
Method Article
Internalização e Observação de Fluorescent Biomoléculas em microorganismos vivos via Eletroporação
Neste Artigo
Resumo
Studies of biomolecules in vivo are crucial for understanding molecular function in a biological context. Here we describe a novel method allowing the internalization of fluorescent biomolecules, such as DNA or proteins, into living microorganisms. Analysis of in vivo data recorded by fluorescence microscopy is also presented and discussed.
Resumo
The ability to study biomolecules in vivo is crucial for understanding their function in a biological context. One powerful approach involves fusing molecules of interest to fluorescent proteins such as GFP to study their expression, localization and function. However, GFP and its derivatives are significantly larger and less photostable than organic fluorophores generally used for in vitro experiments, and this can limit the scope of investigation.
We recently introduced a straightforward, versatile and high-throughput method based on electroporation, allowing the internalization of biomolecules labeled with organic fluorophores into living microorganisms. Here we describe how to use electroporation to internalize labeled DNA fragments or proteins into Escherichia coli and Saccharomyces cerevisiæ, how to quantify the number of internalized molecules using fluorescence microscopy, and how to quantify the viability of electroporated cells. Data can be acquired at the single-cell or single-molecule level using fluorescence or FRET. The possibility of internalizing non-labeled molecules that trigger a physiological observable response in vivo is also presented. Finally, strategies of optimization of the protocol for specific biological systems are discussed.
Introdução
A maioria dos estudos de fluorescência dentro de células vivas depender de fusões de proteínas com proteínas fluorescentes (PQ), tais como 1 GFP. Estes marcadores fluorescentes permitir estudos do número de cópias, padrão de difusão ou localização de proteínas envolvidas em processos, tais como a expressão de genes ou de transporte de membrana 2-7. PQ oferecem alta especificidade rotulagem, de fácil aplicação, e estão disponíveis em um grande estoque de variantes com vários photophysical e propriedades químicas 1. No entanto, fluorophores orgânicos continuam a ser a principal escolha para experiências in vitro, devido à sua maior fotoestabilidade (até 100 vezes mais estável que o PQ) 8,9, tamanho pequeno (até 100 vezes menor do que o volume PQ) e facilidade de rotulagem intramolecular (principalmente através do uso de resíduos de cisteína). Todos estes factores são particularmente importantes para uma única molécula de fluorescência e FRET estuda 10.
Vários métodos de internalização combining as vantagens da rotulagem orgânica e na detecção vivo foram introduzidas ao longo da última década; No entanto, tais métodos ou empregar polipéptidos relativamente grandes etiquetas (tags por exemplo, TMP, halo, ou 20 kDa SNAP) 11-14, requerem a utilização de aminoácidos não naturais 15, ou estão limitadas a células eucarióticas, grandes único à membrana (por ex. , raspe o carregamento, seringa carregamento, microinjeção) 16-19.
Este protocolo descreve um novo, simples e de alto rendimento método internalização que acopla as vantagens de fluoróforos orgânicos com observação in vivo. Para o desenvolvimento desta técnica, foi adaptado do procedimento de electroporação comumente usado para transformar células com o plasmídeo de ADN 20,21 a fim de carregar os microrganismos, tais como E. coli ou S. cerevisiae com biomoléculas rotulados como orgânicos. O protocolo consiste em 4 passos simples: a incubação de células com biomoléculas marcadas,electroporação, a recuperação de células, e lavagem das células para remover biomoléculas não internalizado. Aqui, apresentamos este protocolo eletroporação, bem como os processos de imagem de células e de análise de dados para o estudo de fluorescência baseada em células e única molécula e traste sinais.
A electroporação depende de descarregar um campo eléctrico de alta tensão através de uma suspensão de células de força iónica baixa para formar poros da membrana através da qual transientes biomoléculas podem entrar nas células (Figura 1) 20,21. Assim como com a transformação de bactérias ou de leveduras com DNA de plasmídeo, as células têm que ser preparada antes da electroporação a garantir a sua electrocompetency. Este procedimento, que consiste em vários passos de lavagem com água, aumenta a permeabilidade da membrana e diminui a força iónica da solução de células para evitar a formação de arco eléctrico na cuvete de electroporação. Neste protocolo, as células podem ser preparadas como descrito abaixo (Veja PROTOCOLO: 1.1) ou comprados a partir de provedor comercials.

Figura 1: Representação esquemática do protocolo internalização Da esquerda para a direita:. Adicionar alguns microlitros de biomoléculas marcadas para a alíquota de células electrocompetentes (fragmentos de DNA duplamente marcada e bactérias, neste exemplo); incubar 1 a 10 min em gelo e transferir para uma cuvete de electroporação de pré-arrefecida; electroporate e depois adicionar 0,5-1 ml meio rico para as células imediatamente após; incubar a 37 ° C (ou a temperatura requerida pelo organismo, por exemplo, 29 ° C durante levedura) para permitir que as células recuperar; executar 5 passos de lavagem para remover qualquer excesso de moléculas não-internalizados marcados; ressuspender o sedimento final em 100-200 ul de tampão PBS e pipeta de 10 uL sobre uma almofada de agarose; cobrir o teclado com uma lamela e imagem limpa em um microscópio de fluorescência (no modo de campo largo ou modo HILO).
células electrocompetentes são incubadas com as biomoléculas marcadas pouco antes eletroporação, que podem ser executadas usando electroporators padrão encontrados na maioria dos laboratórios de bioquímica. Imediatamente após a electroporação, as células são incubadas em meio enriquecido que permita a sua recuperação antes da lavagem (Figura 1). O excesso de biomoléculas marcadas não-internalizados é removido em primeiro lugar por lavagem com um tampão contendo uma concentração razoavelmente elevada de sal e um pouco de detergente (Ver PROTOCOLO: 3,3). A presença de sal perturba interacções electrostáticas não-específicos formados por não internalizados biomoléculas marcadas que de outra forma podem colar na membrana exterior. De igual modo, a presença de detergente no tampão de lavagem rompe as interacções hidrófobas não específicas.
Enquanto internalização DNA é simples (Figura 2), as precauções devem ser tomadas quando internalizar proteínas marcadas utilizando eletroporação. Em primeiro lugar, a amostra estoque de proteína organicamente marcado ainda pode conter uma pequena percentagem de corante livre. As moléculas livres de corantes são muito menores do que as proteínas e pode, portanto, ser internalizada preferencialmente. Para assegurar que a vasta maioria das moléculas fluorescentes internalizadas observados correspondem à proteína de interesse, a amostra de proteína inicial deve conter menos de ~ 2% de corante livre (Figura 5) 22. O excesso de proteínas marcadas não-internalizadas também pode furar a membrana exterior da célula após a electroporação; este fenómeno é específico para a proteína e deve ser verificada para cada nova proteína. Propomos várias opções que permitem a remoção de proteínas não-internalizado a partir da amostra celular carregado (Ver PROTOCOLO: 3.3.3).
Finalmente, as células são ressuspensas num pequeno volume de tampão de fosfato e pipetou-se para uma almofada de agarose, permitindo a sua imagem através de um microscópio de fluorescência. A imobilização sobre almofadas de agarose é um simple e forma eficiente de imagiologia células em uma lamela sem prejudicar sua integridade. A almofada deve conter um meio de cultura de baixa fluorescência.
Imagens de células pode ser realizada tanto em microscopia widefield, o total de fluorescência reflexão interna (TIRF) ou utilizando HILO (altamente inclinadas e laminado folha óptica). Na configuração Hilo, o feixe de laser penetra mais profundamente do que a amostra em TIRF, ainda não ilumina toda a amostra como para campo amplo, permitindo uma maior razão sinal-para-ruído de 23. Dependendo da resolução de energia laser e tempo de utilização, biomoléculas internalizados podem ser contadas (utilizando a análise de fotobranqueamento-gradual, Figura 3), localizada, ou rastreados 24-28. Internalização de construções duplamente marcados com um par de fluoróforos FRET permite a quantificação de FRET tanto a uma única célula ou níveis de molécula única (Figura 6).
Diferentes parâmetros podem ser variadosdependendo do resultado desejado e o sistema biológico estudado. Em primeiro lugar, a quantidade de material internalizados por células pode ser ajustado através da alteração da concentração de biomoléculas marcadas adicionado à electroporação as células antes (Figura 2). A intensidade do campo electroporação também irá influenciar tanto a eficiência do carregamento e a viabilidade celular; como esperado, enquanto que a eficiência aumenta com o aumento da carga de intensidade de campo, a viabilidade das células electroporadas diminui (Figura 4A). Ambos os parâmetros podem ser quantificados por registo da percentagem de células em divisão e carregado após a electroporação. Este ensaio de viabilidade juntamente com imagens de fluorescência também verifica a observação de biomoléculas internalizados em células vivas e permite a observação contínua ao longo de várias gerações (Figura 4B).
Em resumo, este protocolo permite que a internalização de DNA e marcado por fluorescência em moléculas proteicasE. coli ou S. cerevisiae 26. Moléculas individuais marcadas com fluoróforos orgânicos podem ser rastreados com alta resolução espaço-temporal para escalas de tempo uma ordem de magnitude maior do que PQ. Finalmente, este método é compatível com widefield, TIRF e detecção confocal, bem como regimes de excitação por impulsos, tais como Alex (alternando excitação laser 28,29).
Protocolo
1. preparação celular
- Preparação de bactérias electrocompetentes feita em laboratório
- Prepare uma pré-cultura 5-10 ml durante a noite a partir de uma única colônia da estirpe de E. coli de interesse em um meio de baixa fluorescência como M9 ou EZ rico Definido Medium.
- De manhã, inocular uma nova cultura de 400 ml com a pré-cultura durante a noite de modo a que OD 600 nm começa a 0,02. Adicionar a 400 ml de baixa fluorescência média 2,5 ml de 1 M de MgSO4, e 2,5 ml de 1 M de MgCl2.
- Crescer a 37 ° C e 250 rpm até DO600 nm atingir 0,4 a 0,6.
- Pare o crescimento da cultura por arrefecimento num banho de gelo-água durante 10-15 min.
Nota: A partir de agora, realizar todas as etapas, a 4 ° C (no gelo). - Centrifuga-se a cultura de 15 min a 1000 x g. Descartar o sobrenadante e ressuspender o sedimento de células em 250 ml estéreis e refrigerada destilada H 2 O.
- Repetir a centrifugação e ressuspensão passos twice, diminuindo o volume de água para 100 ml e em seguida 50 ml.
- Centrifuga-se a cultura a 10 min a 1000 x g. Descartar o sobrenadante e ressuspender o sedimento celular em 25 ml de H destilada estéril gelada e H2O + 10% de glicerol.
- Repetir a centrifugação e ressuspensão passos três vezes, diminuindo o volume de solução de glicerol a 10% a 10 ml, 5 ml, e finalmente para 500 ul.
- Alíquota as células em alíquotas de 20 ul de cada, de flash por congelação em azoto líquido e armazenar a -80 ° C.
- Células bacterianas electrocompetentes comerciais
- Dilui-se uma alíquota de células comercial 1: 1 com água destilada estéril gelada. Faça alíquotas de 20 ul e armazenar a -80 ° C.
- Preparando levedura electrocompetentes
Nota: electrocompetentes S. cerevisiae células são preparadas antes de cada experiência de electroporação e não pode ser armazenada a -80 ° C como para E. coli.- Para começar, inocular 5Médio 0 ml YPD com uma única colônia da estirpe desejada de interesse.
- Incubar a 30 ° C e 250 rpm até a densidade óptica a 600 nm atingir 0,6 a 0,8.
- Células de centrifugação a 1000 xg durante 5 min a 4 ° C.
- Ressuspender o sedimento em 25 ml estéreis e refrigerada destilada H 2 O.
- Repetir os passos de lavagem duas vezes ressuspensão em 25 ml de água e duas vezes ressuspensão em 2 ml de uma solução gelada de sorbitol a 1 M.
- Ressuspender as células em 250 ul de sorbitol 1 M e separar as células em alíquotas de 50 ul.
2. Agarose preparação pad
- Para remover fundo partículas fluorescentes, queimar uma lamela em um forno a 500 ° C durante 1 hora. "Limpo queimado" lamelas pode ser armazenada durante semanas à temperatura ambiente, coberto com folha de alumínio.
Nota: Outros métodos de limpeza comuns, tais como a limpeza de plasma ou solução piranha pode ser usado, desde que o fundofluorescência das lâminas limpas permanece quase nulo. - Prepara-se uma solução de agarose de baixa fluorescência por fusão num forno de microondas um de agarose 2% - solução de água destilada (70 ° C). Imediatamente adicionar 500 mL da solução clara 2% agarose para 500 mL de meio 2X baixa cultura de fluorescência e misture delicadamente.
- Antes que arrefeça e endureça, prontamente pipetar este agarose - solução de meio em lâminas de microscópio (n 1,5 de espessura), a fim de formar uma camada de cerca de 2 cm de diâmetro e uma altura de poucos milímetros. Evite bolhas e pop-los com pontas de pipeta, se necessário.
- Alise a almofada com uma segunda lamela "queimado" (Sem 1,5 de espessura, veja a Figura 1).
Nota: Esta lamela superior ajuda a formar um bloco homogêneo plano e proteger da poeira e secagem, enquanto as células estão sendo preparados. Meio mínimo M9 ou como meio rico tal como o EZ rico meio definido foram testados quanto à sua baixa fluorescência.
3. Electroporatíon
- Incubação
- Adicione até 5 l de moléculas marcadas armazenados em um buffer de baixo teor de sal ( Nota: A concentração de moléculas marcadas por fluorescência nas soluções de reserva e, assim, a quantidade de moléculas marcadas adicionadas à célula antes da electroporação está directamente correlacionada com a eficiência de carga (Figura 3, e Discussão). Como algumas proteínas são menos compatíveis com a condição de baixa concentração salina, concentração de sal no tampão de armazenagem pode ser aumentada, mas o volume de moléculas marcadas adicionadas à electroporação as células anteriores, em seguida, precisa de ser diminuído.
- Transferir a mistura de células e biomoléculas marcadas numa cuvete de electroporação de pré-gelada (0,1 e 0,2 centímetros de espaçamento para as bactérias e leveduras, respectivamente). Bata levemente a tina no banco para remover quaisquer bolhas potenciais da solução.
- Coloque o cuVette no electroporator e aplicar um pulso elétrico de alta tensão para a solução (0,9-1,8 kV / cm, veja a discussão para mais detalhes sobre a escolha de tensão). Uma tal forma de impulso poros transientes nas membranas celulares, permitindo biomoléculas marcadas para difundir para dentro das células.
- Verifique se a constante de tempo exibido na electroporator é entre 4 a 6 ms. As constantes de tempo mais baixas são muitas vezes devido à concentração muito alta de sal e / ou a presença de bolhas na cuvete, e vai levar a muito baixa ou nenhuma carga das células.
- Recuperação
- Imediatamente após a electroporação, adicionar 500 mL de meio rico tal como SOC, Meio Definido EZ rico, YPD ou qualquer meio rico para as células.
- Incubar a amostra a 37 ° C para bactérias e 29 ° C durante levedura para 2 a 10 min. Para medições de viabilidade, em que o utilizador deseja avaliar a percentagem de células em crescimento e divisão após a electroporação, utilização de um tempo de recuperação mais longo (até 1 hora) como we observar tais tempos de atraso antes da primeira divisão celular.
- As etapas de lavagem
- Lavam-se as células para remover quaisquer biomoléculas não interiorizado por centrifugando as células durante 1 min a 3300 xg e 4 ° C. Descartar o sobrenadante e ressuspender as células em 500 ul de PBS.
Nota: Para cada amostra, preparar um controlo negativo das células incubadas com a mesma quantidade de biomoléculas marcadas mas não electroporadas e lavados exactamente da mesma maneira que a amostra principal. - Repita os passos anteriores 3 vezes.
- No caso de internalização proteína, optimizar o processo de lavagem de acordo com as propriedades e o comportamento da proteína marcada de interesse. Os passos seguintes são exemplo de possíveis otimizações:
- Executar os primeiros três ciclos de lavagem com PBS contendo NaCl 100 mM e 0,005% de Triton X100 para remover proteínas não-internalizado que pode furar a membrana exterior da célula 22, 26.
- Filtrar as células electroporadas com um filtro de diâmetro de poro de 0,22 um instalados no interior do tubo de microcentrífuga de 1,5 ml, pipetando as células electroporadas para dentro do filtro. Centrifugue durante 3 min a 800 xg e 4 ° C. Descartar o fluxo de passagem. Adicionar 500 mL novo PBS sobre as células e colocá-los novamente como antes e repetir estes passos uma vez 22.
- Adicionar uma pequena quantidade de protease K (10 ng em 500 ul de PBS) durante o primeiro ciclo de lavagem, para permitir a digestão de qualquer proteína não-internalizado.
- Executar os primeiros três ciclos de lavagem com PBS contendo NaCl 100 mM e 0,005% de Triton X100 para remover proteínas não-internalizado que pode furar a membrana exterior da célula 22, 26.
- Girar as células durante 1 minuto a 3300 x g e 4 ° C. Descartar o sobrenadante e ressuspender as células em 150 ul de PBS.
- Espalhar a solução da célula carregada sobre a almofada de agarose através da remoção da lamela superior e espalhando 10 ul de suspensão de células a gota por gota. Substituir uma lamela não utilizado limpo queimada (n 1,5 espessura, combinando com o microscópio especificação objetiva) em tele parte superior do teclado e pressionar muito suavemente no slide.
- Proteger as células eletroporados da luz, armazenando as almofadas em uma caixa opaca enquanto imaginando diferentes amostras.
- Lavam-se as células para remover quaisquer biomoléculas não interiorizado por centrifugando as células durante 1 min a 3300 xg e 4 ° C. Descartar o sobrenadante e ressuspender as células em 500 ul de PBS.
Aquisição de dados 4. Microscópio
Nota: em uma única célula e a microscopia de fluorescência molécula única em microrganismos vivos pode ser realizada em qualquer microscópio de fluorescência apropriado (ou comercial construído por encomenda).
- Definições
- Widefield ou HILO iluminação
- Amostras de imagem com qualquer TIRF / única molécula microscópio.
Nota: Como exemplo, usamos em laboratório um microscópio invertido personalizado com uma TIRF set-up. Vigas de um 532-nm e um laser de diodo 637 nm são combinados e colimada antes de se concentrar no plano focal de trás da objetiva. Fluorescência da amostra é coletada através do mesmo objectivo, separado da luz de excitação usando um passa-tempo e um filtro de corte, e dividido em red e verdes canais utilizando um espelho dicróico. Os dois canais são gravadas em metades separadas do chip de um (EM-CCD) com dispositivo de acoplamento de carga da multiplicação de electrões. Os vídeos são gravados utilizando o modo de cinética. Imagens de luz branca são obtidos usando uma lâmpada de luz branca e um condensador ligado ao microscópio como uma fonte de iluminação. - Para observação geral de uma única molécula, defina o modo de iluminação do microscópio para TIRF ou HILO 23 (veja a discussão para mais detalhes sobre TIRF contra HILO imaging). Para definir o modo de HILO num microscópio TIRF, diminuir ligeiramente o ângulo de incidência da luz de excitação para mudar o foco ligeiramente maior do que a superfície da lamela (imagem do interior da célula, em vez de a sua membrana inferior em contacto com a lamela, ver 4.5.4 ).
- Para a análise em nível de célula, de longa molécula única rastreamento experiências ou análise passo a passo fotodegradação, defina o modo de iluminação do microscópio para a ensuri modo widefieldng a observação contínua de todo o volume da célula e, por conseguinte, de todas as moléculas marcadas internalizadas.
- Amostras de imagem com qualquer TIRF / única molécula microscópio.
- Normalmente, os poderes uso de excitação em torno de 0,5-3 mW (~ 50-400 W / cm 2).
Nota: poderes de laser mais baixas são úteis para alcançar a observação de fluorescência de longa duração e de rastreamento (mais de 1 minuto), enquanto as potências superiores a laser pode ser necessária para maior resolução espaço-temporal ou análise fotodegradação passo a passo. - Use tempos de exposição variando de 15 ms para rastrear experimentos a 100 ms para a observação mais geral e intensidade quantificação. Nota: Outras taxas de quadro e modos podem ser utilizados, tais como a iluminação estroboscópica, particularmente para o estudo de espécies de difusão rápida 30.
- No microscópio TIRF, gravar o canal de fluorescência com uma câmara CCD de multiplicação de electrões (EMCCD) com uma ampliação, resultando em um comprimento de pixel de ~ 100 nm / pixel. A configuração TIRF é descrever em maiores detalhes na referência 26.
- Widefield ou HILO iluminação
- A aquisição de dados
- Desligar ou bloquear a iluminação por laser até que o início da experiência. Alterne o ganho da câmera EMCCD off para evitar danos na câmara devido a superexposição.
- Coloque a almofada de agarose sanduíche de cabeça para baixo sobre a platina do microscópio, com o lado coberto de células virada para baixo, a fim de trazer a célula perto do objectivo. Definir o foco sobre as células no modo de microscopia de luz transmitida 28. Grave uma imagem de cada célula de vista sob luz branca de imagem, a fim de localizar o celular esboça antes de desligar a luz branca.
- Proteger a amostra de laboratório luz ambiente.
- Para o modo de excitação Hilo, ajustar o ângulo do feixe de excitação para cada proporção máxima de sinal-para-ruído através da iluminação apenas a secção da amostra perto da superfície da lamela.
- Para alcançar a iluminação Hilo, concentrar o feixe de laser para o plano focal traseira de um 100x NA 1,4 objetivo 28 (maior numeRical aberturas tais como 1,45 ou 1,49, também são adequados). Ao deslocar a lente de focagem perpendicular ao feixe, o foco é movido para além do centro da objectiva, de modo que o feixe de sai da objectiva com um ângulo.
- Ajustar a posição da lente, a fim de maximizar a relação sinal-para-ruído, a intensidade de fluorescência intracelular contra o sinal de fundo extracelular.
- Alterne o ganho da câmera e começar a aquisição de dados antes de ligar o laser.
- Durante a gravação de dados, adquirir uma imagem de cada FOV de luz branca, antes ou depois de gravar os dados de fluorescência; Isso ajuda a identificar os limites da célula nos canais de fluorescência.
- Para a estimativa da viabilidade
- Utilizar um meio rico de baixa fluorescência na almofada de agarose para permitir que as células a crescer após a electroporação.
- Equilibrar a microscópio para a temperatura óptima para o microrganismo estudado (37 ° C para E. coli, de 29 ° C para S. cerevisiae) Com um sistema objectivo aquecedor.
- Grave tanto luz e de fluorescência imagens em branco a cada 30 min, certificando-se de permanecer exatamente no mesmo campo de visão durante toda a gravação de dados. A defasagem de ≈1 hora é tipicamente observado antes de células começam a se dividir.
- Contando o número de biomoléculas internalizadas por célula
- Defina a potência do laser para valores elevados (2-3 MW) e longo tempo de exposição (100 ms).
- Defina o modo de iluminação para widefield, a fim de iluminar toda a célula.
- Grave filmes como descrito no passo 4.2 certificando-se de gravar quadros complementares (50-100 frames) após a conclusão da fotodegradação de fluorescência.
Análise 5. Dados
- Análise geral
- Analisar as imagens gravadas e filmes, tanto em luz branca e de fluorescência excitações, usando um software de imagem, como o ImageJ software livre.
- Em ImageJ, abra as imagens ou filmes gravados no microscópio (formato TIF) em File> Open> Seu local do arquivo.
- Para comparar qualitativamente intensidades de fluorescência em uma tela de computador, certifique-se de que todas as imagens de fluorescência são exibidos com as mesmas configurações de brilho e contraste em Image> Adjust> Brightness / Contrast. Ajuste manual ou automaticamente as configurações de uma imagem selecionada, pressione "Set" e selecione a opção "propagar para todas as outras imagens".
- Defina o tipo de informação para extrair: Analisar> Definir Medidas e selecione (pelo menos) "Área", "desvio padrão", "Min & valor de cinza max" e "Mean valor de cinza".
- Para comparar intensidades de fluorescência celular, selecione a área s de interesse utilizando o botão de seleção de Freehand ImageJ e extrair as intensidades celulares em Analisar> Medida. A tabela resultante contém os valores de medição epodem ser salvas e / ou copiado para outro software. O valor "médio" corresponde à intensidade média por pixel na área seleccionada e pode ser directamente comparada entre as células ou entre uma célula e o fundo.
- Em uma amostra de células electroporadas, uma célula é considerado carregado se a sua intensidade média por pixel é maior do que a intensidade média por pixel do controlo negativo mais três vezes o seu desvio padrão (Av (I carregado célula)> Av (I -EP) + 3 * Desvio Padrão (I -EP)).
- Construir falso-coloridas imagens de fluorescência de sobreposição e filmes, a fim de avaliar a qualidade e carregamento das amostras.
- Em ImageJ, imagens de sobreposição, como o da imagem de fluorescência correspondente ao mesmo FOV em Imagem> Cores de imagem e luz branca> Mesclar canais. Selecione uma cor para cada imagem (C4 (cinza) para a luz branca, C1 (vermelho) para o canal vermelho, C2 (verde para verde canal ... etc.).
- Verificarna imagem de sobreposição que a fluorescência está localizado dentro dos limites da célula (imagem de luz branca), e que a fluorescência de fundo é baixa e homogênea (não há pontos brilhantes fora dos limites da célula).
- Antes de analisar um grande número de células, veja qualitativamente as imagens correspondentes à amostra negativa são semelhantes para as imagens das células vazias e exibir intensidades muito mais baixas do que as células electroporadas.
- Para os experimentos de viabilidade, contar manualmente os percentuais de divisão, não de divisão, mas visivelmente intacta (idênticos) e células (mortos) danificados a partir do mesmo campo de visão crescente ao longo do tempo (ver 4.2.6).
- Avaliar a viabilidade de pelo menos 200 células por amostra (electroporado, controle negativo e células vazias), a fim de reunir estatísticas suficientes.
- Analisar as imagens gravadas e filmes, tanto em luz branca e de fluorescência excitações, usando um software de imagem, como o ImageJ software livre.
- Análise baseada em celular
- Contando o número de biomoléculas internalizadas por célula por meio de análise de fotodegradação passo a passo
- Células do segmento por selecting a área de interesse, utilizando o botão de seleção de Freehand ImageJ e desenhar uma forma envolvente precisamente o celular (equivalente a membrana celular).
- Extraia as intensidades de células ao longo do tempo em Imagem> Pilhas> Perfil do eixo Z Plot. O gráfico resultante representa a intensidade média por pixel para a área dentro do limite de uma célula em relação a cada quadro de filme resultando em uma curva de fotobranqueamento para essa célula específica. Ele contém uma diminuição exponencial inicial da intensidade de células alcançar uma assíntota inferior (fundo de fluorescência). Os valores de medição e podem ser salvas e / ou copiado para outro software, clicando em "Salvar" ou "Copiar".
- Copie e cole os valores de branqueamento em uma coluna da planilha (matéria-I).
- Calcule o autofluorescência média por pixel remanescente após a fotodegradação (auto I) pela média dos valores brutos I obtidos nos últimos 50-100 quadros (a assimptota inferior).
- Subtract a autofluorescência média por pixel remanescente após a fotodegradação para essa célula a partir da curva de fotodegradação inicial: I = I branqueamento raw - I auto.
- Use a fotodegradação TimeTraces (I branqueamento contra frame) mostrando a menos de 10 passos quantificados para avaliar o tamanho médio passo (intensidade fluorophore unitária), devido ao branqueamento de um único fluorophore 26 subtraído-base.
- Avaliar o número de moléculas por célula internalizadas dividindo a intensidade inicial de células-subtraída da linha de base (I branqueamento em t = 0) pela intensidade fluoróforo unitária.
- Eficiências FRET de uma única célula
- Medir a intensidade média de células por pixel em ambos os canais de emissão de dador e aceitador (mediante doador de excitação) e no interior do limite da célula, para cada canal, como explicar em 5.1.1.4.
- Medir a intensidade média do pixel para o fundo de cada um pcAnnel de uma área em branco do slide.
- Subtrair esta intensidade de fundo a partir da intensidade média por pixel. Use estas intensidades de fluorescência subtraído-fundo para calcular FRET para cada célula através do cálculo da intensidade aceitante subtraído-fundo dividido pelo total (aceitante + doador) intensidade subtraído-fundo em cima do doador de excitação: Eu ACEITADOR / (I ACEITADOR + I doador)
- Contando o número de biomoléculas internalizadas por célula por meio de análise de fotodegradação passo a passo
- Análise de uma única molécula
- Single-molécula de rastreamento e análise de difusão
Nota: O protocolo para o acompanhamento difusão moléculas fluorescentes em células vivas e avaliar o seu coeficiente de difusão aparente foi descrita 26, 28.- Resumidamente, se encaixam as imagens de fluorophores de solteiro em cada quadro por um Gaussian elíptica 2D. Link localizada moléculas para uma faixa se eles apareceram em quadros consecutivos dentro de uma janela de 5-7 pixels (0,48-0,67 m). Use um memory parâmetro de um quadro para explicar o desaparecimento transiente de um fluoróforo ou intermitente devido à localização dispensada.
- Análise in vivo smFRET
- Identificar manualmente moléculas localizadas no interior das células a partir de filmes, passando por um filme ImageJ e identificar moléculas de imóveis (ou bastante imóveis) no canal FRET (receptor).
- Para extrair as intensidades no canal aceitador e dador que corresponde a uma molécula imóvel, seleccionar a área em torno da molécula em cada canal utilizando o botão de selecção "oval" de ImageJ (círculo em torno de cada fluoróforo única, usando um raio ~ 3-pixel) e extrair as intensidades de moléculas em Analisar> Medida. A tabela resultante contém os valores de medição e podem ser salvos e / ou copiado para outro software.
- Calcule os valores de fundo por canal a partir da intensidade média dos pixels de um círculo do mesmo tamanho em uma área em branco do slide sobre todos os quadros analisados.
- Use valores de fluorescência subtraído-fundo nos doador e receptor canais (em cima do doador de excitação) para a fluorescência e se aflige vestígios de tempo, como no de uma única célula FRET caso (ver 5.2.1.7).
Nota: análise e algoritmos automatizada e robusta foram descritas em Referências 26-28, 31.
- Single-molécula de rastreamento e análise de difusão
Resultados
A preparação das amostras
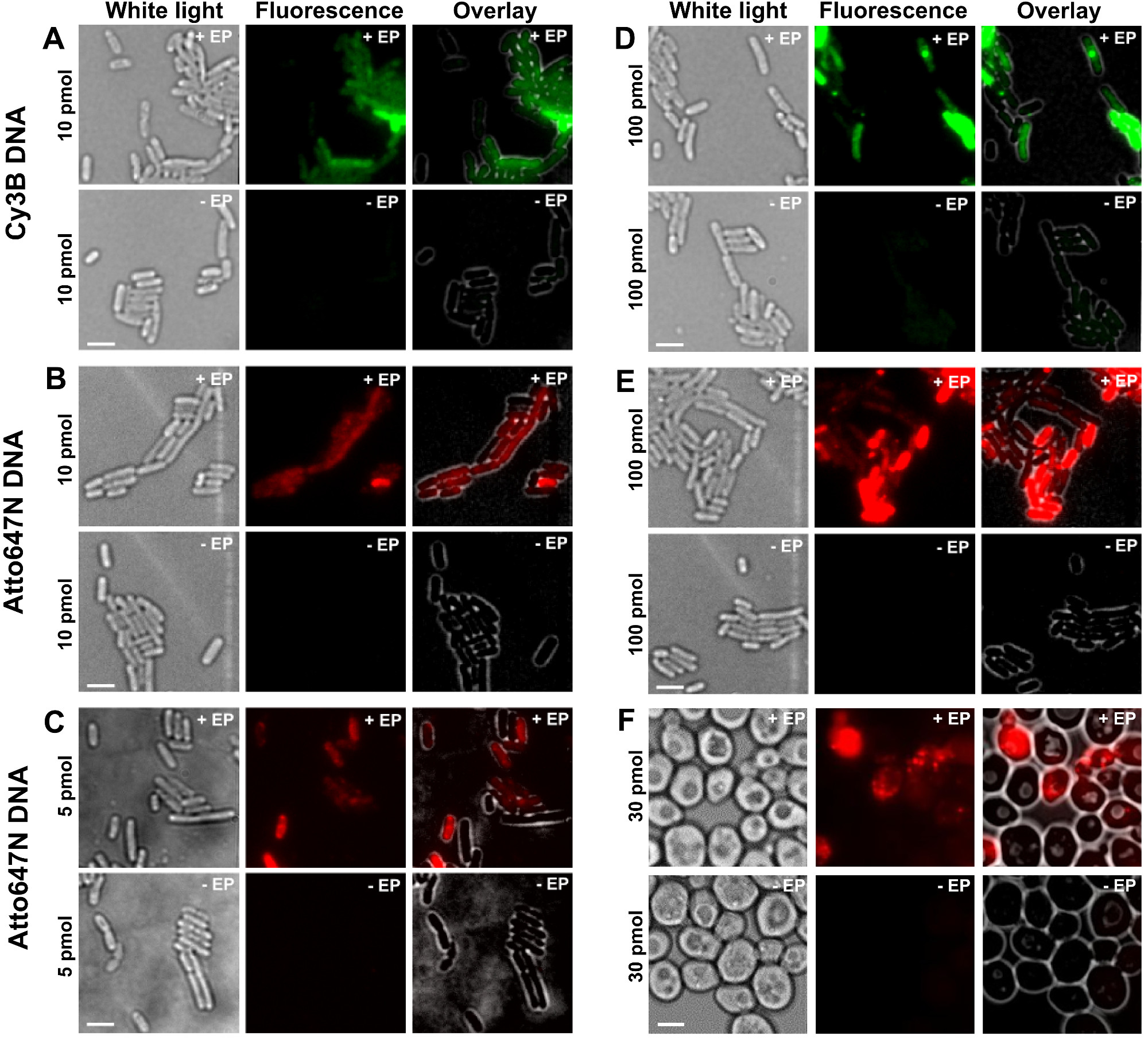
As diferentes etapas do protocolo, são apresentados como esquemas na Figura 1. A título de exemplo, que representado o carregamento de bactérias duplamente marcada com (dador e corantes aceitadores) fragmentos de ADN. Os resultados representativos para a internalização de DNA estão apresentados na Figura 2. Para cada amostra, a electroporação, os dados para as células vazias e de células não-electroporadas foram também registada (Figura 2). "As células vazias" correspondem a electrocompetentes células não incubadas com biomoléculas fluorescentes nem electroporado; sua intensidade no canal fluorescente reflecte o nível de autofluorescência sob condições idênticas experimentais (potência de laser, de resolução de tempo, temperatura, etc.). "Células não-electroporadas" (também chamado -EP, ou seja, menos do PE) correspondem a um controlo negativo no qual electrocompetentes células foram incubadas com o biomolec fluorescenteules mas não electroporado. Estas células não electroporadas deve exibir um nível de fluorescência semelhante ao autofluorescência das células vazias e significativamente menor do que a intensidade de fluorescência exibida por células electroporadas carregados. Isto confirma a remoção de quaisquer biomoléculas marcadas não-internalizadas que poderia ter aderido à membrana exterior da célula.

Figura 2: Os resultados representativos para a internalização de dsDNA marcados com diferentes fluoróforos em diferentes concentrações em bactérias (AE) e levedura (F) Da esquerda para a direita:. De luz branca, de fluorescência e de sobreposição de imagens. - / + EP indica incubação sem / com eletroporação. Barras de escala: 3 um. A. Cy3B dsDNA, 10 pmol, E. coli. B. ATTO647N dsDNA, 10 pmol, E. coli. C. Alexa647 dsDNA, 5 pmol, E. coli. D. Cy3B dsDNA, 100 pmol, E. coli. E. ds ATTO647NADN, 100 pmol, E. coli. F. ATTO647N dsDNA, 30 pmol, levedura. Esta figura foi modificado a partir da referência 26. Por favor, clique aqui para ver uma versão maior desta figura.
{kind=link}
Contando o número de biomoléculas internalizadas por célula
O procedimento para estimar o número de biomoléculas marcadas internalizados por células utilizando análise de fotobranqueamento é apresentado na Figura 3 e complementar Filme 2, juntamente com os resultados representativos obtidos com diferentes concentrações de ADN marcado. Carregamento celular eficiência aumenta com a quantidade inicial de DNA marcado incubadas, permitindo que o usuário ajustar o número de moléculas marcadas por célula a partir de um nível de "single-molécula" (<10, Recurso Filme 2B) para um nível "ensemble" (> 10 Complementar 2A Filme). Uma maneira robusta de estimar a percentagem de células carregadas é to contar o número de células electroporadas indica uma intensidade de células acima da intensidade de célula média de células não-electroporadas e mais 3 vezes o desvio padrão (isto é, Av (I-PE) + 3 * Desvio Padrão (I-PE) onde Av = média, I = intensidade por pixel, Std. Dev. = desvio padrão = não e -EP-electroporadas) como mostrado na Figura 3.

Figura 3:. A contagem do número de moléculas internalizadas por meio de análise de fotobranqueamento (A) em uma única célula fotobranqueamento análise. Exemplos de TimeTraces intensidade de fluorescência (azul: dados brutos; vermelho: ajuste; inserir: WL e imagens de fluorescência de E. coli carregado com marcado com ATTO647N dsDNA antes e após o clareamento). Top: evento de branqueamento de etapa única. Médio: célula contendo ± 3 moléculas que mostram branqueamentoe piscando. Parte inferior: célula contendo> 10 etapas correspondentes a pelo menos 10 moléculas (B) Histograma de passo único de intensidades de altura a partir de um algoritmo de ajuste de passo automatizado de 57 células contendo menos de 6 etapas distintas.. Fit Single-Gaussian está centrada a 11 ± 3 au, o que corresponde a uma intensidade fluorophore unitário de 8.100 fótons por segundo. O asterisco marca o compartimento de recolha de todas as alturas do patamar superior ou igual a 50 au (C) Histograma de moléculas internalizados por células electroporadas com diferentes quantidades de ATTO647N dsDNA, calculadas após dividindo a intensidade de fluorescência inicial pela intensidade fluoróforo unitária. De cima para baixo: as células vazias (ie, não incubadas com moléculas fluorescentes e não eletroporados), não-electroporado (mas incubadas com moléculas fluorescentes, -ep chamado), e as células eletroporados incubadas com 10 e 100 pmol dsDNA (nomeado + EP). As células vazias e não correspondem electroporadoa autofluorescência, ao passo que as células eletroporados mostrar uma ampla distribuição de moléculas internalizadas, com uma maior proporção de células altamente carregadas em 100 pmol (≥ 4 moléculas, consulte marcado asterisco bin). A eficiência de internalização (fracção de células com Int.> Média + 3x Std. Dev. De amostra não -EP) para os 10 e 100 pmol amostras foi de 94% e 90%, respectivamente. 121 ± 106 moléculas para 10 pmol dsDNA, e 176 ± 187 moléculas para 100 pmol dsDNA: número de moléculas internalizadas por célula média. Configurações: 100 ms de exposição, iluminação widefield. Barras de escala: 1 um. Esta figura foi modificado a partir da referência 26. Por favor, clique aqui para ver uma versão maior desta figura.
{kind=link}
Carregamento e viabilidade celular
Para além de alterar a quantidade de biomoléculas marcadas adicionadas às células antes da electroporação, autilizador pode ajustar a quantidade de moléculas internalizadas, escolhendo diferentes intensidades de campo durante a eletroporação (Figura 4, filme Suplementar 1). Intensidades de campo mais elevados levar a uma maior eficiência de internalização, mas levar a uma ligeira diminuição da viabilidade celular. Para internalização proteína, a utilização de um passo de filtração pode ajudar a remover as proteínas não-internalizados marcados (ver 3.3.3.1.1). Em tais casos, a filtração de célula assegura que as proteínas fluorescentes observadas são de facto internalizado no interior do citoplasma bacteriano; podemos notar, no entanto, que a filtração também tem um impacto negativo sobre a viabilidade celular (para mais detalhes, consulte REF 22).

Figura 4: Influência da tensão de electroporação após o carregamento e viabilidade celular (A) Gráfico de barras que representa o efeito de força de campo de electroporação em carga effic.iency (barras vermelhas) e a viabilidade das células (barras verdes). 84 ± 8% das células não-electroporadas (0 kV / cm) depois de 1 h fractura numa almofada de agarose a 37 ° C. Sob as mesmas condições, 78 ± 3% e 49 ± 3% de células electroporadas a 0,9 kV / cm e 1,8 kV / cm, respectivamente, dividir após 1 hr. Para uma maior eficiência de carregamento, 73 ± 8% das células são carregadas a 0,9 kV / cm, enquanto que 92 ± 6% das células são carregadas com 1,8 kV / cm. As barras de erro representam o desvio padrão calculados a partir de três medições independentes; mais do que 200 células foram analisadas para cada amostra e cada repetição. Eficiência de carregamento aumentos globais com tensão eletroporação para a ligeira detrimento da viabilidade celular. (B) medições de fluorescência baseados em células ao longo de várias gerações mostram que a intensidade global de fluorescência é dividido igualmente entre as duas células-filhas. Célula 1 e 2 refere-se ao número de células na imagem de luz branca (esquerda) e imagem de fluorescência a t = 0. Escalabar: 1 m). Esta figura foi modificado a partir da referência 26.
Internalização Protein
Os resultados representativos para a internalização de proteína estão em 5A & B. É particularmente importante remover tanto quanto do corante livre restante (não reagido) a partir da amostra de proteína quanto possível antes da electroporação. Nos exemplos nas Figuras 5A e B, o Cy3b marcado amostra Fragmento Klenow (Cy3b-KF, onde KF é o fragmento de Klenow de E. coli ADN polimerase I, 66 kDa) apenas contém corante livre 1%; tal contribuição para o corante de carga global da célula é negligenciável. As comparações da amostra de interesse electroporadas com ambas as células não-electroporadas (incubadas com a mesma quantidade de proteína marcada), bem como células electroporadas com a quantidade equivalente de corante livre constituem dois controlos necessários para assegurar que as moléculas fluorescentes observadossão, de facto internalizado proteínas marcadas.

Figura 5: internalização proteína em bactérias vivas (A) campo sobreposição fluorescência Representante de pontos de vista.. Células electroporado na tensão de 1,4 kV com 50 pmol RNAP ω subunidade partir de uma solução estoque de proteína que continha corante Cy3b livre apenas 1%. Não electroporadas (não -EP) e células vazias são definidos como anteriormente. Corante livre foi internalizado na mesma concentração que no RNAP ω amostra electroporada. Imaging no modo de campo amplo, de excitação de 532 nm a 1 mW, 50 ms exposição. (B) Distribuição de intensidade média de células não corrigidos para amostras em (A), dada em percentagem do número total de células. Mais de 400 células por amostra foram segmentadas. Esta figura foi modificado a partir da referência 22. (C) Internalização dos UNLABeled polimerase de ARN T7 (T7 RNAP, 98 kDa) em electrocompetentes DH5a carregando a pRSET-EmGFP plasmídeo que codifica GFP esmeralda (EmGFP) sob o controlo de um promotor de T7. Esquerda: Esquema do ensaio. Médio: sobreposição de fluorescência. Direita: histogramas baseados em células de intensidades de fluorescência para a amostra não electroporadas (topo) e as células incubadas e electroporadas com T7 RNAP (inferior); aproximadamente 11% das células electroporadas mostram uma elevada intensidade de fluorescência (fracção de células com Int.> média + 3x Std. Dev. de amostra não -EP) indicando expressão de EmGFP. Os asteriscos indicam bins reunindo todas as intensidades acima ou igual bar Scale 1100 au: 3 um. Esta figura foi modificado a partir da referência 26.
Figura 5C apresenta outra aplicação de eletroporação proteína. Aqui, a proteína é a electroporação não marcado, mas a sua internalização desencadeia uma resposta fluorescente observável. Esta experiência confirma a présença e funcionalidade das proteínas electroporadas no citoplasma da célula. Polimerase de ARN T7 não marcado (98 kDa) foi internalizado em E. estirpe de E. coli DH5a contendo um plasmídeo que codifica para a proteína fluorescente EmGFP sob o controlo de um promotor T7 26. Como o gene para a ARN-polimerase T7 está ausente em DH5a, expressão EmGFP nas nossas experiências que requer funcional polimerase de ARN de T7 é introduzido nas células através de electroporação (Figura 5C). Após a electroporação com 1 pmol de T7 RNAP,> 11% das células (azul barra, Figura 5C) apresentaram fluorescência maior do que o controlo negativo (incubadas com a mesma quantidade de T7 RNAP, mas não electroporadas). Este resultado estabelece que uma proporção das moléculas T7 RNAP internalizadas por eletroporação manter a sua integridade in vivo e pode executar as funções pretendidas no citoplasma da célula.
FRET in vivo na molécula únicae os níveis de uma única célula
Finalmente, a internalização e análise de espécies marcadas duplamente em bactérias vivas é apresentado na Figura 6 e complementar 3. Filme de fusões de proteínas fluorescentes Como não são ideais para estudos in vivo smFRET, a capacidade de entregar biomoléculas duplamente marcadas em células vivas utilizando electroporação é um dos grandes ativos desse método. Figura 6A apresenta uma única célula FRET análise de bactérias carregadas com padrões de ADN FRET diferentes (usando Cy3B e fluorophores ATTO647N como doador-receptor de par FRET). As células são electroporado com 20 pmol de três padrões dsDNA FRET curtos duplamente marcada com eficiência FRET aparentes (E *) de 0,17, 0,48, e 0,86 in vitro (previamente determinado 26). Todos os DNAs entrar nas células de forma eficiente (Figura 6A, à esquerda) e o pico principal de cada E distribuição de uma única célula * concorda bem com os resultados in vitro (Figura 6A, À direita). Nas amostras de intermediário e de alto FRET, populações de células com menor do que o esperado E * são observados, presumivelmente devido a uma combinação de branqueamento aceitador e inactividade fotofísicas, célula de carga variável (assim, a relação sinal-para-ruído variável) e degradação do DNA .

Figura 6:.. Os resultados representativos para uma única célula e única molécula FRET observação em bactérias que vivem Ensemble e smFRET estudos em bactérias individuais (A) Análise de células carregadas com 20 pmol de cada três padrões DNA FRET expositoras baixo (~ 0,17), intermediário (~ 0,48) e alta (~ 0,86) FRET (medida in vitro utilizando medições única molécula, ver REF 26). Esquerda: luz branca e vermelha imagens verdes / (FRET) sobreposição de fluorescência (barra de escala: 3 m). Exemplos de valores de FRET de células diferentes estão indicadas (branco). Equipamento ht (cima para baixo):. FRET baseado em células não corrigido (E *) histogramas para doador só (verde escuro), baixo (verde claro), intermediário (amarelo) e alto (vermelho) FRET padrões de ADN (B-D) In vivo smFRET. As células são carregadas com 0,25 pmol de ADN intermediária-FRET (painel B), 0,25 pmol de DNA de alta-FRET (painel C), e 5 pmol duplamente marcada KF (painel D). Coluna da esquerda: / sobreposição de fluorescência vermelha verde do único quadro antes e depois aceitante fotodegradação. Coluna do meio: os traços de tempo correspondentes à molécula no círculo amarelo. Eficiências preocupe, intensidades de emissão do doador e intensidades de emissão aceitante são exibidos em azul, verde e vermelho, respectivamente. Coluna da direita: histogramas casa da doadora apenas moléculas (verde) e moléculas de doadores-receptores (amarelo, vermelho e cinza) a partir de 20 traços de tempo para cada amostra. Barras de escala: 3 mm para A, de 1 um para B-D. Esta figura foi modificado a partir da referência 26."Target =" _ 208fig6large.jpg blank "> Clique aqui para ver uma versão maior desta figura.
Para observar smFRET in vivo para amostras de ADN ou de proteína, quantidades baixas (0,25 pmol) de padrões de ADN intermediário e de alto FRET (Figura 6B, C) ou 5 pmol de fluoróforos KF (Alexa647 / Cy3B dupla etiquetado como par de FRET, Figura 6D) são electroporados em E. coli. Tais concentrações levou a muitas células carregadas com poucas (n = 1-10) moléculas marcadas, permitindo a localização direta, rastreamento, monitoramento e se aflige por moléculas individuais. Algumas moléculas difundem-se livremente, enquanto outros aparecem imóvel ou difundir lentamente (Filme Complementar 3). TimeTraces de biomoléculas imobilizadas duplamente marcada (Figura 6, no meio) duram 1 a 30 s, e mostram as marcas da smFRET: mudanças anticorrelated na doadora e receptora de fluorescência em cima do receptor de branqueamento (por exemplo, t ~ 16 seg; Figura 6B, médio ), seguido por uma únicabranqueamento Passo-doador (por exemplo, t ~; 19 seg; Figura 6B). Distribuições FRET gerados por esses TimeTraces (Figura 6, à direita) resultam em um valor médio que está em excelente acordo com publicado em estudos in vitro 26,31,32. Estes resultados estabelecem a capacidade para estudos quantitativos sobre smFRET DNAs internalizadas e proteínas, e sugerem que as proteínas manter sua integridade e estrutura em cima de eletroporação e internalização (financiada pelos experimentos T7 RNAP internalização).
Filme Suplementar. 1: A viabilidade celular Esquerda: Imagens luz branca. Imagens de fluorescência: Certo. Animated GIF animado mostrando a divisão após a eletroporação (1,8 kV / cm) de bactérias carregadas de DNA marcado 10 pmol Atto647. A aparente diminuição global da fluorescência é em função da diluição de ADN marcada depois da divisão celular e também parcialmente para o foto-branqueamento que ocorre durante cadamedição.
Filme Complementar 2: estudos de fotodegradação baseados em células A.. Exemplo representativo de uma célula carregado (contendo> 100 moléculas de DNA marcadas com ATTO647N). Imagem de luz branca da célula de interesse (retângulo vermelho) Top esquerda,. Superior direito, filme das células carregadas mostrando seu decaimento de fluorescência ao longo de vários minutos. Inferior, traçado de tempo de decaimento da intensidade de fluorescência global da célula de interesse. Fluorophores orgânicos podem apresentar fotodegradação vidas 2 ordens de magnitude maior do que PQ (aqui, ~ 41 seg para ATTO647N). B. Exemplo representativo de uma célula carregadas com menos de 10 moléculas marcadas (3 neste caso). Topo, mesmo que o painel A. inferior, traço do tempo da intensidade de fluorescência global da célula de interesse mostrando único passo de branqueamento e / ou a piscar correspondente a fluoróforos orgânicas individuais. A altura média destes passos corresponde a uma única molécula intensidade unitária (aqui ~12 au) utilizado para calcular o número inicial de moléculas internalizadas por célula. Filmes sob contínua excitação laser vermelho em 300 mW de potência e 100 ms por quadro.
Filme Complementar 3: Eu n vivo única molécula FRET Top:. Células carregadas com 0,25 pmol high-FRET DNA (como na Figura 6C) continuamente monitorizado a 50 ms por quadro sob iluminação nTIRF usando um verde mW (532 nm) laser. Cada quadro é a / vermelho sobreposição (FRET) fluorescência verde de cada canal. Difusão e vermelho imóvel (intacto) e verde (etiqueta ativa único) moléculas de DNA podem ser observados. Traço do tempo correspondente à molécula no círculo amarelo: Inferior. Eficiências preocupe, intensidades de emissão do doador e intensidades de emissão aceitante são exibidos em azul, verde e vermelho, respectivamente. Evento Anti-correlacionados branqueamento receptor (transições vermelho-verde) corresponde à assinatura de uma única molécula FRET.
Discussão
Muitos parâmetros podem variar durante a electroporação de células e de aquisição de dados, dependendo do sistema biológico de interesse e a natureza exacta da experiência (de nível celular ou análise de uma única molécula). Por exemplo, quando electroporating ADN em bactérias de 0,25 a 5 pmol de fragmentos de dsDNA marcados conduz a uma baixa eficiência de internalização, permitindo que uma única molécula de detecção directa (isto é., Sem a necessidade de fotobranqueamento previamente). Acima de 5 pmol dsDNA, as células tendem a ser muito carregado, um regime mais adequado para a análise de uma única célula. Todos os DNAs marcados devem também ser previamente, a fim de eliminar qualquer vestígio de corante livre purificado em gel (fluoróforo não reagiu) a partir da solução de DNA. Além disso, possíveis problemas com a degradação do DNA, especialmente para experimentos smFRET, podem ser tratadas usando DNAs com ácidos nucleicos não naturais, ou motivos que protegem terminais exonuclease acessível como loops hairpin.
Outra adjustable em parâmetro de electroporação é a força do campo aplicado durante electroporação. Força Low campo (~ 1 kV / cm) vai levar a uma baixa eficiência de carga apropriado para estudos de molécula única. Maior força de campo (até 1,8 kV / cm) irá aumentar a eficiência de carregamento; No entanto, existe uma correlação inversa entre a intensidade do campo e a viabilidade celular após a electroporação (ver Figura 4). Para referência, uma intensidade de campo normal utilizado para bactérias e leveduras electroporação é ~ 1,5 kV / cm. A constante de tempo, que representa o comprimento deste deterioração, é um parâmetro conveniente para seguir, uma vez que as gotas de constantes de tempo, assim que qualquer formação de arco fenómeno ocorre na cuvete. Em configurações normais, a constante de tempo deve ser superior a 4 ms; valores mais baixos levará a baixa eficiência de carga ou células danificadas, mesmo não-carregados. A maioria dos outros electroporators oferecer graus de liberdade (tais como "truncagem de impulso" ou "forma de pulso") que pode ser modificado para ajustar tantocarregamento e viabilidade celular. Aplicou-se este método para ambas as bactérias e leveduras, contudo procedimentos semelhantes também deve permitir a internalização de biomoléculas marcadas em células de mamíferos usando o electroporador configurações adequadas desde a sua membrana é, na verdade, menos complexo (única bicamada lipídica), e uma vez que já electroporação foi usado com tais células 21.
Quando internalizar proteínas marcadas, todos corante livre precisa ser removido a partir da solução de proteína marcada de electroporação anteriores. As moléculas livres de corantes, devido ao seu menor tamanho, podem ser internalizadas preferencialmente sobre as proteínas de interesse, e são difíceis de discriminar durante a análise de dados (não obstante o seu esperado difusão mais rápida). Como um guia, para uma amostra de proteína marcada orgânica como sendo adequados para a electroporação, a quantidade de corante remanescente livre deve ser inferior a 2% (detectado utilizando um varrimento fluorescente de um SDS-PAGE) 22. Este processo é particularmente importante,como algumas moléculas pode aderir às membranas externas de bactérias ou leveduras a electroporação. A este respeito, a amostra de controlo negativo devem exibir a intensidade de fluorescência por célula claramente mais baixo do que as células electroporadas, idealmente tão baixa quanto o nível de autofluorescência das células vazias (células que não foram incubadas com quaisquer biomoléculas marcadas com fluorescência ou a electroporação, Figura 2).
Tal como acontece com dsDNA, a eficiência de internalização proteínas marcadas está relacionada com o valor de biomoléculas adicionados às células antes da electroporação. No entanto, outros parâmetros, tais como o tamanho e carga, desempenhar um papel na internalização. As pequenas proteínas apresentam alta eficiência de internalização, enquanto que as proteínas maiores (até 98 kDa) podem ser internalizados com êxito, mas com uma eficiência mais baixa (Figura 5) 26. O ponto isoelétrico da proteína, potenciais interacções com a membrana celular e os outros parâmetros físico-químicos tambémcélula de carregamento influência durante a electroporação. Como resultado, os usuários precisam otimizar experiências para o seu próprio sistema, sabendo que a concentração inicial de alta proteína marcada (> 50 M) vai lhe dar a melhor chance para o carregamento de sucesso. A electroporação também oferece uma nova ferramenta para perturbar e analisar a função celular mediante a introdução de proteínas e outras biomoléculas para as células (ou marcados ou não marcados). As experiências de ARN polimerase de T7 (Figura 5C) apresentam um exemplo de uma experiência em que se pode introduzir uma biomolécula que pode alterar a expressão de genes in vivo utilizando electroporação.
Ao realizar experiências de fluorescência de molécula única, iluminação TIR é geralmente favorecido sobre os outros modos de iluminação, uma vez que oferece a melhor relação sinal-para-ruído de apenas fluoróforos excitantes dentro de uma secção fina sobre a superfície lamela (~ 100 nm). No entanto imagem rotulada biomoléculas difundem dentro microorganismos vivos pode require iluminação mais profundas (até 0,8 mm para E. coli). Iluminação mais profundo é alcançado no modo de Hilo, preservando ao mesmo tempo uma elevada relação de sinal-para-ruído. Por outro lado, a imagem de campo largo é particularmente importante para a análise de fotobranqueamento passo a passo, em que o utilizador está a estimativa do número de moléculas internalizadas por fotobranqueamento toda uma célula carregada com elevado poder de laser e dividindo a intensidade de fluorescência celular inicial pela intensidade unitária produzida por uma única molécula (fotobranqueamento passo único, Figura 3). Widefield imagiologia também é necessário para a molécula de seguimento de longo prazo, a fim de localizar as moléculas de difusão de interesse mesmo que as suas trajectórias cobrir o volume da célula inteira.
Neste protocolo, apresentamos como eletroporação, uma técnica padrão para biólogos e bioquímicos para a entrega de ácidos nucleicos em células, constitui um método simples para a entrega de biomoléculas fluorescentes em vários tipos de células. Thé nova, a técnica de alto rendimento oferece uma ferramenta única para observar moléculas marcadas em seu ambiente nativo. Em adição biomoléculas marcadas com fluoróforos que abrange uma ampla gama de comprimentos de onda, a electroporação pode entregar moléculas modificadas com vários grupos químicos, tais como nucleótidos e aminoácidos não naturais, quelantes de metais, agentes de reticulação, e grupos obtido confinando. Se o sistema biológico de interesse não é essencial para o desenvolvimento celular, o gene que codifica para a proteína alvo pode também ser eliminado (ou batido para baixo), assegurando que as proteínas observadas após a internalização representam todos (ou mais) do banco de proteína intracelular . Em essência, eletroporação pode "transplante" a flexibilidade do in vitro bioconjugates em células vivas e, portanto, beneficiar esforços em biologia sintética, biologia de sistemas, e in vivo de detecção de uma única molécula.
Divulgações
The authors have nothing to disclose.
Agradecimentos
We thank Stephan Uphoff for discussions.
R.C. was supported by Linacre College, Oxford University. A.P. was supported by the German Academic Exchange Service (DAAD), the German National Academic Foundation and EPSRC. M.S. was supported by the Wellcome Trust. A.N.K. was supported by a UK BBSRC grant (BB/H01795X/1), and a European Research Council Starter grant (261227).
Materiais
| Name | Company | Catalog Number | Comments |
| ElectroMax DH5-alpha Comptent cells | Invitrogen | 11319-019 | or any other commercial or lab-mage electrocompetant bacteria or yeast. |
| EZ Rich Defined Madia | Teknova | M2105 | low fluorescence rich media |
| MicroPulser Electroporation Apparatus | Biorad | 165-2100 | or any classical electroporator for microorganism transformation |
| Certified Molecular Biology agarose | Biorad | 161-3100 | low fluorescence agarose for agarose pad |
| Microscope coverslips No 1.5 thickness | Menzel | BB024060SC | remove background particles by heating slides in furnace at 500 °C for 1h |
| Single-molecule fluorescence microscope | Home-built | described in REFs | |
| Localization software | Custom-written, available online | MATLAB and C++ software package that can be adapted for localization analysis. | |
| Tracking software | Available online | MATLAB implementation by Blair and Dufresne. |
Referências
- Tsien, R. Y. The green fluorescent protein. Annu Rev Biochem. 67, 509-544 (1998).
- Leake, M. C., et al. Stoichiometry and turnover in single, functioning membrane protein complexes. Nature. 443, 355-358 (2006).
- Taniguchi, Y., Kawakami, M. Application of HaloTag protein to covalent immobilization of recombinant proteins for single molecule force spectroscopy. Langmuir. 26, 10433-10436 (2010).
- Xie, X. S., Choi, P. J., Li, G. W., Lee, N. K., Lia, G. Single-molecule approach to molecular biology in living bacterial cells. Annual review of biophysics. 37, 417-444 (2008).
- Lee, J. H., et al. Highly multiplexed subcellular RNA sequencing in situ. Science. 343, 1360-1363 (2014).
- Miesenbock, G., De Angelis, D. A., Rothman, J. E. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 394, 192-195 (1998).
- Sauer, M. Localization microscopy coming of age: from concepts to biological impact. J Cell Sci. 126, 3505-3513 (2013).
- Dempsey, G. T., Vaughan, J. C., Hao Chen, K., Zhuang, X. Evaluation of fluorophores for optimal performance in localizationbased super-resolution imaging. Nat Meth. 8, 1027-1041 (2011).
- Shaner, N. C., Steinbach, P. A., Tsien, R. Y. A guide to choosing fluorescent proteins. Nat Meth. 2, 905-909 (2005).
- Landgraf, D., Okumus, B., Chien, P., Baker, T. A., Paulsson, J. Segregation of molecules at cell division reveals native protein localization. Nat. Methods. 9, 480-482 (2012).
- Jaitin, D. A., et al. Massively Parallel Single-Cell RNA-Seq for Marker-Free Decomposition of Tissues into Cell Types. Science. 343, 776-779 (2014).
- Aldridge, S., et al. AHT-ChIP-seq: a completely automated robotic protocol for high-throughput chromatin immunoprecipitation. Genome Biol. 14, R124 (2013).
- Keppler, A., et al. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat Biotechnol. 21, 86-89 (2003).
- Wombacher, R., et al. Live-cell super-resolution imaging with trimethoprim conjugates. Nat. Methods. 7, 717-719 (2010).
- Zhang, Z., et al. A new strategy for the site-specific modification of proteins in vivo. Biochemistry. 42, 6735-6746 (2003).
- McNeil, P. L., Murphy, R. F., Lanni, F., Taylor, D. L. A method for incorporating macromolecules into adherent cells. J Cell Biol. 98, 1556-1564 (1984).
- Clarke, M. S., McNeil, P. L. Syringe loading introduces macromolecules into living mammalian cell cytosol. J Cell Sci. 102, 533-541 (1992).
- Sakon, J. J., Weninger, K. R. Detecting the conformation of individual proteins in live cells. Nat. Methods. 7, 203-205 (2010).
- Taylor, L. S. Electromagnetic syringe. IEEE Trans. Biomed. Eng. 25, 303-304 (1978).
- Dower, W. J., Miller, J. F., Ragsdale, C. W. High efficiency transformation of E. coli by high voltage electroporation. Nucleic Acids Res. 16, 6127-6145 (1988).
- Neumann, E., Schaefer-Ridder, M., Wang, Y., Hofschneider, P. H. Gene transfer into mouse lyoma cells by electroporation in high electric fields. EMBO J. 1, 841-845 (1982).
- Sustarsic, M., et al. Optimized delivery of fluorescently labeled proteins in live bacteria using electroporation. Histochem Cell Biol. , (2014).
- Tokunaga, M., Imamoto, N., Sakata-Sogawa, K. Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat. Methods. 5, 159-161 (2008).
- Sinha, A., et al. A cascade of DNA-binding proteins for sexual commitment and development in Plasmodium. Nature. 000, 1-5 (2014).
- English, B. P., et al. Single-molecule investigations of the stringent response machinery in living bacterial cells. Proc Natl Acad Sci U S A. 108, E365-E373 (2011).
- Crawford, R., et al. Long-lived intracellular single-molecule fluorescence using electroporated molecules. Biophys J. 105, 2439-2450 (2013).
- Uphoff, S., Reyes-Lamothe, R., Garza de Leon, F., Sherratt, D. J., Kapanidis, A. N. Single-molecule DNA repair in live bacteria. Proc Natl Acad Sci U S A. 110, 8063-8068 (2013).
- Uphoff, S., Sherratt, D. J., Kapanidis, A. N. Visualizing Protein-DNA Interactions in Live Bacterial Cells Using Photoactivated Single-molecule Tracking. J Vis Exp. , (2014).
- Hohlbein, J., Gryte, K., Heilemann, M., Kapanidis, A. N. Surfing on a new wave of single-molecule fluorescence methods. Phys Biol. 7, 031001 (2010).
- Xie, X. S., Yu, J., Yang, W. Y. Perspective - Living cells as test tubes. Science. 312, 228-230 (2006).
- Santoso, Y., et al. Conformational transitions in DNA polymerase I revealed by single-molecule FRET. Proc Natl Acad Sci U S A. 107, 715-720 (2010).
- Hohlbein, J., et al. Conformational landscapes of DNA polymerase I and mutator derivatives establish fidelity checkpoints for nucleotide insertion. Nature communications. 4, 2131 (2013).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados