussiana per Sigma 1 e la soglia. (D) Un esempio di un rilevamento di successo con una vista ingrandita di un cordone rilevato correttamente. (E) Segmentazione con troppi falsi positivi e più rilevamenti di un unico tallone. Barra della scala in (C) rappresenta il 50 micron. Clicca qui per vedere una versione più grande di questa figura.
{kind=link}
- Salvare di nuovo dataset in formato HDF5
- Per salvare nuovamente l'intero set di dati, selezionare tutti i file con Ctrl / Mela + A e fare clic destro. Quindi selezionare set di dati Salvare nuovamente e come HDF5.
- Apparirà una finestra che mostra un messaggio di avviso che tutte le viste del set di dati corrente saranno salvare di nuovo. Premere Sì.
NOTA: Il programma continuerà a salvare nuovamente i file .czi a HDF5 aprendo ogni file e il successivo salvataggio dei diversi livelli di risoluzione del formato HDF5. Si confermerà con 'fatto' al termine. resaving usually prende un paio di minuti per timepoint (vedi tabella 1).
NOTA: Dal momento che i file nel formato HDF5 possono essere caricate molto veloce, è ora possibile visualizzare il set di dati non registrati dal 'clic destro' nella esploratore e commutando 'esposizione a BigDataViewer (on / off)'. Una finestra BigDataViewer apparirà con la visualizzazione selezionata (Figura 1C). Le funzioni principali del BigDataViewer 33 sono illustrate nella tabella 2 e http://fiji.sc/BigDataViewer.
- Rilevare i punti di interesse
- Selezionare tutti i punti di tempo con Ctrl / Mela + A e fare clic destro selezionare rilevare i punti di interesse.
- Selezionare Difference-di-gaussiano 34 per il tipo di rilevamento di punti di interesse. Dal momento che la registrazione tallone-based è qui usato, tipo "perle" in campo per i punti di interesse etichetta. Attiva 'Downsampling immagini prima di segmentazione'.
- Nella finestra successiva, osservare il detImpostazioni ezione. Per 'subpixel localizzazione' uso 'in forma quadratica 3 dimensioni' 34 e per determinare i valori differenza-di-gaussiana e raggio per l'uso tallone segmentazione 'interattivo' nella specifica punto i'nterest '.
- Per 'Downsampling XY' uso 'Match Z Risoluzione (meno downsampling)' e per 'Downsampling Z' uso 1 ×. La dimensione z passo è più grande della dimensione dei pixel xy, quindi semplicemente verso il basso di campionamento xy per corrispondere alla risoluzione z è sufficiente. Seleziona 'calcolare sulla CPU (JAVA)'. Premere 'OK'.
- In una finestra pop-up, selezionare una vista per testare i parametri dal menu a discesa. Una volta caricato vista, regolare la luminosità e il contrasto della finestra con le Figi> Immagine> Regola> Luminosità / Contrasto o Ctrl + Shift + C. Selezionare la casella 'look per maxima (verde)' per la rilevazione tallone.
- Osservare la segmentazione in anelli come verdi 'viewSetup' around le rilevazioni quando alla ricerca di massimi e minimi rosso per. La segmentazione è definito da due parametri, i valori di differenza-di-gaussiana per Sigma 1 e la soglia (Figura 1D). Regolarle alla segmentare il maggior numero di perle intorno al campione e il minor numero di falsi positivi rilevazioni all'interno del campione il più possibile (Figura 1D). Rilevare ogni perla sola volta e non più volte (Figura 1E). Dopo aver determinato i parametri ottimali, premere 'fatto'.
NOTA: Il rilevamento inizia caricando ciascun vista del punto di tempo e segmentare le perline. Nel file di registro, il programma emette il numero di perline è rilevato per view. Il riconoscimento deve essere fatto in pochi secondi (vedi Tabella 1). - Premere Salva quando la quantità di rilevazioni è appropriato (600 a diverse migliaia per view).
Nota: Una cartella verrà creata nella directory dei dati, che conterrà il information sulle coordinate dei branelli rilevati.
- Registrati Usando Punti di interesse
- Selezionare tutti i punti di tempo con Ctrl / Mela + una, fare clic destro e selezionare 'Registra utilizzando Punti di interesse'.
- Utilizzare '3d hashing geometrica veloce (rotazione invariante)' per la rilevazione tallone come 'algoritmo di registrazione'.
NOTA: Questo algoritmo assume alcuna conoscenza circa l'orientamento e posizionamento dei diversi punti di vista rispetto all'altro. - Per la registrazione delle viste incastrarsi tra di loro, selezionare 'registrare timepoints individualmente' come 'tipo di registrazione'. Per "punti di interesse" nel canale selezionato, l'etichetta precedentemente specificato per i punti di interesse dovrebbe essere visibile (cioè "perle").
- Nella finestra successiva, utilizzare i valori impostati pre per la registrazione. Fissare la prima vista selezionando 'piastrelle Fix: Fix prima piastrella e uso Non mappare indietro' (utilizzare questo se tilES non sono fissi) nella sezione 'Mappa indietro piastrelle'.
- Utilizzare un 'modello di trasformazione affine' con 'regolarizzazione'.
NOTA: L'errore consentito per RANSAC sarà 5px e 'importanza per una partita descrittore' sarà 10. Per 'regolarizzazione' utilizzare un 'modello rigido' con un lambda di 0,10, il che significa che la trasformazione è del 10% rigida e 90 % affine 35. Premere OK per avviare la registrazione.
NOTA: Come visualizzato nella finestra di log, prima ogni visualizzazione è abbinato con tutti gli altri punti di vista. Poi il consenso campione casuale (RANSAC) 36 verifica le corrispondenze ed esclude i falsi positivi. Per una registrazione robusta, il valore RANSAC deve essere superiore al 90%. Quando si trovano un numero sufficiente di candidati veri corrispondenti tra i due punti di vista, un modello di trasformazione è calcolato tra ogni partita con lo spostamento medio in pixel. Poi l'ottimizzazione globale iterativa viene effettuata e tutte le viste sono registrati sul panorama fisso. Con la registrazione di successo, un modello di trasformazione viene calcolato e visualizzato con il suo ridimensionamento e lo spostamento in pixel. L'errore medio dovrebbe essere in modo ottimale sotto di 1 px e la scala della trasformazione vicino a 1. La registrazione viene eseguita in pochi secondi (vedi tabella 1). - Verificare che non vi è alcun passaggio tra diverse viste come osservato su strutture sottili all'interno del campione, ad esempio membrane cellulari. Quindi salvare la trasformazione per ogni vista nel file .xml.
NOTA: Ora i punti di vista legale si sovrappongono l'un l'altro nel BigDataViewer (Figura 2A) e le immagini di perline vanno sovrapponendo così (Figura 2B). - Rimuovere le trasformazioni selezionando i punti temporali tasto destro del mouse su di essi e quindi selezionare Rimuovi Trasformazioni> Ultime Novità Trasformazione /.
re 2 "src =" / files / ftp_upload / 53966 / 53966fig2.jpg "/>
Figura 2:. I risultati della ricostruzione MultiView (A) sovrapposto viste iscritti, ognuno in un colore diverso per dimostrare la sovrapposizione tra di loro. (B) Visualizzazione ingrandita che mostra la sovrapposizione delle PSF di perline immaginati dai diversi punti di vista. (C) Primo piano di una perlina dopo la fusione, il PSF è una media dei diversi punti di vista. (D) PSF dello stesso tallone come in (C) dopo multivisione deconvoluzione mostrando che la PSF collassa in un unico punto. (E) sezione xy e (F) Sezione yz di un'unica vista di una vescicola ottica, in cui le membrane sono etichettati con GFP che mostra la degradazione del segnale in z profondo nel tessuto. (G) sezione xy e (H) sezione yz della stessa vista dopo la fusione ponderata-media di 4 viste circa 20 gradi l'una dall'altra, con un po 'più Degrarisoluzione xy ded generale, ma una maggiore z-risoluzione. (I) sezione xy e (J) Sezione yz degli stessi dati dopo multiview deconvoluzione, mostrando un significativo aumento della risoluzione e contrasto del segnale sia in xy e z. Immagini in (EJ) sono singole fette ottici. Barra della scala rappresenta 50 micron (A, B, EJ) e 10 micron (C, D). Clicca qui per vedere una versione più grande di questa figura.
{kind=link}
- Time-lapse registrazione
- Selezionare l'intero lasso di tempo, fare clic destro e selezionare 'Registra usando interesse nelle vicinanze' per stabilizzare il time-lapse nel corso del tempo.
- Nella 'finestra di parametri di base Registrazione selezionare Partita contro un certo punto il tempo di riferimento (senza ottimizzazione globale) per il tipo di registrazione'. Ke ep L e altre impostazioni lo stesso come nella registrazione dei singoli punti di tempo.
- Nel prossimofinestra, selezionare il punto di tempo da utilizzare come riferimento, tipicamente un punto temporale in mezzo al time-lapse. Barrare la casella 'considerare ogni timepoint come unità rigida', dal momento che le singole viste all'interno di ogni punto di tempo sono già registrati su ogni altro.
- Barrare la casella per la 'Mostra statistiche timeseries'. Gli altri parametri di registrazione rimangono come prima di includere la regolarizzazione. Premere OK.
NOTA: Nella finestra di log, la stessa uscita verrà visualizzata come nel singolo punto di registrazione tempo. Se la registrazione per i singoli punti di tempo ha avuto successo e robusto, il RANSAC è ora 99-100% e la media, minima e massima di errore è di solito inferiore a 1 px. Salva questa registrazione prima di procedere.
7. Multiview Fusion
NOTA: Le trasformazioni derivanti dalla procedura di registrazione vengono utilizzati per calcolare una pila isotropo fusa fuori dei molteplici punti di vista. Questa pila has aumento del numero di fette z rispetto ai dati originali, perché la spaziatura z è ora uguale alla dimensione dei pixel originali in xy. La fusione può essere eseguita tramite un content-based multiview fusione 21,31 o bayesiana basata multiview deconvoluzione 31, che sono entrambi implementato nell'applicazione ricostruzione multivista.
- Rettangolo di selezione
NOTA: Fusion è un processo costoso computazionalmente (vedi Tabella 1), riducendo così la quantità di dati definendo un riquadro aumenta notevolmente la velocità di elaborazione.- Selezionare tutti i punti di tempo fare clic destro e selezionare Definisci 'rettangolo di selezione'. Utilizzare 'Definire con BigDataViewer' e scegliere un nome per il riquadro di delimitazione.
- Spostare il cursore per 'min' e 'Max' in ogni asse per determinare la regione di interesse e premere 'OK'. Verranno visualizzati i parametri del riquadro di delimitazione.
NOTA: Il riquadro conterrà tutto all'internola scatola verde che è ricoperto di uno strato di colore magenta trasparente.
- Basato sul contenuto Multiview Fusion
NOTA: multivista fusione 21-based contenuto prende le differenze di qualità dell'immagine sopra la pila (cioè degradazione del segnale in z) in considerazione e applica pesi elevati per una migliore qualità dell'immagine anziché utilizzare una semplice media.- Selezionare il punto di tempo (s) che dovrebbero essere fuse nel 'ViewSetup Explorer', fare clic destro e selezionare 'Immagine Fusion / Deconvoluzione'.
- Nella finestra di fusione di immagini, selezionare 'Media ponderata fusion' dal menu a tendina e selezionare 'Usa predefiniti rettangolo di selezione per rettangolo di selezione'. Per l'uscita dell'immagine fusa selezionare 'Aggiunge al progetto XML corrente', che scrive i nuovi file HDF5 ai file già esistenti HDF5 e permette di utilizzare i punti di vista non fuse registrati e l'immagine fusi insieme. Premere 'OK'.
- Poi nel 'pre-definire rettangolo di selezione & #39; finestra pop-up selezionare il nome del riquadro definito in precedenza e premere 'OK'.
- Osservare i parametri del riquadro nella finestra successiva. Per la fusione rapida, applicare un campionamento in giù sul set di dati fuso.
NOTA: Se la pila fusa è al di sopra di una certa dimensione, il programma consiglia di utilizzare un più efficiente della memoria 'containe'r ImageLib2. Passare dalla 'ArrayImg' al 'PlanarImg (immagini di grandi dimensioni, facile da visualizzare) o CellImg (grande)' contenitore, che consentono l'elaborazione dei dati di grandi dimensioni. In caso contrario, utilizzare le impostazioni predefinite e applicare 'fusione miscelazione e content-based'. Procedere premendo 'OK'. - Osservare le impostazioni HDF5 nella finestra successiva. Utilizzare i parametri predefiniti e avviare il processo di fusione. Assicurarsi che Fiji ha assegnato memoria sufficiente a Modifica> Opzioni> Memoria & thread.
- Multiview Deconvoluzione
NOTA: Multiview deconvoluzione 31 è anotil suo tipo di fusione multivista. Qui aggiunta alla fusione, la PSF del sistema di imaging viene presa in considerazione per deconvolve la risoluzione dell'immagine e resa maggiore e contrasto del segnale (confrontati in Figura 2C-J, Figura 5 e film 3).- Selezionare il punto di tempo (s) da deconvolved, fare clic destro e premere 'Immagine Fusion / Deconvoluzione'.
- Seleziona 'Multiview Deconvoluzione' e uso 'il rettangolo di selezione pre-definiti e aggiungere al progetto XML corrente'. Selezionare la casella di delimitazione e continuare.
NOTA: I parametri predefiniti per la deconvoluzione sono un buon punto di partenza. Per il primo utilizzo di prova '20 iterazioni 'e valutare l'impatto della deconvoluzione utilizzando la' modalità debug '. Infine impostare il calcolo a in 512 x 512 x 512 blocchi. - Nella finestra successiva, utilizzare le impostazioni predefinite. Impostare la 'modalità di debug' per visualizzare i risultati di ogni '5 iterazioni '.
NOTA: L'uscita del deconvoluzione sono dati a 32 bit, ma il BigDataViewer attualmente supporta solo dati a 16-bit. Per aggiungere l'output del deconvoluzione al dataset HDF5 esistente deve essere convertito a 16-bit. - Per la conversione, eseguire 'Usa min / max della prima immagine (potrebbe saturare intensità nel corso del tempo)'.
NOTA: La deconvoluzione sarà poi iniziare il caricamento delle immagini e li prepara per la deconvoluzione.
- BigDataServer
- Al fine di condividere il grande XML / set di dati HDF5 utilizzano il server http BigDataServer 33. Un'introduzione a come impostare e connettersi a tale server può essere trovato alla http://fiji.sc/BigDataServer.
- Per connettersi a una BigDataServer esistente aperto Fiji> Plugin> BigDataViewer> BrowseBigDataServer.
- Inserire l'URL tra cui il porto nella finestra.
NOTA: I film descritti in questa pubblicazione sono accessibili tramite questo annunciovestito: http://opticcup.mpi-cbg.de:8085 - Osservare una finestra che permette di selezionare solo i film. Fare doppio clic per aprire la finestra BigDataViewer e visualizzare i dati come descritto in precedenza.
Supplementare: Lista di controllo di materiale necessario prima di iniziare
- embrioni di zebrafish / larve che esprimono proteine fluorescenti (Mantenere gli embrioni nel medio E3 zebrafish senza blu di metilene. Per le fasi di età superiore a 24 ore contrastare la pigmentazione con l'aggiunta di PTU ad una concentrazione finale di 0,2 mm.)
- stereoscope fluorescente
- capillari (20 volumi microlitri, con un segno nero) e gli stantuffi appropriate (Non riutilizzare i capillari. Gli stantuffi, invece, possono essere riutilizzati per diversi esperimenti.)
- 1,5 ml tubi di plastica
- pinzette taglienti
- vetro (fuoco lucido) o pipette di plastica (plastica può essere utilizzato per 24 ore e di embrioni più vecchi)
- diametro vetro o piatti di plastica 60 mm (plastica può essereutilizzato per 24 ore e di embrioni più vecchi)
- due bicchieri 100 ml
- 50 ml Luer-Lock siringa con tubo di prolunga 150 centimetri per infusione (Il tubo e la siringa deve essere mantenuto completamente asciutto tra esperimenti per evitare la contaminazione da microrganismi.)
- plastilina
- basso punto di fusione (LMP) agarosio
- E3 medio (5 mM NaCl, 0,17 mM KCl, 0,33 mM CaCl 2, 0,33 mM MgSO 4)
- MS-222 (Tricaine)
- feniltiourea (PTU)
- microsfere fluorescenti (qui indicati come perline)
- doppia H 2 O distillata (DDH 2 O)
Risultati
LSFM è un metodo ideale per l'imaging processi di sviluppo attraverso le scale. Diverse applicazioni sono compilate qui che mostra l'imaging sia a breve che a lungo termine delle strutture intracellulari, così come le cellule ei tessuti interi. Questi esempi dimostrano anche che LSFM è uno strumento utile a vari stadi di sviluppo dell'occhio dalla formazione tazza ottico al neurogenesi nella retina. Film 1 serve come illustrazione dell'approccio generale LSFM, prima che mostra una vista ingrandita di un embrione intatto nel incorporamento cilindro agarosio in campo chiaro e successivamente mostra una vista dettagliata della retina nel canale di fluorescenza.

Film 1:. LSFM della retina zebrafish Per illustrare l'approccio LSFM, questo film mostra prima nel campo chiaro che l'embrione è ripreso intatto all'interno di ungarose cilindro prima di passare a fluorescenza. Successivamente si è dimostrato che il grande campo di vista permette l'osservazione di tutta la retina. Successivamente, il film mostra una piccola regione ingrandita della retina per evidenziare la buona risoluzione subcellulare. Un Ath5: gap-GFP 37 transgenico zebrafish è stato utilizzato per l'imaging. Questo transgene etichette diverse neuroni della retina (principalmente cellule gangliari e precursori dei fotorecettori). La parte di fluorescenza del film è stato catturato come una singola registrazione con illuminazione a doppia faccia con l'obiettivo 40X / 1.0 W con 5 min intervalli di vista. La proiezione massima intensità di un grosso volume di 30 micron viene mostrato. Clicca qui per scaricare questo file.
Movie 2 dimostra, come gli eventi intracellulari molto veloci possono essere catturati con alta risoluzione; in questo caso la crescita dei microtubuli ala loro più finisce nelle cellule progenitrici neuronali della retina. Le informazioni contenute nel film consente per il monitoraggio e la quantificazione dei microtubuli, più fine la crescita.

Movie 2:. Dinamica dei microtubuli in una singola cella Questo film consente di catturare in crescita più punte di microtubuli etichettati dal più la punta proteina marcatore EB3-GFP 38. La proteina è espressa in una singola cellula progenitrice retinica. I microtubuli sono in crescita prevalentemente nella direzione da apicale al basale (dall'alto in basso). La velocità media delle comete Eb3 stata misurata come 0.28 ± 0.05 micron / sec. Il punto luminoso nella parte apicale della cellula espositiva attività nucleazione dei microtubuli alto è il centrosoma. Il tipo di embrione selvaggio è stato iniettato con HSP70: EB3-GFP plasmide DNA. Il film è stato acquisito 4 ore dopo shock termico (15 minuti a 37 ° C) a circa 28 hrpost fertilizzione (HPF) come una singola registrazione con illuminazione singola faccia vista utilizzando gli intervalli di tempo oggettivo e 1 sec 63X / 1.0 W. La singola cellula è stata ritagliata da un campo visivo che copre la maggior parte della retina. L'intensità massima è indicata la proiezione di due fette. Cliccate qui per scaricare questo file.
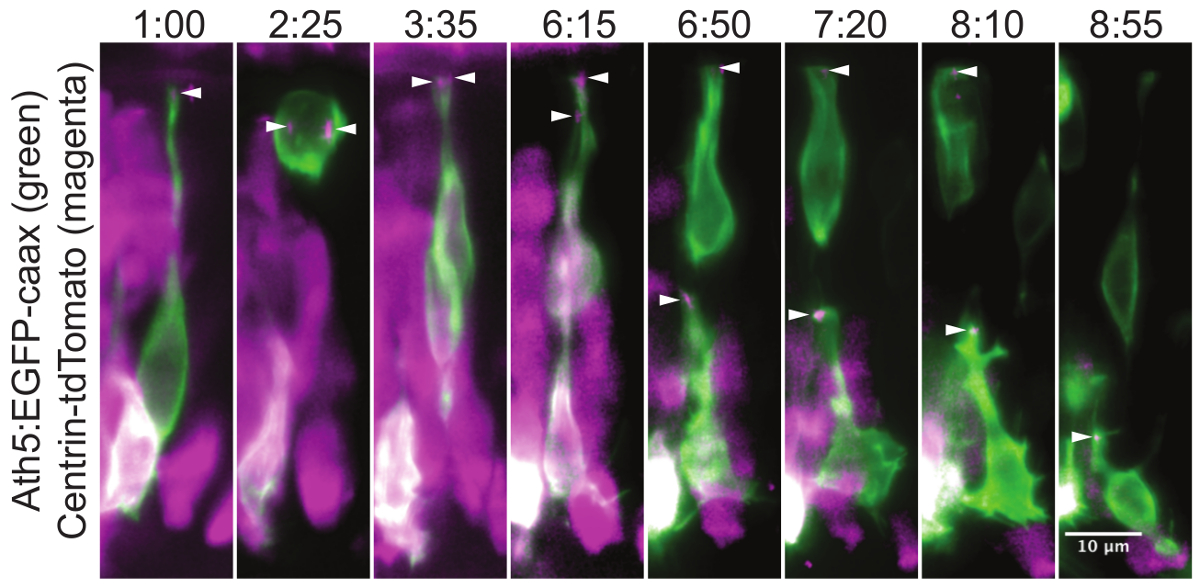
La figura 3 mostra, come le strutture intracellulari possono essere seguite per molte ore. Qui, il centrosoma entro translocating cellule gangliari della retina (RGCs) viene catturato.

Figura 3:. Localizzazione Centrosome durante la traslocazione cellule gangliari retiniche Questo montaggio di un esperimento time-lapse mostra la posizione del centrosoma durante la maturazione della retina cellule gangliari (RGC).Nei progenitori neuronali centrosoma è localizzata nella punta estrema del processo apicale (01:00). Durante la divisione cellulare, i due centrosomi servono come pali per il fuso mitotico (2,25). Questa divisione si traduce in una cellula figlia che si differenzia in un RGC e una seconda cellula figlia che diventa un precursore dei fotorecettori. Dopo divisione, il corpo cellulare del RGC trasloca nel lato basale della retina, mentre il processo apicale rimane attaccato al lato apicale. Una volta che la RGC raggiunge il lato basale, il suo processo apicale stacca e il centrosoma viaggia con esso (6,15). Il centrosoma può essere seguita mentre gradualmente ritraendo insieme con il processo apicale (6:50, 7:20, 8:10). Nel l'ultimo fotogramma (08:55) il cellule gangliari sta crescendo un assone dal suo lato basale, mentre il centrosoma è ancora localizzato apicale. L'espressione del mosaico è stata realizzata per iniezione plasmide DNA in tipo selvaggio embrione in uno stadio delle cellule. Le cellule vengono visualizzati dal Ath5: GFP-CAAX (verde) construct, che etichette RGCs e altri neuroni. I centrosomi (frecce) sono etichettati da Centrin-tdTomato 29 espressione (magenta). Il lato apicale della retina è nella parte superiore dell'immagine e il lato basale in basso. Viene mostrata la proiezione massima intensità di un grosso volume di 30 micron. Le immagini sono state ritagliate da un film che copre l'intera retina. Il film inizia a circa 34 ore dopo la fecondazione (HPF). A z pila è stata acquisita ogni 5 minuti con l'obiettivo 40X / 1.0 W. Il tempo è mostrato in hh: mm. Barra della scala rappresenta 10 micron. Clicca qui per vedere una versione più grande di questa figura.
{kind=link}
In figura 4 è mostrato, come singola comportamento cellulare può essere estratto dai dati catturando tutto il tessuto come nel film 1. Traslocazione di RGC può essere facilmente tracciati e il suo processo apicale e basalees seguito.

Figura 4:. Translocation di una singola cellula gangliare retinica La traslocazione RGC da apicale a lato basale della retina avviene dopo la mitosi terminale come descritto in figura 3 L'RGC è etichettato con espressione del Ath5:. Gap-GFP 37 transgene. Viene mostrata la proiezione massima intensità di un grosso volume di 30 micron. Le immagini sono state ritagliate da un film che copre l'intera retina. Il film inizia a circa 34 HPF. A z pila è stata acquisita ogni 5 minuti con l'obiettivo 40X / 1.0 W. Il tempo è mostrato in hh: mm. Barra della scala rappresenta il 5 micron. Clicca qui per vedere una versione più grande di questa figura.
{kind=link}
Figura 5 dimostra la capacità di MultiView LSFM per catturare sc tessutiprocessi morfogenetici ale con risoluzione cellulare sull'esempio di ottica morfogenesi tazza, durante il quale ottica vescicole si trasforma in una coppa ottica. La qualità dell'immagine può essere notevolmente migliorata mediante imaging multiview, quando l'immagine finale è combinata dalle informazioni su 5 viste diverse (in questo caso) in una z stack con risoluzione isotropica. Questa figura illustra il miglioramento della qualità di immagine dopo la fusione medio ponderato e l'ulteriore aumento di contrasto e risoluzione dopo MultiView deconvoluzione. La figura mostra due fette ottici in diversi orientamenti attraverso il set di dati. Inoltre, un montaggio dal set di dati deconvolved mostra la morfogenesi coppa ottica nel corso del tempo. Tutti i punti di tempo sono poi mostrati in Movie 3.

Figura 5: Confronto tra qualità dell'immagine tra visualizzazione singola e due metodi di multiview fusione. (A) Una fetta ottica di visualizzare i dati riportati da singoli vista laterale e (B) vista dorsale. artefatti banda e il degrado del segnale più all'interno del campione sono evidenti. Anche una parte dell'immagine visibile nei dati fuse (CF) non è stato catturato in questa vista particolare. (C) La stessa fetta ottica, ora come MultiView fuso i dati riportati dalla vista laterale e (D) vista dorsale. Si noti che gli artefatti banda sono soppressi e strutture profonde nel campione sono meglio risolti. (E) La stessa fetta ottica ora come MultiView deconvolved dati mostrati alla vista laterale e (F) vista dorsale. Si noti la maggiore contrasto e la risoluzione in modo che le singole membrane cellulari e nuclei possono essere ben distinti. La risoluzione non si deteriora notevolmente più profonda all'interno del campione. Il set di dati è stata acquisita con illuminazione a doppia faccia da 5 viste di circa 20 gradi. Lo Z pile di abil 100 micron con 1,5 micron dimensione del passo sono stati acquisiti ad ogni vista in intervalli di 10 min per 10 ore, con l'obiettivo 20X / 1.0 W. immagini di ingresso per la fusione MultiView e deconvoluzione sono stati giù campionati 2 × per accelerare l'elaborazione delle immagini. 15 iterazioni del deconvoluzione multivista sono stati eseguiti. (G) Il montaggio mostra una parte ritagliata del vista dorsale dai dati deconvolved per evidenziare gli eventi morfogenetici durante lo sviluppo dell'occhio presto dalla vescicola ottica alla fase coppa ottica. I due strati della vescicola ottica, che sono inizialmente simili epiteli colonnare, si differenziano in distinte popolazioni cellulari. Lo strato distale vicino l'epidermide diventa neuroepitelio della retina (RN) e lo strato prossimale vicino al tubo neurale diventa l'epitelio pigmentato retinico (RP). Le cellule del allungato RN e invaginate per formare la coppa ottica (1:40-05:00); Allo stesso tempo le cellule RP appiattiscono. L'ectoderma superficiale è indotto a formare una lente (01:40), whinvaginates ich più tardi (05:00). Il film inizia alle 17 HPF. Il tempo è mostrato in hh: mm. Tutte le membrane cellulari sono etichettati dal β-actina: ras-GFP transgene e tutti i nuclei sono etichettati dal HSP70: H2B-RFP transgene. barra della scala rappresenta 30 micron. FB prosencefalo, lente LE, OP placode olfattiva, RN neuroepitelio retina, RP dell'epitelio pigmentato retinico. Si prega di cliccare qui per vedere una versione più grande di questa figura.
{kind=link}

Film 3: Optic tazza morfogenesi mostrato con un'unica vista e due metodi di fusione MultiView Il filmato time-lapse illustra il processo completo di ottica morfogenesi tazza dalla vescicola ottica alla fase coppa ottica.. Essa mostra una sola fetta ottica dalla vista laterale (in alto) e una sola fetta ottica dalla vista dorsale (in basso). Le cellule del sviluppo vescicola ottica subiscono riarrangiamenti complessi per formare finalmente la coppa ottica emisferica con neuroepitelio retina interna ed esterna dell'epitelio pigmentato retinico. Una lente è formata da ectoderma superficie. Si invagina insieme al neuroepitelio e si siede in tazza ottica. Tutte le membrane cellulari sono etichettati dalla espressione della β-actina: ras-GFP (green) transgene ed i nuclei sono etichettati con HSP70: H2B-RFP (magenta). Il film inizia a circa 17 HPF. Il set di dati è stata acquisita con illuminazione a doppia faccia da 5 vedute circa 20 gradi l'uno dall'altro e az pile di circa 100 micron sono stati acquisiti ogni 10 minuti con l'obiettivo 20X / 1.0 W. Il tempo è mostrato in hh: mm. Barra della scala rappresenta 50 micron. Clicca qui per scaricare questo file.
Discussione
1. passaggi critici e risoluzione di problemi per l'acquisizione dei dati
Le impostazioni tipiche di imaging per un GFP e RFP esprimere campione si trovano nella Tabella 3. Nella configurazione microscopio descritto il foglio leggero è statica, formata da una lente cilindrica. I due obiettivi illuminazione sono lenti aria e l'obiettivo di rilevamento è una lente di acqua-dipping. Zoom 1.0 con 20X / 1.0 o 40X / 1.0 obiettivi dà 230 nm e 115 nm dimensione dei pixel e un campo visivo di 441 x 441 micron o 221 x 221 micron, rispettivamente. Si consiglia di utilizzare lo spessore della lamiera luce di default con il centro a confine rapporto di 1: 2. Per 20X / 1.0 questo spessore corrisponde a 4,5 micron e 40X / 1,0-3,2 micron di centro. Se la velocità di imaging non è la priorità primaria, utilizzare tracce separate nel caso di un campione multicolore per evitare la diafonia di emissione di fluorescenza tra i canali. La massima velocità di acquisizione è limitata a 50 msec per z passo dalvelocità di movimento del z-driver. Se l'obiettivo è quello di raggiungere la velocità massima di imaging in caso di, ad esempio, due piste con illuminazione doppia faccia, il tempo di esposizione devono essere impostati in modo che la somma di tutte le immagini scattate per z passo è inferiore a 50 msec. D'altra parte, se solo una singola immagine viene acquisita per z passo, non è vantaggioso per impostare il tempo di esposizione più breve di 50 msec.
| dimensioni immagine 1920 x 1920 |
| 16-bit |
| Pivot scansione su |
| illuminazione lato doppio con la fusione in linea |
| Obiettivo / 0,2 illuminazione 10X |
| 20X obiettivo rilevamento / 1.0 W Plan-Apochromat |
| Traccia 1: eccitazione 488 nm normalmente dal 2% di 100 mW laser, 550 nm filtro per le emissioni SP |
| Traccia 2: eccitazione 561 nm tipicamente 3% di 75 mW laser, 58filtro per le emissioni LP 5 nm |
| Tempo di esposizione fino a 100 msec |
| Z spessore pila 50-100 micron |
| 1-1,5 micron dimensioni passo z in modalità z unità continua |
| L'incubazione a 28,5 ° C |
Tabella 3: Imaging Impostazioni.
Ispezione del campione dopo l'esperimento
È importante garantire che il campione è ancora sano alla fine dell'esperimento. Come prima lettura, controllare il battito del cuore del campione sotto uno stereoscopio. Con una coppia di pinze taglienti campione può essere estratto dalla agarosio e spostato nella incubatore di sviluppare ulteriormente per verificare se è stato influenzato dal imaging. In alternativa, può essere fissato per colorazione anticorpale.
Montaggio e deriva
È essenziale per mantenere l'osmolarità della camera di soluzione vicino alla osmolarità della agarosio embedding, altrimenti rigonfiamento / restringimento del agarosio e conseguente instabilità del campione si verificherà. Pertanto, utilizzare la stessa soluzione (media E3 senza blu di metilene) per riempire la camera e preparare le 1% bassa temperatura di fusione aliquote punto agarosio. Inoltre, non lasciare l'agarosio nel blocco di riscaldamento C 70 ° per più di 2 ore, in quanto può perdere le sue proprietà gelificanti.
Non incorporare il pesce in agarosio troppo caldo, come questo può portare a risposta heat shock o la morte dell'embrione. In caso di dubbi circa l'effetto di agarosio caldo sugli embrioni, controllare che la coda non si piega e che la frequenza cardiaca non rallenta. In questo caso, utilizzare un embrione diversa per l'esperimento.
Mantenere la lunghezza complessiva della colonna agarosio con il campione breve (circa 2 cm) e montare il zebrafish con la testa orientata verso la punta dello stantuffo. Allo stesso modo, il cilindro agarosio estrusa dal capillary deve essere il più breve possibile. Tali misure garantiranno la stabilità del campione in tutto il film. Allo stesso tempo, la colonna agarosio deve essere sufficientemente lungo in modo che il capillare vetro stesso non raggiunge nel percorso ottico, in quanto ciò comporterebbe maggiore rifrazione e riflessione.
La deriva iniziale del campione è causato dalle variazioni di volume del cilindro agarosio stesso. Scorrimento dello stantuffo non è la ragione per questo. Pertanto, non aiuta a risolvere il pistone con la plastilina o smalto. L'embrione potrebbe cambiare la sua posizione durante il film per la sua crescita naturale troppo. Pertanto, si consiglia di centrare la regione di interesse nel centro del campo visivo e mantenere un certo spazio ai bordi per accogliere questi movimenti.
ridotta quantità di mezzo di inclusione nel percorso della luce
Orientare il campione correttamente aiuta a ottenere la migliore qualità d'immagine possibile 15. Generally, di eccitazione e di emissione di luce deve viaggiare attraverso il meno tessuto e mezzo di montaggio possibile. La soluzione ottimale è il montaggio agarosio-libera. Questo è stato ottenuto per esempio in una configurazione per l'imaging Arabidopsis radice laterale 14, in cui la radice principale era montato in phytagel e le radici laterali sono stati successivamente lasciato crescere su colonna di gel completamente. Montaggio senza-agarosio è stato sviluppato anche per l'imaging della embriogenesi completa di Tribolium coleottero in due giorni 12. miglioramento della qualità delle immagini non è stata una motivazione primaria in quel caso. embrioni Tribolium semplicemente non sopravvivono all'interno del agarosio abbastanza a lungo. Un montaggio senza mezzi assolutamente incorporamento non è stato raggiunto per l'imaging a lungo termine in zebrafish. Ancora, possiamo sfruttare il fatto che quando solidifica agarosio, la maggior parte embrioni sono posizionati diagonalmente nel capillare con un occhio situato in profondità nel agarosio e il secondo occhio essendo vicino alla superficie della colonna incorporamento. Tegli occhio più vicino alla superficie fornisce immagini di qualità superiore e, pertanto, deve essere ripreso preferenzialmente.
la concentrazione agarosio e l'imaging a lungo termine
La concentrazione di agarosio per il montaggio è un compromesso tra stabilità del campione e la possibilità di ospitare crescita dell'embrione e diffusione di ossigeno ad esso. Non vi è alcun ulteriore guadagno nella stabilità del campione quando si utilizzano concentrazioni agarosio superiori all'1%. Come punto di partenza per ottimizzare gli esperimenti si consiglia 0,6% agarosio, che è anche adatto per gli embrioni di età inferiore ai 24 HPF che sono troppo delicati per essere montato in 1% di agarosio. Per anestetizzare gli embrioni più anziani e le larve, la concentrazione di MS-222 può essere portato a 200 mg / ml, senza effetti collaterali 13.
Nel caso in cui gli embrioni in via di sviluppo vengono esposte per più di ± 12 ore, il montaggio agarosio non è consigliabile, perché limita la crescita dell'embrione e causes deformazione coda. Questo problema è stato risolto per zebrafish montando gli embrioni in tubi di polimero FEP con indice di rifrazione simile all'acqua 13,39. Embrioni di topo, invece, possono essere immobilizzati in bombole agarosio cave 40 o in fori di una barra acrilica attaccato ad una siringa 41. FEP montaggio tubo non è raccomandato come metodo predefinito se, perché la parete del tubo rifrange luce poco più di agarosio.
allineamento foglio di Luce
Per una buona qualità delle immagini è fondamentale per eseguire l'allineamento automatico foglio di luce prima di ogni esperimento. Soprattutto se le impostazioni di zoom sono state modificate, gli obiettivi sono stati portati via, o di un liquido diverso è stato utilizzato nella camera.
Illuminazione
La scansione perno del foglio leggero deve sempre essere attivato. Per grandi campioni di scattering, è necessario applicare l'illuminazione doppia faccia con f lineausione per ottenere un'illuminazione uniforme in tutto il campo di vista. Doppia illuminazione lati diminuisce anche un problema specifico di imaging dell'occhio, che è la rifrazione del foglio leggero arrivo dalla lente dell'embrione. I più piccoli, i campioni meno di scattering possono essere esposte in modo efficiente utilizzando l'illuminazione lato singolo, che riduce il tempo di imaging della metà e può portare a leggermente migliore qualità delle immagini rispetto a illuminazione a doppia faccia. Questo perché i percorsi di luce per i due bracci di illuminazione sono sempre diversi e quella più efficiente può essere scelto. Inoltre, i due fogli leggeri provenienti da ciascun lato sono mai perfettamente in un piano, che causa lieve sfocatura dopo la fusione. Per gli eventi intracellulari molto veloce, come le crescenti microtubuli (Film 2), l'illuminazione a doppia faccia non è appropriato, dal momento che le immagini con illuminazione da sinistra e destra sono acquisite in sequenza, che potrebbe tradursi in motion blur.
photobleachinge fototossicità
Meno photobleaching fluoroforo viene spesso citato come uno dei principali vantaggi del LSFM. Noi sosteniamo che l'obiettivo dovrebbe essere photobleaching a tutti. Se c'è photobleaching evidente nel esperimento di imaging in tempo reale, il campione è probabilmente già fuori della sua gamma fisiologica di esposizione laser tollerato. Quando l'imaging gli embrioni di zebrafish al microscopio disco rotante, nella nostra esperienza, alta fototossicità può stallo sviluppo dell'embrione prima ancora che gli sbiancanti segnale fluorescente notevolmente. Pertanto, le impostazioni di imaging nella LSFM devono essere regolati in modo che si osserva poco o nessun photobleaching. Anche se LSFM è dolce al campione, è prudente usare solo la potenza del laser e tempo di esposizione come necessario per ottenere un rapporto segnale-rumore sufficiente per la successiva analisi dei dati.
Z-stack, intervalli di tempo e dimensioni dei dati
I file generati da LSFM sono generalmente di grandi dimensioni; a volte nella gamma terabyte. Spesso è necessario fare un compromesso tra qualità dell'immagine e dimensione dei dati. Questo è particolarmente vero per z spaziatura delle pile e gli intervalli delle acquisizioni time-lapse. Per definire gli intervalli z, il pulsante ottimale nella scheda utensile Z-stack dovrebbe idealmente essere utilizzato, soprattutto se il set di dati verrà deconvolved seguito. Si calcola la distanza per raggiungere il 50% di sovrapposizione tra le fette ottici vicini. Ancora, gli intervalli di z po 'più grandi di solito sono accettabili. Essi riducono il tempo necessario per acquisire la z pila nonché le dimensioni del file finale. Il campionamento tempo ottimale dipende dal processo di interesse. Per gli occhi di sviluppo globale 5-10 minuti a intervalli di solito sono accettabili. Se alcune strutture devono essere monitorati automaticamente, i punti di tempo successivi devono essere sufficientemente simili.
perline fluorescenti
perline fluorescenti servono principalmente come marcatori fiduciali per la registrazione dei diversi punti di vistadi un set di dati multivista uno sull'altro. vortice sempre le soluzioni di perline accuratamente prima dell'uso. Non riscaldare le perline come questo può portare alla perdita del colorante fluorescente. La concentrazione ottimale tallone per la registrazione MultiView deve essere determinato sperimentalmente. Il plugin funziona meglio descritto con circa 1.000 perline rilevate nel corso di ogni punto di vista. Più grandi (500 nm o 1000 nm) perline vengono rilevati più robusto di piccole perle (inferiore a 500 nm). Questo perché perline più grandi sono più luminoso e sono più facili da settore senza false rilevazioni positive di strutture nel campione. Lo svantaggio delle perle più grandi è che sono molto importanti nell'immagine fuso e deconvolved finale. Per ogni nuovo marcatore fluorescente, il formato del branello appropriato e fluorescenza devono essere ottimizzati. Per fare un esempio di campione dalla Figura 5 e Movie 3, 100 nm perline di emissione verdi hanno dato troppi rilevazioni di falsi positivi nel canale membrana GFP, ma 1000 nm perline emissione rosso sono stati robustamente rilevati nel canale H2B-RFP con pochissime rilevazioni positive all'interno del campione. Se il rilevamento tallone fallisce nel canale con il marcatore fluorescente, un canale separato contenente solo perline può essere acquisita, ma questo non è molto pratico. Le perle di dimensioni sub-risoluzione danno una lettura diretta della funzione di diffusione di punto (PSF) del microscopio, che può essere utilizzato per deconvoluzione (Figura 2C-D). Se la registrazione e la fusione funziona meglio con perline più grandi (ad esempio, 1000 nm), un immagine separata del PSF può essere acquistato con sub-risoluzione, ad esempio, 100 perle nm. Utilizzando perline multicolore è utile durante la registrazione di acquisizione multicanale e di verificare che i canali di sovrapposizione perfetta.
L'aggiunta di perline fluorescenti non è necessaria quando l'imaging da una singola vista, senza successiva registrazione MultiView e fusion. Tuttavia, anche in questi casi perline possono essere utili durante il initial adeguamento foglio leggero per controllare la qualità del foglio leggero ed in generale per rivelare aberrazioni ottiche. Tali aberrazioni ottiche possono provenire da varie fonti come obiettivi danneggiati o sporchi, vetri sporchi della camera o disomogeneità nella agarosio. Beads possono essere utilizzati anche per la correzione della deriva dalla registrazione MultiView Fiji plug-20.
Multiview
Ai fini ricostruzione Multi View, è meglio acquisire un numero dispari di vista 3, 5 e così via, che non si oppongono a vicenda. Questo migliora deconvoluzione dal momento che le PSF vengono esposte da diverse direzioni. E 'anche importante confermare all'inizio dell'acquisizione lasso di tempo che vi sia sufficiente sovrapposizione tra i punti di vista. Questo è fatto meglio empiricamente, cioè, subito conferma che il punto di vista del primo punto di volta possono essere registrati con successo. Quando l'obiettivo dell'acquisizione multiview è quello di aumentare la risoluzionedi un'immagine di un grande esemplare di scattering, non è consigliabile per l'immagine dell'intero campione in ciascuna vista, ma per fermare intorno al centro del campione, in cui il segnale si deteriora. L'acquisizione di bassa qualità dalla seconda metà del campione non aggiungere informazioni utili alla ricostruzione multivista.
2. passaggi critici e risoluzione di problemi per l'elaborazione dei dati
Attualmente, esistono diverse possibilità per l'elaborazione dei dati MultiView da un microscopio foglio di luce che sono ben documentati e relativamente facile da adottare. Usiamo l'applicazione ricostruzione MultiView, che è un software open source implementato in Fiji 32 (Stephan Preibisch inedito, di collegamento 1a e Link1b nel l'elenco dei materiali). Questo plugin è una riprogettazione importante della SPIM precedente plug-in di registrazione 20, recensioneda Schmied et al. 42, integrando la BigDataViewer e il suo formato XML e HDF5 33 con il flusso di lavoro di registrazione SPIM (Figura 1B, Link 2 , Link 3 ). Questa applicazione può essere anche adattato per cluster di calcolo ad alte prestazioni, che accelera in modo significativo il trattamento 43. Questa domanda di registrazione MultiView è attivamente sviluppato ulteriormente e continua a migliorare. In caso di problemi o caratteristiche richieste per il software descritto, si prega di presentare le questioni sulle rispettive pagine GitHub ( Link 4 per Multiview la ricostruzione e di collegamento 5 per BigDataViewer).
La seconda opzione è quella di utilizzare il software commerciale disponibile insieme con il microscopio. Questa soluzione funziona bene e dà lavoro lo stesso principio di utilizzare perline fluorescenti per registrare i diversi punti di vista. Tuttavia, manca la possibilità di visualizzare l'intero set di dati veloce come con il BigDataViewer. Inoltre il software non può essere adattato al cluster e inoltre i blocchi di elaborazione del microscopio per altri utenti, a meno licenza aggiuntiva per il software viene acquistato.
La terza opzione, che è anche un software open source, è stato recentemente pubblicato dal laboratorio Keller 44 e fornisce un quadro completo per l'elaborazione e l'analisi a valle dei dati del foglio di luce. Questo software utilizza le informazioni all'interno del campione per eseguire la fusione multivista, quindi non richiede la presenza di perline fluorescenti intorno campione. Ma allo stesso tempo assume orientamento ortogonale delle viste di imaging (obiettivi), quindi non può essere utilizzato per i dati acquisiti da angolazioni arbitrarie 44.
requisiti hardware
S copi "> L'hardware utilizzato per la lavorazione può essere trovato in tabella 4. Ci deve essere la capacità di archiviazione sufficiente e una chiara conduttura per l'elaborazione dei dati disponibili, in vista del esperimento vero e proprio. L'acquisizione delle immagini è più veloce di successiva analisi ed è facile per ottenere inondato di dati non elaborati. spesso è realistico per memorizzare tutte le immagini crude, ma piuttosto una versione ritagliata o immagini elaborate come viste fuse, proiezioni di massima intensità o proiezioni sferiche 45.| Processore | Due processore Intel Xeon E5-2630 (sei core, 2,30 GHz Turbo, 15 MB, 7,2 GT / sec) |
| Memoria | 128 GB (16 × 8 GB) 1600 MHz DDR3 ECC RDIMM |
| Disco rigido | 4 × 4 TB 3.5inch Serial ATA (7.200 rpm) Disco rigido |
| controller HDD | PERC H310 SATA / SAS Controller per Dell Precision |
| Configurazione HDD | C1 SATA da 3,5 pollici, 1-4 dischi rigidi |
| Grafica | Dual 2 GB NVIDIA Quadro 4000 (2 carte w / 2 DP & 1 DVI-I ciascuno) (2 DP-DVI e 2 adattatore DVI-VGA) (MRGA17H) |
| Rete | Intel X520-T2 Dual Port 10 GbE Network Interface Card |
Tabella 4: Requisiti hardware.
la velocità di elaborazione dei dati
Il tempo necessario per l'elaborazione dei dati dipende dalle dimensioni dei dati e dell'hardware utilizzato. Nella Tabella 1, forniamo una panoramica del tempo necessario per i passaggi chiave in lavorazione un esempio 8,6 GB MultiView set di dati che consisteva di 1 punto di tempo con 4 punti di vista e 2 canali.
| Processing step | Tempo | passo protocollo |
| Salvare nuovamente come HDF5 | 6 min 30 sec | 6.3 |
| Rilevare i punti di interesse | 20 sec | 6.4 |
| Registrati utilizzando i punti di interesse | 3 sec | 6.5 |
| Fusion MultiView basato sul contenuto | 4 ore | 7.2 |
| Multiview deconvoluzione (CPU) | 8 ore | 7.3 |
| Multiview deconvoluzione (GPU) | 2 ore | 7.3 |
Tabella 1: Dati tempo di lavorazione.
formati di dati di ingresso per la ricostruzione MultiView
Il Fiji plugin Multiview la ricostruzione in grado di supportare .czi, .tif e formati ome.tiff. A causa della struttura dei dati del formato .czi, set di dati non sono discontinuisupportato senza pre-elaborazione. Discontinua significa che la registrazione doveva essere riavviato (ad esempio per regolare le posizioni dovuti alla deriva del campione). In questo caso i file .czi devono essere risalvato come .tif. Per .tif file ogni vista e la direzione di illuminazione deve essere salvato come file separato.
Taratura di dimensione dei pixel
Il software microscopio operatorio calcola la calibrazione per la dimensione xy pixel in base all'obiettivo selezionato. Tuttavia, la dimensione dei pixel in z è definita indipendentemente dalla dimensione del passo. Se un obiettivo sbagliato è specificato nel software xy z per il rapporto non è corretto e la registrazione fallirà.
Nuova registrazione
Dopo aver definito il set di dati il numero di registrazioni sarà 1 e il numero di punti di interesse sarà 0 nella ViewSetup Explorer. La registrazione iniziale rappresenta la taratura del set di dati. Sia il numero oregistrazioni F e punti di interesse aumenteranno durante la lavorazione.
Giù campionamento per il rilevamento dei punti di interesse
Utilizzando giù campionamento è consigliata, poiché il caricamento di file e la segmentazione sarà molto più veloce. E 'comunque importante notare che i parametri di rilevamento cambieranno a seconda del campionamento giù, trasferendo così impostazioni di rilevamento tra diverse impostazioni del campione verso il basso non è possibile.
Rilevamento di punti di interesse
Si consiglia di segmento come molti veri perle come possibile in ogni vista, anche a costo di ottenere alcune rilevazioni di falsi positivi, perché non ostacolano notevolmente la registrazione. rilevazioni spurie, se pochi di numero, sono esclusi durante la registrazione (Vedere Registrazione di punti di interesse). Tuttavia, massicci rilevazioni di falsi positivi rappresentano un problema per l'algoritmo. Esso non solo riduce le prestazioni per la rilevazione ela registrazione, dal momento che ci vuole molto più tempo per segmentare l'immagine così come confrontare queste perle tra i punti di vista, ma riduce anche la precisione della registrazione. Questo può essere affrontato utilizzando i parametri di rilevamento più stringenti. Inoltre, nel corso di segmentazione delle perle (vale a dire più rilevazioni su un tallone Figura 1E) è dannoso per la registrazione e deve essere evitato.
Registrazione dei punti di interesse
Per registrare il punto di vista le une sulle altre, la posizione di ogni tallone in ogni vista è descritta dalla sua posizione rispetto ai suoi tre perle vicini più vicini. Queste costellazioni stanno formando un descrittore geometrica locale e permettono il confronto ogni perla tra i punti di vista. Perline con corrispondenti descrittore tra due punti di vista sono quindi considerati come corrispondenze candidati. Si noti che questo funziona solo per perline distribuiti in modo casuale, per i quali i descrittori locali sono in genere unico per ciascuna sfera.Si possono utilizzare altre strutture come nuclei nel campione per la registrazione. Tuttavia, al fine di rilevare nuclei, che sono distribuiti casualmente non nel campione, applicare altri metodi 20,21.
Le corrispondenze candidati vengono poi testati contro RANSAC 36 al fine di escludere i falsi positivi. Ogni corrispondenza suggerisce un modello di trasformazione per sovrapporre i punti di vista incastrarsi tra di loro. I veri corrispondenze sarebbero probabilmente d'accordo su un modello di trasformazione, mentre i valori anomali farebbe ogni punto ad un altro. I veri corrispondenze vengono poi utilizzati per calcolare un modello di trasformazione affine tra i due punti di vista a confronto. Una ottimizzazione globale con un algoritmo di ottimizzazione iterativa viene quindi eseguita, in cui tutti i punti di vista sono registrati sulla prima vista con l'obiettivo di spostamento minima tra le viste 20,21.
registrazione Time-lapse
A causa di muoversizione del movimento del motore agarosio e imprecisa del palco microscopio, la posizione di ciascuna pila varia moderatamente nel tempo. Considerando che la registrazione del punto di tempo individuale elimina la differenza tra il punto di vista di questo punto di tempo, il time-lapse ha anche bisogno di essere registrato nel suo complesso. A tal fine, ciascun punto di tempo individuale è registrata sul punto di tempo di riferimento.
punto di tempo di riferimento
Se una serie temporale con molti punti di tempo viene elaborato, un punto temporale rappresentante viene selezionato come riferimento di solito dal centro della serie temporale poiché le intensità tallone può degradare nel tempo a causa imbianchimento. Su questo riferimento, i parametri per la determinazione del punto di interesse, di registrazione, riquadro di delimitazione e la fusione può essere determinato. Questi parametri vengono poi applicati all'intera time lapse calcolare un modello di trasformazione specifico per ciascun punto di tempo individuale. Durante la registrazione time-lapse tutti gli altri punti di tempo sono anche registEred spazialmente su questo punto di tempo di riferimento. Così i parametri dell'imballo per l'intera registrazione dipendono da questo specifico punto di tempo.
registrazione multicanale
Quando l'imaging più canali, idealmente le stesse perline fluorescenti devono essere visibili in tutti i canali creata l'immagine. Il rilevamento e la registrazione possono essere eseguite su ogni canale, che tiene conto dell'influenza delle diverse lunghezze d'onda di luce sulla trasformazione. Spesso questo non è possibile, perché le perline non sono visibili in tutti i canali o le perline dominano troppo l'immagine in un canale e sono troppo debole nell'altro canale (s). La soluzione tipica è quella di utilizzare perle visibili solo in un canale per il rilevamento e la registrazione e il modello di trasformazione acquisita (cioè dopo il rilevamento, registrazione e registrazione time-lapse) viene poi applicato gli altri canali da Fiji> Plugin> Multiview Reconstruzione> Elaborazione batch> Strumenti> Duplicate Trasformazioni. Nel menu a tendina per applicare la trasformazione di selezionare un canale ad altri canali. Nella finestra successiva selezionare l'XML e fare clic su OK. Quindi selezionare il canale che contiene le perle come canale di origine e di destinazione come canale di selezionare il canale (s) senza perline. Per i duplicati che utilizzano le trasformazioni Sostituire tutte le trasformazioni e premere OK. Le trasformazioni vengono poi copiati su tutti gli altri canali e salvate nel formato XML. Per vedere le nuove trasformazioni nel ViewSetup Explorer, riavviare il MultiView Ricostruzione applicazione.
Rettangolo di selezione
La fusione di molteplici punti di vista è computazionalmente molto intenso. Tuttavia, le grandi immagini sono tipicamente acquisite per accogliere non solo il campione, ma anche le perline intorno ad esso. Una volta che il parametro di registraziones sono estratti dalle perline, non sono più utili come parte dell'immagine. Pertanto, per aumentare l'efficienza della fusione, solo le parti delle pile di immagini contenenti il campione dovrebbero essere fusi insieme. Una regione di interesse (riquadro di delimitazione) dovrebbe essere definito per contenere il campione e il meno della agarosio circostante possibile. Per l'esempio in figura 2 una fusione media ponderata su tutto il volume con 2229 × 2136 × 2106 px richiederebbe 38,196 MB di RAM, mentre con un rettangolo di selezione del volume si riduce a 1634 × 1746 × 1632 px e i requisiti di memoria sono ridotti a 17.729 MB.
Fusion MultiView basato sul contenuto
La sfida in fusione di dati Multi View è che il punto di vista di solito finiscono bruscamente e non contengono la stessa qualità di immagine per gli stessi voxel. Semplice calcolo della media dei punti di vista sarebbe quindi portare ad artefatti di fusione e degradazione dell'immagine inutili. MultiView fu basato sul contenuto sione prende entrambi questi problemi in considerazione. In primo luogo, fonde le diverse viste, in cui un'immagine finisce e l'altro inizia e secondo, che valuta la qualità dell'immagine locale e applica un peso maggiore qualità d'immagine superiore nella fusione 21. Rispetto ad una singola vista c'è un miglioramento della risoluzione in z con un leggero degrado del segnale in xy (Figura 2E-H, Figura 5A-D).
Multiview deconvoluzione
deconvoluzione Multiview è un altro approccio per ottenere la fusione delle viste. Con questo metodo anche i PSF delle diverse viste sono presi in considerazione per recuperare l'immagine che è stata convoluta dall'ottica del microscopio. Questo metodo migliora drasticamente la qualità delle immagini, eliminando l'effetto mosso e aumentando la risoluzione e il contrasto del segnale 31 (Figura 2C-D, figura 2I-J, Figura 5E-G, Film 3).
ve_content "> deconvoluzione è computazionalmente molto intenso (vedi tabella 1), utilizzando quindi una GPU per l'elaborazione aumenta la velocità di questo processo. Può anche essere necessario eseguire la deconvoluzione sui dati giù campionati. Per esempio verso il basso, utilizzare Fiji> Multiview ricostruzione > Elaborazione batch> Strumenti> applicare trasformazioni. Questo si applica un nuovo modello di trasformazione sui punti di vista che verranno salvati nel file XML.formato di file HDF5 per BigDataViewer e BigDataServer
Il BigDataViewer 33 consente una facile visualizzazione dei dati terabyte. Come controllare la BigDataViewer è sintetizzata nella tabella 2. Uno screencast del funzionamento di base del programma è anche disponibile come supplemento nella sua pubblicazione originale 33. Il BigDataViewer è centrato su un file XML, che contiene i metadati, e un file HDF5, che contiene i dati di immagine. I dati di immagine sono present nel HDF5 in molteplici livelli di risoluzione in blocchi 3D. I molteplici livelli di risoluzione permettono di visualizzare i dati più velocemente con risoluzione inferiore, prima che la piena risoluzione viene caricato. I singoli blocchi vengono caricati in memoria solo quando necessario. Così, il formato di immagine HDF5 permette la visualizzazione diretta e veloce dei dati tramite la BigDataViewer 33. Si accelera l'elaborazione poiché il caricamento dei file viene effettuata in modo più efficiente. Pertanto, si consiglia resaving l'insieme di dati in questo formato, anche se non è strettamente necessario per la lavorazione. Per ulteriori spiegazioni del formato dei dati, si rimanda al link 3 . Inoltre, i dati possono essere condivisi con i collaboratori o il pubblico con il BigDataServer 33 ( collegamento 6 ).
| chiave | effetto |
| F1 | mostra la Guida in linea con una breve descrizione del BigDataViewer e il suo funzionamento di base |
| <> O la rotella del mouse | movimento z |
| su e giù freccia | zoom in e out |
| tasto destro del mouse e trascinate | si muove campione nel visualizzatore |
| click sinistro e trascinare | ruota del campione attorno al cursore |
| cursore nella parte inferiore del visualizzatore o Tab e freccia sinistra o destra | si muove su asse del tempo |
| premendo Inoltre spostamento | movimento veloce o la rotazione lungo un asse |
| x e poi freccia sinistra e destra | ruota intorno all'asse x |
| freccia Y e poi a sinistra e destra | ruota attorno all'asse y |
| z e poi a sinistra e freccia a destra | ruota attorno all'asse z |
| spostamento e x | orienta la vista lungo l'asse x |
| spostamento e y | orienta la vista lungo l'asse Y |
| spostamento e Z | orienta la vista lungo l'asse Z |
| io | commuta tra le diverse modalità di interpolazione (cioè vicini e trilineari più vicino) |
| s o Impostazioni> Luminosità e contrasto | modifica il colore dei canali, luminosità e contrasto |
| F6 o Impostazioni> Visibilità e raggruppamento | cambia i gruppi visualizzati, permette il raggruppamento di sovrapponendo diversi gruppi e chiamando i gruppi mediante i tasti numerici |
| F10 o Strumenti> Record di film | acquisisce una serie temporale della fetta attualmente visualizzato |
Tabella 2: Big Data Viewer.
3. Limitazioni di attuazione descritta di LSFM
bassa produttività
In un tipico esperimento LSFM un solo campione per ogni esperimento è ripreso. Ancora, nella nostra esperienza, molte informazioni utili può essere estratto da quel singolo campione. Imaging ad alta produttività di più embrioni è stato raggiunto recente incorporata casa configurazioni LSFM 46-48, anche se in genere a scapito della libertà di posizionamento del campione e la rotazione.
profonda penetrazione insufficiente nei tessuti
Anche se gli embrioni di zebrafish sono trasparenti, la qualità dell'immagine ottenuta si sta deteriorando rapidamente quando l'imaging più profonda nel tessuto a causa della dispersione e l'assorbimento. In parte ciò è un effetto di dispersione e l'assorbimento della fluorescenza emessa dal campione e non può essere corretto nella configurazione attuale. Un'altra fonte di qualità di immagine irregolare è l'illuminazione irregolare. Tegli foglio di luce incidente dal lato sinistro o destro ed eventuali oggetti nel suo percorso rifrangono esso, che si traduce in manufatti banda e sfocatura. La doppia faccia di illuminazione e MultiView fusione può ridurre gli artefatti nell'immagine finale. Infine, la qualità dell'immagine tende ad essere leggermente peggio verso il margine del campo di vista a causa della geometria naturale del foglio leggero, che sta diventando più spessa verso i bordi.
manipolazione chimica limitata
L'uso di farmaci o inibitori è molto diffusa nel campo della ricerca zebrafish. In questo uso del microscopio di farmaci è vincolato, a causa del grande volume della camera del campione e considerazioni di altri utilizzatori dello strumento, che condividono la stessa camera. L'utilizzo di una camera in più dedicato alla sperimentazione di farmaci in grado di risolvere questo problema. Riempire la camera parzialmente con perle di vetro riduce il volume di liquido che è richiesto.
No fotomanipolazione
Currently vi è alcuna possibilità di manipolazione ottica localizzata come fotoconversione, o ablazione laser in questo microscopio. Tuttavia, costruita casa configurazioni possono essere usati per tali applicazioni specifiche.
4. La domanda di significato e future
LSFM è il metodo migliore ad oggi disponibili per un veloce l'imaging di grandi volumi di embrioni vivi. La maggior parte degli esperimenti concepibili su un microscopio confocale può anche essere eseguita su un microscopio foglio leggero con vantaggi suddetti. Nel caso di imaging sviluppo dell'occhio, la velocità di LSFM non è il parametro fondamentale. Invece, la bassa fototossicità e flessibilità nel posizionamento del campione sono i vantaggi decisivi.
I dati LSFM hanno un elevato SNR, che aiuta a ottenere buoni risultati deconvoluzione ed è anche utile per l'analisi delle immagini automatizzata e monitoraggio oggetto. In conclusione, LSFM è un grande strumento per la generazione di dati quantificabili sullo sviluppo embrionale e overall cellule e le caratteristiche del tessuto per la successiva modellazione e descrizioni fisiche dei processi in questione.
Divulgazioni
La pubblicazione di questo video-articolo è supportato da Zeiss.
Riconoscimenti
We want to thank Tobias Pietzsch for providing his powerful open source software BigDataViewer. We thank the Light Microscopy Facility of the Max Planck Institute of Molecular Cell Biology and Genetics (MPI-CBG), namely Jan Peychl, Sebastian Bundschuh and Davide Accardi for technical assistance, perfect maintenance of the microscopes used in the study and for comments on the manuscript and H. Moon (MPI-CBG Scientific Computing Facility) for the BigDataServer. We thank Julia Eichhorn for assembling the Movie 1. The Norden lab members and Svea Grieb provided many helpful comments on the manuscript. Jaroslav Icha, Christopher Schmied and Jaydeep Sidhaye are members of the International Max Planck Research School for Cell, Developmental and Systems Biology and doctoral students at TU Dresden. Pavel Tomancak is supported by the ERC Starting Grant: Quantitative Analysis of the Hourglass Model of Evolution of Development and Human Frontier Science Program Young Investigator grant RGY0093/2012. Caren Norden is supported by the Human Frontier Science Program (CDA-00007/2011) and the German Research Foundation (DFG) [SFB 655, A25].
Materiali
| Name | Company | Catalog Number | Comments |
| Lightsheet Z.1 microscope | Carl Zeiss Microscopy | ||
| Low melting point agarose | Roth | 6351.1 | |
| Low melting point agarose | Sigma | A4018 or A9414 | |
| Ethyl 3-aminobenzoate methanesulfonate (MESAB/MS-222/Tricaine) | Sigma | E10521 | |
| N-Phenylthiourea (PTU) | Sigma | P7629 | |
| 500 nm red fluorescent beads F-Y 050 | Millipore (Estapor) | 80380495 | |
| 20 μl (1 mm inner diameter, marked black) capilllaries | Brand | 701904 | sold as spare part for transferpettor |
| Teflon tip plungers for 20 μl capillaries | Brand | 701932 | sold as spare part for transferpettor |
| Circular glass coverslips diameter 18 mm, selected thickness 0.17 mm | Thermo scientific (Menzel-Glaser) | ||
| O-rings for chamber windows 17×1.5 mm | Carl Zeiss Microscopy | ||
| Tweezers, style 55 | Dumont | 0209-55-PO | |
| 50 ml Luer-Lock syringes | Becton Dickinson | 300865 | |
| 150 cm extension cable for infusion compatible with Luer-Lock syringes | Becton Dickinson | 397400 | |
| Links | |||
| Link 1 Multiview reconstruction application | https://github.com/bigdataviewer/SPIM_Registration http://fiji.sc/Multiview-Reconstruction | ||
| Link 2 BigDataViewer | https://github.com/bigdataviewer | ||
| Link 3 BigDataViewer | http://fiji.sc/BigDataViewer | ||
| Link 4 Multiview reconstruction application-issues | https://github.com/bigdataviewer/SPIM_Registration/issues | ||
| Link 5 BigDataViewer-issues | https://github.com/bigdataviewer/bigdataviewer_fiji/issues | ||
| Link 6 BigDataServer | http://fiji.sc/BigDataServer |
Riferimenti
- Pantazis, P., Supatto, W. Advances in whole-embryo imaging: a quantitative transition is underway. Nat Rev Mol Cell Biol. 15 (5), 327-339 (2014).
- Truong, T. V., Supatto, W. Toward high-content/high-throughput imaging and analysis of embryonic morphogenesis. Genesis. 49 (7), 555-569 (2011).
- Keller, P. J. Imaging Morphogenesis: Technological Advances and Biological Insights. Science. 340 (6137), 1234168-1234168 (2013).
- Huisken, J., Swoger, J., Del Bene, F., Wittbrodt, J., Stelzer, E. H. K. Optical Sectioning Deep Inside Live Embryos by Selective Plane Illumination Microscopy. Science. 305 (5686), 1007-1009 (2004).
- Voie, A. H., Burns, D. H., Spelman, F. A. Orthogonal-plane fluorescence optical sectioning: three-dimensional imaging of macroscopic biological specimens. J Microsc. 170 (Pt 3), 229-236 (1993).
- Jemielita, M., Taormina, M. J., DeLaurier, A., Kimmel, C. B., Parthasarathy, R. Comparing phototoxicity during the development of a zebrafish craniofacial bone using confocal and light sheet fluorescence microscopy techniques. J Biophoton. 6 (11-12), 920-928 (2012).
- Stelzer, E. H. K. Light-sheet fluorescence microscopy for quantitative biology. Nat Meth. 12 (1), 23-26 (2015).
- Chen, B. C., Legant, W. R., et al. Lattice light-sheet microscopy: Imaging molecules to embryos at high spatiotemporal resolution. Science. 346 (6208), 1257998-1257998 (2014).
- Keller, P. J., Ahrens, M. B. Visualizing Whole-Brain Activity and Development at the Single-Cell Level Using Light-Sheet Microscopy. Neuron. 85 (3), 462-483 (2015).
- Pampaloni, F., Chang, B. -. J., Stelzer, E. H. K. Light sheet-based fluorescence microscopy (LSFM) for the quantitative imaging of cells and tissues. Cell Tissue Res. 360 (1), 129-141 (2015).
- Weber, M., Mickoleit, M., Huisken, J. Light sheet microscopy. Quantitative Imaging in Cell Biology. , 193-215 (2014).
- Strobl, F., Stelzer, E. H. K. Non-invasive long-term fluorescence live imaging of Tribolium castaneum embryos. Development. 141 (11), 2361-2361 (2014).
- Kaufmann, A., Mickoleit, M., Weber, M., Huisken, J. Multilayer mounting enables long-term imaging of zebrafish development in a light sheet microscope. Development. 139 (17), 3242-3247 (2012).
- Maizel, A., von Wangenheim, D., Federici, F., Haseloff, J., Stelzer, E. H. K. High-resolution live imaging of plant growth in near physiological bright conditions using light sheet fluorescence microscopy. Plant J. 68 (2), 377-385 (2011).
- Swoger, J., Muzzopappa, M., Lòpez-Schier, H., Sharpe, J. 4D retrospective lineage tracing using SPIM for zebrafish organogenesis studies. J Biophoton. 4 (1-2), 122-134 (2010).
- Keller, P. J., Schmidt, A. D., Wittbrodt, J., Stelzer, E. H. K. Reconstruction of zebrafish early embryonic development by scanned light sheet microscopy. Science. 322 (5904), 1065-1069 (2008).
- Wu, Y., Ghitani, A., et al. Inverted selective plane illumination microscopy (iSPIM) enables coupled cell identity lineaging and neurodevelopmental imaging in Caenorhabditis elegans. Proc Natl Acad Sci USA. 108 (43), 17708-17713 (2011).
- Sarov, M., Barz, C., et al. A genome-wide resource for the analysis of protein localisation in Drosophila. bioRxiv. , 028308 (2015).
- Amat, F., Lemon, W., et al. Fast, accurate reconstruction of cell lineages from large-scale fluorescence microscopy data. Nat Meth. 11 (9), 951-958 (2014).
- Preibisch, S., Saalfeld, S., Schindelin, J., Tomancak, P. Software for bead-based registration of selective plane illumination microscopy data. Nat Meth. 7 (6), 418-419 (2010).
- Preibisch, S., Rohlfing, T., Hasak, M. P., Tomancak, P. Mosaicing of single plane illumination microscopy images using groupwise registration and fast content-based image fusion. SPIEMed Imaging. 6914, 69140E (2008).
- Reynaud, E. G., Peychl, J., Huisken, J., Tomancak, P. Guide to light-sheet microscopy for adventurous biologists. Nat Meth. 12 (1), 30-34 (2015).
- Keller, P. J., Schmidt, A. D., Wittbrodt, J., Stelzer, E. H. K. Digital Scanned Laser Light-Sheet Fluorescence Microscopy (DSLM) of Zebrafish and Drosophila Embryonic Development. Cold Spring Harb Protoc. 2011 (10), (2011).
- Pinto-Teixeira, F., Muzzopappa, M., Swoger, J., Mineo, A., Sharpe, J., Lòpez-Schier, H. Intravital imaging of hair-cell development and regeneration in the zebrafish. Front Neuroanat. , (2013).
- Keller, P. J. In vivo imaging of zebrafish embryogenesis. METHODS. , 1-11 (2013).
- Pitrone, P. G., Schindelin, J., et al. OpenSPIM: an open-access light-sheet microscopy platform. Nat Meth. 10 (7), 598-599 (2013).
- Gualda, E. J., Vale, T., Almada, P., Feijò, J. A., Martins, G. G., Moreno, N. OpenSpinMicroscopy: an open-source integrated microscopy platform. Nat Meth. 10 (7), 599-600 (2013).
- Martinez-Morales, J. R., Rembold, M., et al. ojoplano-mediated basal constriction is essential for optic cup morphogenesis. Development. 136 (13), 2165-2175 (2009).
- Strzyz, P. J., Lee, H. O., Sidhaye, J., Weber, I. P., Leung, L. C., Norden, C. Interkinetic Nuclear Migration Is Centrosome Independent and Ensures Apical Cell Division to Maintain Tissue Integrity. Dev Cell. 32 (2), 203-219 (2015).
- Young, L. K., Jarrin, M., Saunter, C. D., Quinlan, R., Girkin, J. M. Using SPIM to track the development of the focal power of the zebrafish lens. SPIE BiOS. 9334, 933408 (2015).
- Preibisch, S., Amat, F., et al. Efficient Bayesian-based multiview deconvolution. Nat Meth. 11 (6), 645-648 (2014).
- Schindelin, J., Arganda-Carreras, I., et al. Fiji: an open-source platform for biological-image analysis. Nat Meth. 9 (7), 676-682 (2012).
- Pietzsch, T., Saalfeld, S., Preibisch, S., Tomancak, P. BigDataViewer: visualization and processing for large image data sets. Nat Meth. 12 (6), 481-483 (2015).
- Brown, M., Lowe, D. G. Invariant Features from Interest Point Groups. Proceedings of the British Machine Vision Conference. , 23.1-23.10 (2002).
- Saalfeld, S., Fetter, R., Cardona, A., Tomancak, P. Elastic volume reconstruction from series of ultra-thin microscopy sections. Nat Meth. 9 (7), 717-720 (2012).
- Fischler, M. A., Bolles, R. C. Random sample consensus: a paradigm for model fitting with applications to image analysis and automated cartography. Commun ACM. 24 (6), 381-395 (1981).
- Zolessi, F. R., Poggi, L., Wilkinson, C. J., Chien, C. -. B., Harris, W. A. Polarization and orientation of retinal ganglion cells in vivo. Neural Dev. 1 (1), 2 (2006).
- Stepanova, T., Slemmer, J., et al. Visualization of microtubule growth in cultured neurons via the use of EB3-GFP (end-binding protein 3-green fluorescent protein). J Neurosci. 23 (7), 2655-2664 (2003).
- Weber, M., Mickoleit, M., Huisken, J. Multilayer Mounting for Long-term Light Sheet Microscopy of Zebrafish. J Vis Exp. (84), e51119 (2014).
- Udan, R. S., Piazza, V. G., Hsu, C. W., Hadjantonakis, A. K., Dickinson, M. E. Quantitative imaging of cell dynamics in mouse embryos using light-sheet microscopy. Development. 141 (22), 4406-4414 (2014).
- Ichikawa, T., Nakazato, K., et al. Live imaging and quantitative analysis of gastrulation in mouse embryos using light-sheet microscopy and 3D tracking tools. Nat Protoc. 9 (3), 575-585 (2014).
- Schmied, C., Stamataki, E., Tomancak, P. Open-source solutions for SPIMage processing. Methods Cell Biol. 123, 505-529 (2014).
- Amat, F., Höckendorf, B., Wan, Y., Lemon, W. C., McDole, K., Keller, P. J. Efficient processing and analysis of large-scale light-sheet microscopy data. Nat Protoc. 10 (11), 1679-1696 (2015).
- Schmid, B., Shah, G., et al. High-speed panoramic light-sheet microscopy reveals global endodermal cell dynamics. Nat Commun. 4, 1-10 (2013).
- Gualda, E. J., Pereira, H., Vale, T., Estrada, M. F., Brito, C., Moreno, N. SPIM-fluid: open source light-sheet based platform for high-throughput imaging. Biomed Opt Express. 6 (11), 4447-4456 (2015).
- McGorty, R., Liu, H., Kamiyama, D., Dong, Z., Guo, S., Huang, B. Open-top selective plane illumination microscope for conventionally mounted specimens. Opt Express. 23 (12), 16142-16153 (2015).
- Jemielita, M., Taormina, M. J., et al. Spatial and Temporal Features of the Growth of a Bacterial Species Colonizing the Zebrafish Gut. mBio. 5 (6), e01751-14-8 (2014).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati