
Method Article
クロマチン免疫沈降分析のための凍結キメラ肝臓組織からのクロマチン抽出
要約
このプロトコルは、スナップ凍結組織からのクロマチン調製に焦点を当てており、架橋クロマチン免疫沈降(X-ChIP)とそれに続く定量的PCR分析(X-ChIP-qPCR)または次世代シーケンシングアプローチ(X-ChIP-seq)のいずれかに適しています。
要約
架橋クロマチン免疫沈降(X-ChIP)は、ヒストンマークのレベルと宿主および/または病原体クロマチン上の転写因子の占有率を評価するために広く使用されている技術です。組織からのクロマチン調製は、細胞培養に使用されるものに匹敵する再現性と信頼性の高いプロトコルを得るために克服する必要がある追加の課題を生み出します。組織の破壊と固定は、クロマチンの効率的なせん断を達成するための重要なステップです。異なる細胞タイプとクラスターが共存すると、最適なフラグメントサイズに到達するために異なるせん断時間が必要になり、せん断の再現性が妨げられる場合があります。この方法の目的は、ChIP-qPCRと次世代シーケンシング(NGS)アプリケーションの両方に適した、凍結組織(肝臓)からの信頼性と再現性のあるホストクロマチン調製を実現することです。液体窒素組織粉砕とホモジナイズの組み合わせは、ホモジナイズのみの場合と比較して、効率的にせん断できる解離した単一細胞からなる懸濁液を提供するため、再現性が向上することを観察しました。さらに、固定ステップは、均質な架橋を提供するために穏やかな回転下で実行する必要があります。固定された材料は、バッファーベースの核分離に適しており、細胞質タンパク質および病原体DNAおよびRNA(該当する場合)の汚染を低減し、時間のかかる遠心分離勾配を回避します。その後の超音波処理は、核溶解を完了し、クロマチンを剪断し、選択された剪断条件に従って特定のサイズ範囲を生成します。サイズ範囲は、NGSアプリケーションの場合は100〜300 ntである必要がありますが、ChIP-qPCR分析の場合はそれより高くなる可能性があります(300〜700 nt)。このようなプロトコルの適応は、凍結組織標本からのクロマチン分析を大幅に改善することができます。
概要
哺乳類細胞におけるエピジェネティック制御は、その発見以来、このようなメカニズムの理解が細胞生物学だけでなく、疾患や腫瘍生物学においても重要な洞察を提供すると考えて、ますます認識されています1。さらに、感染因子も宿主のエピジェネティックな変化2を引き起こす可能性がありますが、宿主細胞の機構は、持続するDNAウイルスなどの病原体のクロマチンにも影響を与える可能性があります3,4。この宿主と病原体の相互作用は、感染の持続性に役割を果たしているようです。2
DNAとの可逆的な会合により、ヒストンタンパク質はヌクレオソームと呼ばれる複合体を形成します。ヌクレオソームは、クロマチンとして知られるより高いレベルの組織化に到達します。クロマチンリモデリングは、遺伝子発現を厳密に制御し、転写因子(TF)へのアクセスを許可または拒否することが知られています5。これらの因子は、RNAポリメラーゼII(PolII)の遺伝子プロモーターへの動員を誘発または阻止し、DNAテンプレート6からのmRNA合成に影響を与える可能性があります。ヒストンタンパク質には、ヒストンフォールドの両端に隣接するテール7が含まれており、翻訳後修飾(PTM)を受ける可能性があり、構造クロマチン変化による遺伝子転写の厳密な制御を可能にします。ほとんどのヒストンPTMは尾N末端に位置し、アセチル化とメチル化が最もよく研究されているPTMですが、リン酸化8、ユビキチン化9 、リボシル化10 も報告されています。このようなタンパク質の特性評価と研究は、遺伝子制御に関する深い洞察を得るために不可欠です。
現在、直接的なDNA-タンパク質相互作用を研究するために利用できるいくつかの確立された方法とツールがあります:電気泳動移動度シフトアッセイ(EMSA)、酵母ワンハイブリッドアッセイ(Y1H)およびDNAフットプリント11。ただし、これらの方法は それ自体 が単一のDNA-タンパク質相互作用に焦点を当てており、ゲノム全体の研究には適用できません。これらの技術の別の制限は、調査されたDNAセグメントとのヒストンの会合の欠如です。したがって、このようなアプローチは、 in vivoでの 転写機構の複雑さを反映することを意図したものではなく、DNAへのタンパク質結合に影響を与える(促進または阻害する)可能性のある重要な構造変化12 または他の必要な酵素/補因子13 を考慮していません。
ホルムアルデヒド(FA)などの薬剤で細胞を固定することで、タンパク質-DNA相互作用の in vivo スナップショットが得られるという考えが、クロマチン免疫沈降アッセイ(ChIP)14の開発の基礎となりました。これは、定量的PCR(qPCR)技術と高度に特異的な抗体の利用可能性とともに、ChIP-qPCRアッセイの開発を可能にしました。その後、コストがより手頃な価格になっている次世代シーケンシング技術(NGS)の出現により、ChIP実験とNGSアプローチ(ChIP-seq)を組み合わせることが認められ、クロマチン調節の調査を可能にする新しい強力なツールが研究者に提供されます。これらのアッセイでは、単離または培養した細胞をジスクシンイミジルグルタル酸(DSG)および/またはFAで固定し、核を単離し、クロマチンを断片化し、目的の抗体によって沈殿させます。今後、DNAを精製し、PCRまたはNGSアプローチで分析します。EMSA、Y1H、およびDNAフットプリントとは対照的に、ChIPアッセイは、細胞内のタンパク質-DNA相互作用のグローバルなスナップショットを提供する能力を備えています。これにより柔軟性が得られ、同じサンプル内の複数の遺伝子座の分析が可能になります。ただし、アッセイの性質上、ChIPは、直接的な相互作用だけでなく、直接的な相互作用も検出し、直接的な相互作用に関心がある場合、上記の方法の精度を提供しない可能性があります。
細胞培養材料からのクロマチン調製プロトコルは十分に確立されており15、再現性が高いため、ユーザーはqPCRとNGSの両方のアプローチに適したクロマチンを1〜2営業日で取得できます。しかし、クロマチンの最適な固定とせん断を達成しながら、組織内の細胞を解離する必要があるため、組織全体から高品質のクロマチンを得ることは依然として課題です。さらに、異なるタイプの組織の組成および形態は様々であり、したがって、既存のプロトコールの調整を必要とする16、17。凍結保存された組織の使用は、新鮮なサンプルと比較して追加の課題を提供します。これは、広範囲の材料損失なしに単一細胞懸濁液を得ることが困難であるためである。これは不適切なせん断につながり、下流の用途を妨げます。それにもかかわらず、新鮮な組織標本ではなく凍結組織標本にアクセスすることは、作業の柔軟性を高めるだけでなく、縦断的または比較研究に由来する標本を扱う研究者にとって唯一の選択肢となる可能性があります。凍結組織用のクロマチン調製プロトコルがいくつか公開されています。これらは主に、試料の解凍とそれに続くミンチ、手動/機械ベースの解離、または液体窒素粉砕ステップ18、19、20に基づいています。
本稿では,凍結未固定肝検体に対して最適化されたクロマチン調製法15 について述べるが,液体窒素中での組織粉砕と乳棒の均質化を組み合わせ,ウイルスゲノムと宿主ゲノムの解析を目的としたX-ChIPアプローチに適した再現性のあるクロマチンせん断法を実現することである。
プロトコル
ヒト肝キメラマウス21 からの組織サンプリングは、欧州連合指令86/609/EECに従って実施され、ヘルシンキ宣言の原則に従ってハンブルク市および州の倫理委員会によって承認された。
1. 試薬の調製
- 脱イオン水中の1.25 Mグリシン溶液を調製します。0.22 μmの細孔サイズフィルターを備えた滅菌フィルター。4°Cで保存してください。
- 5 M塩化ナトリウム(NaCl)溶液を調製します。室温で保管してください。
- CaCl2 溶液を調製する:脱イオン水中の300 mM CaCl2 および10 mM Tris-HCl pH 8。0.22 μmの細孔サイズのフィルターを備えた滅菌フィルターで、RTで保管します。
- 脱イオン水で10%Triton X-100希釈液を調製します。RTで保存してください。
- トリス-EDTAバッファーを調製します:脱イオン水中で1 mM EDTAおよび10 mM Tris pH 8。4°Cで保存してください。
- 必要な量に応じて、次のバッファーを準備します。
- バッファーAを調製します:脱イオン水中の50 mM Hepes-KOH pH 7.5、140 mM NaCl、1 mM エチレンジアミン四酢酸(EDTA)、10%グリセロール、0.5%NP-40および0.25%トリトンX-100。0.22 μmの細孔サイズフィルターを備えた滅菌フィルター。4°Cで保存してください。
- バッファーBを調製する:10 mM トリス塩酸 pH 8、200 mM NaCl、1 mM EDTA、0.5 mM エグタジン酸(EGTA)。0.22 μmの細孔サイズフィルターを備えた滅菌フィルター。4°Cで保存してください。
- バッファーC:1%SDS、10 mM EDTA、および50 mM トリス塩酸pH 8を脱イオン水中で調製します。0.22 μmの細孔サイズフィルターを備えた滅菌フィルター。RTで保存してください。
- クロマチン希釈バッファーを調製します:脱イオン水中で0.01%SDS、1.1%トリトンX-100、1.2 mM EDTA、16.6 mM トリス塩酸pH 8および166 mM NaCl。0.22 μmの細孔サイズフィルターを備えた滅菌フィルター。4°Cで保存してください。
2.材料の準備
- ドライアイス、氷、液体窒素を収集します。
注意: ドライアイスと液体窒素は、火傷をしないように必要な注意を払って取り扱ってください。 - 遠心分離機を4°Cで予冷します。
注:このステップは、クロマチンの品質を低下させるため、洗浄ステップ中のタンパク質の分解と脱架橋を避けるために重要です。 - 滅菌プレートをドライアイスの上に置き、冷まします。
注意: プレートが切断プロセスを容易にするのに十分な大きさであることを確認してください。100 mmのペトリプレート/細胞培養皿をお勧めします。 - グリシン1.25 Mの必要なアリコートを取り出し、RTに到達させます。
- バッファーA、B、PBSの必要なアリコートを取り出します。プロテアーゼおよび/またはデアセチラーゼおよびホスファターゼ阻害剤を1倍の濃度になるまで添加し、氷上に置いておきます。
- 必要なバッファーCのアリコートを取り出し、RTに残します。指定されるまで、プロテアーゼおよび/またはデアセチラーゼおよびホスファターゼ阻害剤を追加しないでください。.
- RT PBSの必要なアリコートを取り出します。
注意: バッファー C には、ドデシル硫酸ナトリウム (SDS) が含まれています。バッファーを準備するときは、適切な安全対策を講じてください。
注:SDSは氷上で沈殿し、プロテアーゼおよびデアセチラーゼ阻害剤はRTで安定していません。 - チャンバー内に液体窒素を注ぐモルタルを予冷し、サプライヤーの指示に厳密に従ってください。金属乳棒をドライアイスで少なくとも5分間冷却します。
注:提案されたモルタルに代わるモルタルを使用することは可能です。しかしながら、このプロトコルで使用される装置は、その独特の構造のために、粉砕プロセス中に実質的な損失なしに少量の組織で作業することを可能にする。 - ダウンスホモジナイザーを関連する乳棒Aとともに氷上で事前に冷却します。
注意: 乳棒Aはホモジナイザーとのゆるいフィット感を持っています。これにより、有意な細胞溶解なしに単一細胞懸濁液を得ることができる。
3.組織架橋
- メスとピンセットを使ってドライアイスの上で皿の上で直接約50 mgの凍結組織を切ります。
注:メスをRTに保つと、切断プロセスが容易になるため、メスをRTに保つことをお勧めします。メスに過度の圧力をかけると、切断領域の外側に組織片が散乱するリスクが高まるため、避けてください。注目すべきことに、50 mgの組織(この場合は肝臓)は約500万個の細胞を生み出すはずです。暖かい刃は刃先を解凍することに注意してください。ただし、組織片のサイズが比較的大きいことを考えると、効果は限られているはずです。小さな断片を切るときは、組織の飛散を避けるために注意を払いながら冷たいメスを使用することが有益かもしれません。 - 切り取ったティッシュを、ドライアイスで冷やした1.5mLチューブに入れます。組織の解凍を避けてください。
- ティッシュが入っているチューブを乳鉢に移し、5分間そのままにします。
注意: サンプルを乳鉢に入れておくと、サンプルの温度が下がります(-80°Cから-196°C)。これにより、靭性が向上し、粉砕ステップが容易になります。 - 固体の崩れが見えなくなるまで、事前に冷やした乳棒の助けを借りてサンプルに圧力をかけます。
注意: サンプルが解凍されるため、過度の回転力による乳棒の加熱を避けることが重要です。すべてのサンプルを粉砕した後、乳棒を70%エタノール(EtOH)で洗浄し、ドライアイスで再び冷やします。 - サンプルが入っているチューブを乳鉢から取り出し、必要な阻害剤を含む950 μLの氷冷PBSを加えます。サンプルが完全に再懸濁されるまで、ゆっくりと上下にピペットで固定します。すぐに手順 3.6 に進みます。
- 組織懸濁液をホモジナイザーに移し、乳棒Aで20〜30ストロークを適用して、より細かい懸濁液を得る。泡立ちを避けてください。
注:ストローク量は、組織の一貫性に応じて最適化する必要があります。この工程は、粉砕後に得られた細胞の小さなクラスターをさらに解離させる。不適切な均質化は、せん断効率に影響を与える可能性があります。 - ホモジネートを、すでに氷上で予冷した新しい1.5 mLチューブに移します。
- 4°Cで1,300 x g で5分間遠心分離し、上清を慎重に除去します。
- 穏やかなピペッティングによりペレットを950 μLのRT PBSに完全に再懸濁し、63.6 μLの16%MeOHフリーFAを加えて最終濃度を1%にします。すぐに手順 3.10 に進みます。
注意: FAは有毒な化学物質です。適切な安全対策を講じたドラフトの下で取り扱ってください。
注:不完全な再懸濁は、固定ステップ中に細胞凝集を引き起こす可能性があります。これは溶解およびせん断プロセスを妨げる。 - RTで10分回転します。すぐに手順3.11に進みます。
注: 集計を避けるために回転が必要です。固定に必要な時間は、対象ターゲットとサンプルタイプに応じて最適化する必要があります。過度の固定時間は適切なせん断を妨げる可能性があることに注意することが重要です。 - RTで113 μLの1.25 Mグリシンを加えて最終濃度125 mMとし、5分間回転させます。
注:グリシンは固定反応を消光し、過剰架橋を回避します。 - 1,300 x g で 4 °C で 3 分間遠心分離します。
- 上清を廃棄し、必要な阻害剤を含む950 μLの氷冷PBSにピペッティングしてペレットを慎重に再懸濁します。
- 1,300 x g で 4 °C で 3 分間遠心分離します。
- 手順3.13〜3.14を繰り返し、すぐにクロマチン分離手順に進みます。
4. クロマチン単離
- 必要な阻害剤を含む950 μLのバッファーAをペレットに加えます。ペレットが完全に再懸濁するまでピペッティングで穏やかに混合し、4°Cで10分間回転させます。
注:このステップでは、核溶解なしで固定単一細胞懸濁液を溶解します。これにより、細胞質タンパク質およびRNAのサンプルを取り除くことができます。溶解時間を長くすることは、溶解が困難な細胞にとって有益である可能性がありますが、組織の取り扱い時間が長くなります。この時点で、トリパンブルー/DAPI染色後の顕微鏡下で調製物を確認し、クラスターのサイズと単一細胞の存在を確認することができます。ただし、単一の核は、組織材料が固定されているため、簡単に理解できない場合があります。 - 2,000 x g で4°Cで5分間遠心分離し、上清を慎重に除去します。
- 必要な阻害剤を含む950 μLのバッファーBをペレットに加えます。ペレットが完全に再懸濁するまでピペッティングで穏やかに混合し、4°Cで10分間回転させます。
注:このステップでは、核調製物から溶解バッファーを洗い流して、さらなる望ましくない溶解を回避します。 - 2000 x g で4°Cで5分間遠心分離します。 一方、必要な阻害剤(ステップ2.5と同じ)をバッファーCに追加します。
- 上清を慎重に取り除きます。
- 300 μLのRTバッファーCをペレットに加え、激しくピペットで移します。
- サンプルを15〜30秒間ボルテックスし、チューブを短時間回転させて蓋の液滴を収集します。
注:このステップは、固定核を遊離して溶解するために重要です。サンプルの完全性を維持し、同時にSDS沈殿を避けるために、9-11°Cの温度を維持するために氷上に保たれたプラスチックラックに超音波処理の前にサンプルを保管してください。
5. クロマチン断片化
- サンプルを3つのきれいな0.65 mL超音波処理認定チューブに移し、チューブあたり100 μLの溶解核懸濁液を確保します。
注:最大容量300μLの1.5mL超音波処理認定チューブを使用することができます。これらのチューブには特定のホルダーが必要です。0.65 mLは、チューブあたりのサンプル量が少ないため、より均質なせん断を提供します。 - クロマチンを30秒オンと30秒オフの設定で高強度で28サイクル超音波処理します。超音波処理器バスが適切に冷却されていることを確認してください(氷または冷却装置)。
注: この手順は、ほとんどの場合、最適化が必要です。ユーザーは、せん断時間を増やすと、より小さく、より均質なフラグメントが得られることを覚えておく必要があります。ただし、これによりクロマチンの品質が低下する可能性が高くなります。必要なフラグメントサイズを提供する最小サイクル数を選択します。このステップの最適化中に、核染色を実行して、サイクル数が核の大部分を溶解するのに十分であったかどうかを確認することは有用です。 - 超音波処理されたクロマチンを、以前に氷上で冷却した新しい1.5mLチューブに移す。
- 30 μLのTriton X-100 10%溶液を加え、5〜10秒間ボルテックスします。
注:Triton X-100はSDSに結合し、4°Cでのさらなる沈殿を防ぎます。 Triton X-100の最終量は常に1%でなければなりません。 - 16,000 x g で4°Cで15分間遠心分離します。
- 上清を氷上で予め冷やした清潔な1.5 mLチューブに移します。
- 注:上清には剪断されたクロマチンが含まれており、透明に見えるはずです。ペレットには「せん断できない」休息が含まれており、非常に小さいままである必要があります(肝臓組織の場合はほとんど茶色)。せん断の失敗の兆候を探します:クロマチン溶液が透明にならなかったこと、およびステップ4.5のものと同様のペレット寸法。
6. DNA精製
- せん断クロマチン10〜25 μLを新しいチューブに移し、バッファーCを加えて最終容量200 μLにします。 残りのクロマチンは、さらに使用するまで-80°Cで保存してください。必要に応じて、このステップで手順を中断し、サンプルを-20°Cで保存できます。
- 8 μLの5 M NaClを添加し、1000 rpmで振とうしながら加熱ブロック内で65°Cで少なくとも6時間インキュベートします。
注:このステップでは、クロマチンを脱架橋します。可能であれば、脱架橋を一晩延長する方が安全です。NaClの存在はプロセスをより効率的にします。 - サンプルをRTで5分間冷却し、2 μLのRNase Aを加えます。
- 1000 rpmで振とうしながら37°Cで1時間インキュベートします。
- 加熱ブロックからサンプルを取り出し、7 μLの300 mM CaCl 2と2 μLのプロテイナーゼKを加えます。
- 加熱ブロックを56°Cに設定し、1000rpmで振とうしながら30分間インキュベートします。一方、サンプルごとに1本の相分離チューブを準備し、16,000 x g で4°Cで1分間遠心分離します。
注:これらの特別なチューブは、核酸フェノール-クロロホルム抽出中の相分離を容易にします。 - 加熱ブロックからチューブを取り外し、RTで3分間平衡化させます。
- 400 μLのサンプルを、予め遠心分離した相分離チューブに移します。
- 400 μLのフェノール-クロロホルム-イソアミルアルコール溶液(PCI)を加え、5秒間ボルテックスします。
注意: PCI は揮発性が高く毒性の高い化合物です。ドラフトの下で必要な安全対策を講じて取り扱ってください。 - 16,000 x g で 4 °C で 5 分間遠心分離します。
- 400 μLのクロロホルムとボルテックスを5秒間加えます。
注意: クロロホルムは非常に揮発性で有毒な化合物です。ドラフトの下で必要な安全対策を講じて取り扱ってください。
注:このステップは、ダウンストリームPCRアプリケーションを妨げる可能性のあるフェノールの残留物を洗い流します。 - 16,000 x g で 4 °C で 5 分間遠心分離します。
- 上相400 μLを新しい1.5 mLチューブに移し、そこで24 μLの5 M NaClと0.75 μLのグリコーゲンを添加しました。簡単に渦。
- 1,055 μLの100%EtOHを加え、完全にボルテックスします。適切な混合を確保してください。
- -80°Cで1時間、または-20°Cで一晩(ON)インキュベートします。
注:このステップは、剪断されたDNAを沈殿させます。収量を最大化するには、ONインキュベーションを選択することをお勧めします。 - 16,000 x g で 4 °C で 30 分間遠心分離します。
- ペレットを脱臼させないように注意しながら上清を慎重に除去します。
- 500 μLの冷たい70%EtOHを加えます。チューブを静かに傾けて、ペレットが確実に洗浄されるようにします。
注:このステップは、核酸と共沈した可能性のある塩の残留物を除去するために不可欠です。塩は、他の下流の用途を妨げる可能性があります。 - 16,000 x g で4°Cで15分間遠心分離します。
- 上清全体を慎重に取り除き、ペレットをRTで乾燥させます。
注意: 37°Cの加熱ブロックでチューブをインキュベートすると、乾燥に必要な時間が短縮されます。 - 50 μLのトリス-EDTA溶液(TEバッファー)を加え、300 rpmで振とうしながら37°Cの加熱ブロックに5〜10分間チューブを置きます。
注:このステップにより、ペレットの溶解が保証されます。プロトコルはここで一時停止することができ、サンプルは4°Cで最大1週間、または-20°Cで長期保存することができます。 - 1%アガロースゲルでDNA分析を行います。
7. DNAサイズ解析
- ランニングバッファー100 mLあたり1 gのアガロース(酢酸トリス-EDTA(TAE)またはホウ酸トリス-EDTA(TBE))を混合して、1%アガロースゲルを調製します。アガロースが完全に溶解するまで懸濁液を加熱する。ゲル溶液を注ぐ前に、アガロース溶液100 mLごとに10 μLのEtBrを加えます。
注意:EtBrは、発がん性があることが知られているDNA挿入剤です。ドラフトの下で必要な安全対策を講じて取り扱ってください。
注:EtBr染色(ゲル中で直接または実行後)を強く推奨します。他のDNAインターカレート色素は、DNA塗抹標本を扱うときに私たちの手の中でうまく機能しませんでした。狭いローディングウェルは、幅の広いウェルと比較してより良い解像度を提供します。 - 10 μLのサンプルを2 μLの6xローディングダイと混合します。次に、10 μLのサンプルをゲルにロードし、ローディング色素の最後のバンドがゲルの2/3になるまで実行します。必ずDNAラダーを追加してください。
- ゲルを画像化し、塗抹標本のサイズが目的の用途の範囲内にあるかどうかを確認します。
クロマチンが品質管理に合格した場合、ダウンストリームアプリケーションに使用できます。
結果
クロマチンの調製は、ChIPを成功させるための重要なステップです。凍結標本から良質のクロマチンを調製するためには、効率的なせん断を妨げる可能性のある組織塊の存在を避けるために、固定前に効率的な組織破壊を確保する必要があります。図 1 に、このプロトコルのパイプラインの概要を示します。粉砕だけでは、さまざまなサイズで単一細胞がほとんどない細胞クラスターを生成するため、組織を完全に解離するには不十分です(図1a)。最初の粉砕ステップをダウンス均質化に関連付けると、組織凝集塊の量が大幅に減少し、残りの凝集塊は少なくなります(図1b)。固定および溶解ステップの後、目に見える単一核の数(図1c)は増加しますが、典型的な球形の外観は失われます。28サイクルの超音波処理の後、核染色(Hoechst 33258/DAPI)はほとんど見えなくなりました。これは確かにせん断が成功した兆候です(図1d)。クロマチンアリコートの脱架橋およびアガロースゲル上のDNAの可視化後、100〜300 bpの範囲の断片の存在によって、せん断の成功を認識することができる(図2a)DNAの量は、調製された組織片の組成に応じて変化し得る。このようなクロマチンは、ChIP-qPCRにうまく使用することができます。図2bに示すように、クロマチンは、H3K4me3、H3K27ac(活性遺伝子関連修飾)およびH3K27me3(サイレンシング遺伝子関連修飾)抗体で正常に沈殿することができました。染色体1オープンリーディングフレーム43(C1orf43)、プロテアソーム20Sサブユニットベータ2(PSMB2)およびグリセルアルデヒド3-リン酸デヒドロゲナーゼ(mGapdh)プロモーター領域は、ホメオボックスC13(HOXC13)、ホメオボックスC12(HOXC12)およびマウスミエリン転写因子1(mMyt1)プロモーター領域と比較して、H3K4me3およびH3K27acに富む結果となった(表1)。これは、C1orf43、PSMB2、およびmGapdhが肝臓で構成的に転写されるのに対し、HOXC13、HOXC12、およびmMyt1はサイレンシングされるためです。H3K27me3は反対の挙動を示し、ChIPアッセイの成功を裏付けています。これらのマウスの肝臓がキメラであるという事実は、我々がマウスとヒトの両方のクロマチンを分析することを可能にした。さらに、同じクロマチンをChIP-seq実験にうまく使用することができました。シーケンシングステップの後、リードをマウスゲノムとヒトゲノムの両方で構成されるインデックスにアラインして、アラインされていないフラグメントの量を減らしました。続いて、リードを種ごとに分離し、EaSeq22でさらに分析した。次に、すべての遺伝子の転写開始部位(TSS)でシグナル強度を測定し、その結果をH3K4me3シグナル強度でソートしました。図3aと図3cは、マウスとヒトの両方のクロマチン内の遺伝子のかなりの部分について、TSSにH3K4me3とH3K27acが顕著に存在することを示しています。それに加えて、H3K27me3はH3K4me3 / H3K27acと反相関します。H3K27me3は、このPTMから予想されるように、TSSだけでなく、遺伝子の全長に存在します。図3bおよび図3dは、H3K27me3に富み、マウスとヒトの両方の肝臓で転写的に不活性であることが知られているHOXC/HoxCクラスターを示しています。H3K4me3とH3K27acのプロファイリングでは、この2つのPTMのピークが示されていますが、H3K27me3の信号強度は低く、より分散している傾向があります。
クロマチン調製の複雑さのために、過剰固定が起こる可能性があり、溶解または超音波処理時間が最適ではない可能性があり、大細胞凝集塊が持続するか、またはサンプルの不適切な取り扱いが不十分である可能性があります。これらはすべて準備の品質に影響を与えるイベントです。場合によっては、正しいサイズ内のクロマチンフラグメントの濃縮がまだ存在するか、より高いサイズにシフトされます。他の場合には、時期尚早の溶解または失敗したせん断のために材料の損失があるかもしれません。 図4 は、このような否定的で最適ではない結果の例をいくつか示しています。レーン3と4は、200 bpから800 bpの間のフラグメントサイズの濃縮を示しています。ただし、フラグメントサイズが100 bpから>10,000 bpの範囲であることは明らかです。レーン5および6では、100〜250 bpの範囲の濃縮が存在し、調製中に材料が明らかに失われます。これは、超音波処理がより小さな断片を生成した理由を説明することができます。レーン7は、フラグメント範囲が増加したわずかに最適ではない調製を示し、レーン8は材料のほぼ完全な損失を示しています。これは、早期の核溶解または不十分な組織解離と、ステップ5.5後の結果としての喪失によって引き起こされる可能性があります。.

図1:クロマチン調製プロトコルの概要。 写真は、組織粉砕後(a)、追加の手動均質化(b)、核溶解後(c)および超音波処理後(遠心分離前)(d)に撮影した。核染色はヘキスト33258/DAPIで行った。スケールバー = 200 μm。 この図の拡大版を表示するには、ここをクリックしてください。
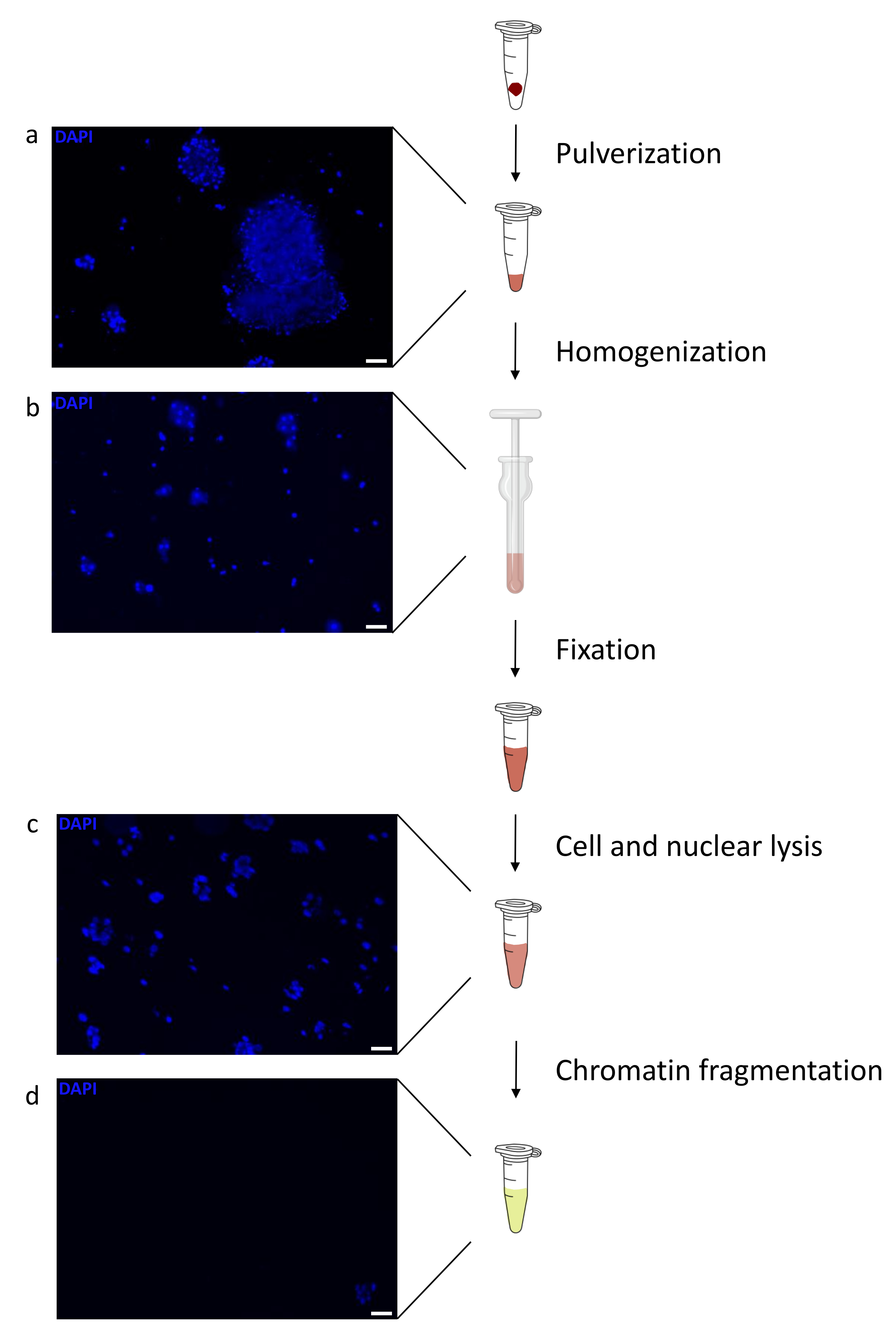{kind=link}

図2:代表的なクロマチンせん断とChIP-qPCRによるその品質評価 。 異なるクロマチン調製物のプロトコルに従って断片化されたクロマチンサンプルを含む1%アガロースゲル。クロマチン/DNA分解を事前に行わないよう、せん断されていないクロマチンのコントロールを添加します (a)。せん断クロマチンは、ChIP-qPCRアッセイを実行して品質がテストされています。H3K4me3、H3K27acおよびH3K27me3抗体を用いて、新たに調製したクロマチンを沈殿させた。( b) qPCR分析は、ヒト(C1orf43およびPSMB2)、マウス(Gapdh)活性プロモーターおよびヒト(HOXC13、HOXC12)、マウス(Myt1)不活性プロモーターについて実施した。 この図の拡大版を表示するには、ここをクリックしてください。
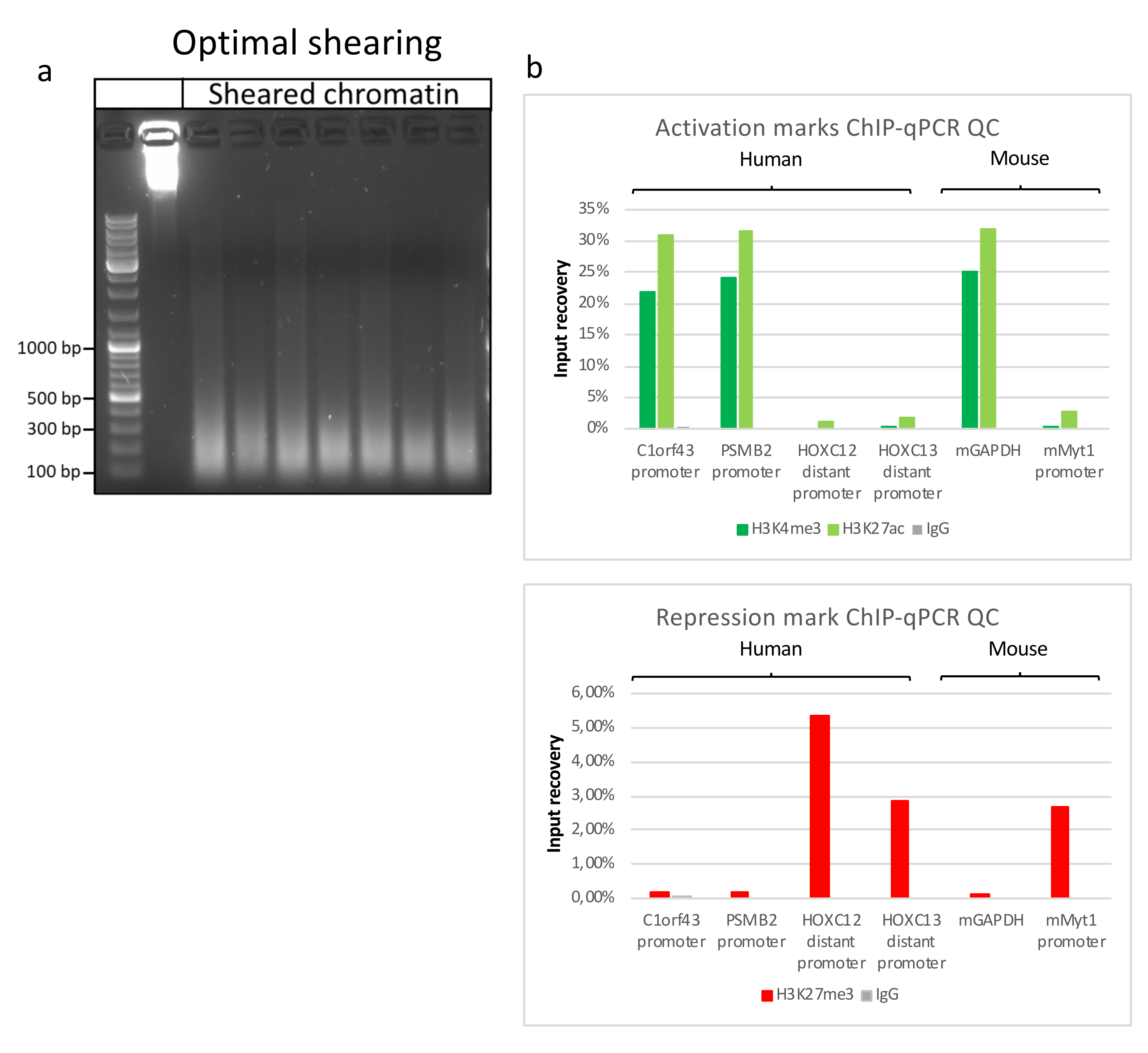{kind=link}

図3:代表的なChIP-seq分析。 リードは、ヒトとマウスの両方のゲノム(hg19およびmm10)で作成されたインデックスにアライメントされています。アライメント後、ヒトおよびマウスリードを分離し、さらに分析した。シグナルをTSSで定量し、H3K4me3強度の降順で示したヒト遺伝子のヒートマップ(a)。活性遺伝子(b)に囲まれた抑制遺伝子のヒト遺伝子クラスター(HOXクラスター)の例。シグナルをTSSで定量し、H3K4me3強度の降順で示したマウス遺伝子のヒートマップ(c)。活性遺伝子(d)に囲まれた抑制された遺伝子のマウス遺伝子クラスター(Hox cluster)の例。表示されているすべてのデータは、100 万回のリード ごとに EaSeq によって正規化されています。 この図の拡大版を表示するには、ここをクリックしてください。
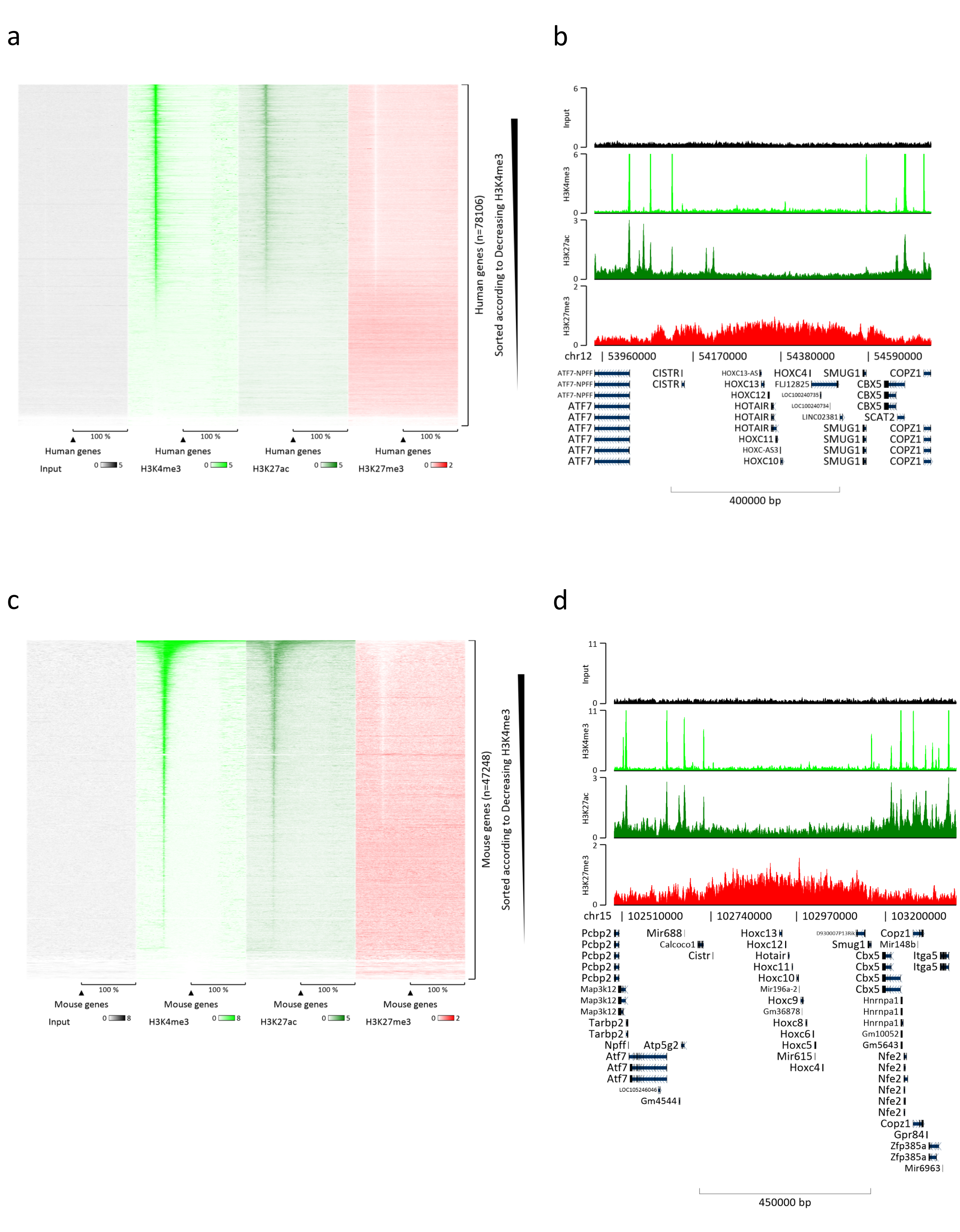{kind=link}

図4:最適ではないクロマチン調製物と失敗したクロマチン調製物。 プロトコルに従って断片化されたクロマチンサンプルを含む1%アガロースゲル。図には、対照として使用されたせん断されていないクロマチン(レーン2)、最適せん断ではない(レーン3-4)、明確な材料損失を伴う最適なせん断(レーン5-6)、最適ではないせん断(レーン7)、および広範な材料損失(レーン8)が含まれています。この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
| プライマー名 | 順序 | |
| C1orf43プロモーター | フォワード | AGTGGGGGGGGAGAATGCAGAC |
| 逆 | ガッタッカッカット | |
| PSMB2プロモーター | フォワード | CTTATTCAACCCCCCGACAAA |
| 逆 | ガ | |
| HOXC13遠位プロモーター | フォワード | GAGCCCGAGATTCACTCAAC |
| 逆 | TTATGCCCAGTTTTTGGTA | |
| HOXC12遠位プロモーター | フォワード | AAAGCTTCCCACTGCAAAGA |
| 逆 | AAATCTGGGGGGGGAACTACT | |
| mGAPDHプロモーター | フォワード | GGTCCAAAGAGAGGAGGAGAGAG |
| 逆 | GCCCTGCTTATCCAGTCCTA | |
| mMYTプロモーター | フォワード | CAGCCCAATTCTAGCCACAT |
| 逆 | CCAAGCAGGGGAGTAGGAG |
表1:ChIP-qPCRアッセイに用いた活性遺伝子および不活性遺伝子のqPCRプライマーリスト。
ディスカッション
スナップ凍結組織からのクロマチン調製は、再現性と信頼性の高い結果を得るために最適化する必要があるステップの数が多いため、依然として課題です。すでに公開されているプロトコル16、23のほとんどは、手動解離(douncing)の前に組織を細かく刻む必要があります。サンプルの固定前にタンパク質の分解を引き起こす可能性のあるステップをできるだけ避けようとしました。粉砕工程は、凍結肝臓調製物24において既に使用されており、手動解離をより容易かつ再現可能にする(図2a参照)。1.5 mLチューブ用に特別に設計されたモルタルを使用することで(プロトコルを参照)、粉砕プロセス中の検体損失が減少し、肝生検検体などの少量の組織を処理できます。原則として、粉砕ステップなしで直接組織均質化を使用することが可能です。しかし、以前の粉砕なしの組織均質化は、我々の経験では再現性が悪く、下流の用途での問題の出現が高かった(データ示さず)。
組織からクロマチンを調製する際に遭遇する問題のほとんどは、これらのサンプルの性質と、細胞クラスターが品質を損なうことなく固定するのに十分小さいかどうかを適切にチェックできないことに起因します。さらに、各ステップで各アリコートをチェックすると時間がかかり、タンパク質分解の可能性が高まります。
固定(ステップ3.9)は、クロマチン調製の基本的かつ重要な部分です。組織の性質上、組織が均質化されるまで固定ステップが遅れています。このような延期された固定工程は、より均質な細胞懸濁液を生成する利点を有する。ただし、特に操作に敏感なターゲットの場合は、ステップ3.6の直前に固定を行う必要があるかもしれないことを認識しています。これは、非常に敏感なタンパク質またはPTMを保護するのに役立ちますが、細胞クラスターのサイズを大きくする可能性がありますが、固定すると不均一なせん断が発生する可能性があります。プロトコルで使用されるFA溶液の濃度は標準ですが、全体的な固定を改善するために変更することができます。ここで選択した固定時間は、現場で一般的に使用される標準条件も反映しています。固定液の濃度が高い場合は、固定時間を短縮できますが、少量の場合は固定時間を短縮する必要があります。オペレーターは、固定時間の変更がサンプルの過剰固定につながるか、タンパク質分解の余地を与える可能性があることを考慮する必要があります。大きな複合体(またはその一部)とTFの沈殿を目的とする場合は、DSG溶液とそれに続くFA溶液25,26を使用して二重段階固定を実行することが有利です。この場合のDSGはタンパク質間相互作用を安定化させますが、ホルムアルデヒドは主に直接的なDNA-タンパク質相互作用に作用します27。
オペレーターは、ステップ6.7から開始し、より高速で有毒な化合物を使用しないDNA精製用のカラムベースのキットを実装する可能性を考慮する必要があります。ただし、失われる結合していないDNAは常に一定量存在します。このため、古典的なフェノール-クロロホルム抽出とそれに続くEtOH沈殿を使用することをお勧めします。さらに、アガロースゲルを実行する前に(ステップ7.2)、DNA濃度を測定し、すべてのウェルに同じ量をロードして、より明確な画像を得ることが有益である可能性があります。
このプロトコルの限界は、ヒト-肝臓キメラマウスに由来する肝臓標本のみを用いてこのプロトコルを探索し、利用したという事実に由来する28。それ自体、肝臓は上皮組織と結合組織で構成されています29。疾患の場合、線維性組織および脂肪組織が存在し得る30,31 組織破壊中にさらなる課題を引き起こす。しかし、我々は、我々のプロトコルは、解離と超音波処理のステップの最適化なしに骨、筋肉や脂肪組織に使用できないかもしれないことを認識しています。注意すべきことは、細胞培養サンプル15のように、それらすべてに適したプロトコルがないため、すべての組織に何らかの最適化が必要であるということです。しかし、最適化がほとんどまたはまったくなくても、このプロトコルは、肺、腸、胃、膵臓、腎臓組織など、組成が肝臓と類似している他の組織にうまく適用できると考えています。
また、HBV共有結合閉鎖DNAエピソーム(cccDNA)32のTFやヒストン修飾の解析にも使用されています。これにより、ヒトサイトメガロウイルス33 (hCMV)やヒトアデノウイルス34 (HAdV)など、肝臓に影響を与える他のウイルスゲノムにこのようなアプローチを適用する機会が開かれます。カポジ肉腫ヘルペスウイルス35 (KHSV)、単純ヘルペスウイルス36 (HSV1 / 2)ポリオーマウイルス、エプスタインバーウイルス37 (EBV)などの他の組織で持続的な感染を確立する他のDNAウイルスを分析できることを排除するものではありません。
開示事項
著者は開示するものは何もありません。
謝辞
この研究は、ドイツ研究財団(DFG)によってMaura Dandri(SFB 841 A5)への助成金によって、ハンブルク州によって研究プログラム(LFF-FV44:EPILOG)によって支援されました。
タッシロ・ヴォルツ博士、イボンヌ・ラディジェス博士、アニカ・ヴォルマリ博士に、技術的な支援と原稿の批判的読書に感謝します。トーマス・ギュンター博士とアダム・グルンドホフ教授は、ChIP-qPCR分析のための非常に役立つ提案とプライマーセットを提供してくれました。
資料
| Name | Company | Catalog Number | Comments |
| 0.22µm sterile syringe filter | Labsolute | 7699822 | |
| 1.5 mL Safeseal tubes | Sarstedt | 7,27,06,400 | |
| 6x orange loading dye | Thermofisher | R0631 | |
| Benchtop refrigerated centrifuge | |||
| Bioruptor NGS | Diagenode | ||
| Blade or Scalpel | |||
| Calcium chloride dihydrate | Carl Roth | HN04 | |
| Chloroform | Sigma Aldrich (Merck) | C2432 | |
| cOmplete Protease Inhibitor Cocktail | Roche | 11697498001 | |
| Deacetylase Inhibitor | Active Motif | 37494 | |
| Dounce tissue grinder set | Sigma Aldrich (Merck) | DWK885300-0001-1EA | |
| EDTA 500 mM solution | PanReac AppliChem | A4892 | |
| EGTA | Sigma Aldrich (Merck) | E4378 | |
| EtBr | Carl Roth | 2218 | Concentration 10mg/mL |
| Ethanol absolute | CHEMSOLUTE | 2273 | |
| Glycerol | Sigma Aldrich (Merck) | G9012 | |
| Glycin | Carl Roth | 0079 | |
| Glycogen | Roche | 10901393001 | Concentration: 20mg/mL |
| Heating block | |||
| HEPES | Sigma Aldrich (Merck) | H4034 | |
| LE Agarose | Biozym | 840000 | |
| Liquid nitrogen cooled mini mortar | Bel-Art | H37260-0100 | |
| MeOH free Formaldehyde 16% | Thermofisher | 28908 | |
| NP-40 | Roche | 11332473001 | |
| PBS 1x | Thermofisher | 10010015 | |
| Pefabloc SC-Protease-Inhibitor | Sigma Aldrich (Merck) | 11429868001 | |
| Phase Lock Gel - Heavy | QuantaBio | 2302830 | |
| Phenol:Chloroform:Isoamyl alcohol 25:24:1 | Sigma Aldrich (Merck) | P3803 | |
| Potassium chloride | Carl Roth | 6781 | |
| Potassium hydroxyde | Merck | 105033 | |
| Proteinase K | Lucigen | MPRK092 | Concentration: 50 µg/µL |
| RNAse A | Lucigen | MRNA092 | Concentration: 5 mg/mL |
| SDS 10% solution | PanReac AppliChem | A3950 | |
| Sodium carbonate anhydrous | Carl Roth | A135 | |
| Sodium chloride | Sigma Aldrich (Merck) | S7653 | |
| Sterile Petri dishes | Sarstedt | 83,39,02,500 | |
| Tris-HCl solution | Sigma Aldrich (Merck) | T2694 | |
| Triton-X100 | Sigma Aldrich (Merck) | X100 |
参考文献
- Waddington, C. H., Pantelouris, E. M. Transplantation of nuclei in newt's eggs. Nature. 172 (4388), 1050-1051 (1953).
- Silmon de Monerri, N. C., Kim, K. Pathogens hijack the epigenome: A new twist on host-pathogen interactions. American Journal of Pathology. 184 (4), 897-911 (2014).
- Knipe, D. M., et al. Snapshots: chromatin control of viral infection. Virology. 435 (1), 141-156 (2013).
- Tropberger, P., et al. Mapping of histone modifications in episomal HBV cccDNA uncovers an unusual chromatin organization amenable to epigenetic manipulation. Proceedings of the National Academy of Sciences of the United States of America. 112 (42), 5715-5724 (2015).
- Sproul, D., Gilbert, N., Bickmore, W. A. The role of chromatin structure in regulating the expression of clustered genes. Nature Reviews Genetics. 6 (10), 775-781 (2005).
- Fischle, W., Wang, Y., Allis, C. D. Histone and chromatin cross-talk. Current Opinion in Cell Biology. 15 (2), 172-183 (2003).
- Ling, X., Harkness, T. A., Schultz, M. C., Fisher-Adams, G., Grunstein, M. Yeast histone H3 and H4 amino termini are important for nucleosome assembly in vivo and in vitro: redundant and position-independent functions in assembly but not in gene regulation. Genes & Development. 10 (6), 686-699 (1996).
- Zhang, L., Eugeni, E. E., Parthun, M. R., Freitas, M. A. Identification of novel histone post-translational modifications by peptide mass fingerprinting. Chromosoma. 112 (2), 77-86 (2003).
- Wang, H., et al. Role of histone H2A ubiquitination in Polycomb silencing. Nature. 431 (7010), 873-878 (2004).
- Hassa, P. O., Haenni, S. S., Elser, M., Hottiger, M. O. Nuclear ADP-ribosylation reactions in mammalian cells: where are we today and where are we going. Microbiology and Molecular Biology Reviews. 70 (3), 789-829 (2006).
- Dey, B., et al. DNA-protein interactions: methods for detection and analysis. Molecular and Cellular Biochemistry. 365 (1-2), 279-299 (2012).
- Hager, G. L., McNally, J. G., Misteli, T. Transcription dynamics. Molecular Cell. 35 (6), 741-753 (2009).
- Nagy, Z., Tora, L. Distinct GCN5/PCAF-containing complexes function as co-activators and are involved in transcription factor and global histone acetylation. Oncogene. 26 (37), 5341-5357 (2007).
- Solomon, M. J., Larsen, P. L., Varshavsky, A. Mapping protein-DNA interactions in vivo with formaldehyde: evidence that histone H4 is retained on a highly transcribed gene. Cell. 53 (6), 937-947 (1988).
- Gunther, T., Theiss, J. M., Fischer, N., Grundhoff, A. Investigation of viral and host chromatin by ChIP-PCR or ChIP-Seq analysis. Current Protocols in Microbiology. 40, 11-21 (2016).
- Cotney, J. L., Noonan, J. P. Chromatin immunoprecipitation with fixed animal tissues and preparation for high-throughput sequencing. Cold Spring Harbor Protocols. 2015 (4), 419(2015).
- Haim, Y., Tarnovscki, T., Bashari, D., Rudich, A. A chromatin immunoprecipitation (ChIP) protocol for use in whole human adipose tissue. American Journal of Physiology-Endocrinology and Metabolism. 305 (9), 1172-1177 (2013).
- Castellano-Castillo, D., et al. Chromatin immunoprecipitation improvements for the processing of small frozen pieces of adipose tissue. PLoS One. 13 (2), 0192314(2018).
- Savic, D., Gertz, J., Jain, P., Cooper, G. M., Myers, R. M. Mapping genome-wide transcription factor binding sites in frozen tissues. Epigenetics Chromatin. 6 (1), 30(2013).
- Perna, A., Alberi, L. A. TF-ChIP method for tissue-specific gene targets. Frontiers Cell Neuroscience. 13, 95(2019).
- Allweiss, L., et al. Proliferation of primary human hepatocytes and prevention of hepatitis B virus reinfection efficiently deplete nuclear cccDNA in vivo. Gut. 67 (3), 542-552 (2018).
- Lerdrup, M., Johansen, J. V., Agrawal-Singh, S., Hansen, K. An interactive environment for agile analysis and visualization of ChIP-sequencing data. Nature Structural & Molecular Biology. 23 (4), 349-357 (2016).
- Perna, A., Alberi, L. A. TF-ChIP Method for Tissue-Specific Gene Targets. Frontiers in Cellular Neuroscience. 13, 95(2019).
- Liang, N., Fan, R., Goni, S., Treuter, E. Preparation of Frozen Liver Tissues for Integrated Omics Analysis. Methods in Molecular Biology. 1951, 167-178 (2019).
- Liu, Z., et al. Proteomic and network analysis of human serum albuminome by integrated use of quick crosslinking and two-step precipitation. Scientific Reports. 7 (1), 9856(2017).
- Singh, A. A., et al. Optimized ChIP-seq method facilitates transcription factor profiling in human tumors. Life Science Alliance. 2 (1), 201800115(2019).
- Aoki, T., et al. Bi-functional cross-linking reagents efficiently capture protein-DNA complexes in Drosophila embryos. Fly. 8 (1), 43-51 (2014).
- Allweiss, L., Dandri, M. Experimental in vitro and in vivo models for the study of human hepatitis B virus infection. Journal of Hepatology. 64, 1 Suppl 17-31 (2016).
- Krishna, M. Microscopic anatomy of the liver. Clinics in Liver Disease. 2, Suppl 1 4-7 (2013).
- Tannapfel, A., et al. Histopathological diagnosis of non-alcoholic and alcoholic fatty liver disease. Virchows Archiv. 458 (5), 511-523 (2011).
- Schuppan, D., Afdhal, N. H. Liver cirrhosis. Lancet. 371 (9615), 838-851 (2008).
- Allweiss, L., et al. Therapeutic shutdown of HBV transcripts promotes reappearance of the SMC5/6 complex and silencing of the viral genome in vivo. Gut. , 322571(2021).
- Gerna, G., Kabanova, A., Lilleri, D. Human cytomegalovirus cell tropism and host cell receptors. Vaccines. 7 (3), (2019).
- Echavarria, M. Adenoviruses in immunocompromised hosts. Clinical Microbiology Reviews. 21 (4), 704-715 (2008).
- Frohlich, J., Grundhoff, A. Epigenetic control in Kaposi sarcoma-associated herpesvirus infection and associated disease. Seminars in Immunopathology. 42 (2), 143-157 (2020).
- Nicoll, M. P., Proenca, J. T., Efstathiou, S. The molecular basis of herpes simplex virus latency. FEMS Microbiology Reviews. 36 (3), 684-705 (2012).
- Thorley-Lawson, D. A., Hawkins, J. B., Tracy, S. I., Shapiro, M. The pathogenesis of Epstein-Barr virus persistent infection. Current Opinion in Virology. 3 (3), 227-232 (2013).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved