
Method Article
Internalizzazione e di osservazione di Fluorescent Biomolecole in Living microrganismi via elettroporazione
In questo articolo
Riepilogo
Studies of biomolecules in vivo are crucial for understanding molecular function in a biological context. Here we describe a novel method allowing the internalization of fluorescent biomolecules, such as DNA or proteins, into living microorganisms. Analysis of in vivo data recorded by fluorescence microscopy is also presented and discussed.
Abstract
The ability to study biomolecules in vivo is crucial for understanding their function in a biological context. One powerful approach involves fusing molecules of interest to fluorescent proteins such as GFP to study their expression, localization and function. However, GFP and its derivatives are significantly larger and less photostable than organic fluorophores generally used for in vitro experiments, and this can limit the scope of investigation.
We recently introduced a straightforward, versatile and high-throughput method based on electroporation, allowing the internalization of biomolecules labeled with organic fluorophores into living microorganisms. Here we describe how to use electroporation to internalize labeled DNA fragments or proteins into Escherichia coli and Saccharomyces cerevisiæ, how to quantify the number of internalized molecules using fluorescence microscopy, and how to quantify the viability of electroporated cells. Data can be acquired at the single-cell or single-molecule level using fluorescence or FRET. The possibility of internalizing non-labeled molecules that trigger a physiological observable response in vivo is also presented. Finally, strategies of optimization of the protocol for specific biological systems are discussed.
Introduzione
La maggior parte degli studi di fluorescenza all'interno delle cellule viventi dipendono fusioni di proteine con proteine fluorescenti (PQ), come la GFP 1. Questi tag fluorescenti permettono studi il numero di copie, modello di diffusione o la localizzazione di proteine coinvolte in processi come l'espressione del gene o il trasporto di membrana 2-7. PQ offrono alta specificità etichettatura, facile implementazione, e sono disponibili in una grande inventario di varianti con diversi fotofisiche e chimiche 1. Tuttavia, fluorofori organici rimangono la prima scelta per gli esperimenti in vitro a causa della loro maggiore fotostabilità (fino a 100 volte più stabile rispetto PQ) 8,9, di piccole dimensioni (fino a 100 volte il volume più piccolo di PQ) e la facilità di etichettatura intramolecular (principalmente attraverso l'uso di residui di cisteina). Tutti questi fattori sono particolarmente importanti per la singola molecola fluorescenza e FRET 10 studi.
Diversi metodi di internalizzazione combining i vantaggi di etichettatura biologica e nel rilevamento vivo sono state introdotte negli ultimi dieci anni; tuttavia, tali metodi o impiegano polipeptidi relativamente grandi etichette (tag ad esempio, TMP, HALO, o 20 kDa SNAP) 11-14, richiede l'uso di aminoacidi naturali 15, o si limitano a grandi, single-membrana delle cellule eucariotiche (ad es. , raschiare il carico, la siringa di carico, microiniezione) 16-19.
Questo protocollo descrive un romanzo, semplice e high-throughput metodo interiorizzazione che le coppie i vantaggi di fluorofori organici con l'osservazione in vivo. Per sviluppare questa tecnica, abbiamo adattato la procedura di elettroporazione comunemente usato per trasformare cellule con DNA plasmidico 20,21 per caricare microrganismi, come E. coli o S. cerevisiae con biomolecole etichettati organicamente. Il protocollo si compone di 4 semplici passi: l'incubazione di cellule con biomolecole marcate,elettroporazione, recupero di cellule, e il lavaggio delle cellule per rimuovere biomolecole non interiorizzato. Qui, vi presentiamo questo protocollo elettroporazione, così come i processi di imaging cellulare e di analisi dei dati per lo studio di fluorescenza delle cellule-based e singola molecola e Fret segnali.
Elettroporazione si basa su scaricando un campo elettrico ad alta tensione attraverso una sospensione di cellule forza ionica basso per formare pori della membrana transitori attraverso cui biomolecole possono entrare cellule (Figura 1) 20,21. Come con la trasformazione di batteri o lieviti con DNA plasmidico, le cellule devono essere preparati prima elettroporazione per garantire la loro electrocompetency. Questa procedura, composti di più fasi di lavaggio con acqua, aumenta la permeabilità della membrana e riduce la forza ionica della soluzione cellulare per evitare la formazione di archi nella cuvetta elettroporazione. In questo protocollo, le cellule possono essere preparati come descritto di seguito (Vedi PROTOCOLLO: 1.1) o acquistati dai provider commerciales.

Figura 1: Rappresentazione schematica del protocollo interiorizzazione Da sinistra a destra:. Aggiungere qualche microlitri di biomolecole etichettati alla aliquota di cellule electrocompetent (frammenti di DNA doppiamente marcati e batteri in questo esempio); incubare 1 a 10 min in ghiaccio e versare in un elettroporazione cuvetta pre-raffreddata; elettroporazione e quindi aggiungere 0,5-1 ml terreno ricco alle cellule immediatamente dopo; incubare a 37 ° C (o la temperatura richiesta dall'organismo, ad esempio, 29 ° C per il lievito) per permettere alle cellule recuperare; effettuare 5 fasi di lavaggio per eliminare ogni-non interiorizzate molecole marcate in eccesso; risospendere il pellet finale in 100-200 ml di tampone PBS e pipetta 10 ml su un pad agarosio; coprire il pad con un coprioggetto pulito e immagine su un microscopio a fluorescenza (in modalità campo largo o modalità HILO).
celle Electrocompetent vengono incubate con le biomolecole marcate poco prima elettroporazione, che possono essere eseguite utilizzando elettroporatori standard che si trovano nella maggior parte dei laboratori di biochimica. Immediatamente dopo l'elettroporazione, le cellule sono incubate in un terreno ricco permettendo loro recupero prima del lavaggio (Figura 1). L'eccesso di biomolecole etichettati non internalizzati viene prima rimosso mediante lavaggio in un tampone contenente un piuttosto elevata concentrazione di sale e detersivo (Vedere PROTOCOLLO: 3.3). La presenza di sale interrompe non specifici interazioni elettrostatiche formate da biomolecole etichettati non internalizzati che altrimenti potrebbero attaccarsi sulla membrana esterna. Analogamente, la presenza di detersivo nel tampone di lavaggio interrompe non specifiche interazioni idrofobiche.
Mentre internalizzazione DNA è semplice (figura 2), le precauzioni devono essere prese quando interiorizzare proteine marcate con elettroporazione. Innanzitutto, il campione di proteina marcata azione biologica potrebbe ancora contenere una piccola percentuale di colorante libero. Molecole di colorante liberi sono molto più piccoli di proteine e possono quindi essere internalizzati preferenzialmente. Per garantire che la stragrande maggioranza delle molecole fluorescenti internalizzati osservati corrispondono alla proteina di interesse, il campione iniziale proteine deve contenere meno di ~ 2% colorante libero (Figura 5) 22. L'eccesso di proteine marcate non internalizzati può anche aderire alla membrana cellulare esterna dopo l'elettroporazione; questo fenomeno è specifico di proteine e deve essere controllata per ogni nuova proteina. Proponiamo diverse opzioni che consentono la rimozione delle proteine non interiorizzato dal campione di cellule caricato (Vedere PROTOCOLLO: 3.3.3).
Infine, le cellule sono risospese in un piccolo volume di tampone fosfato e pipettati su un pad agarosio, permettendo loro di imaging su un microscopio a fluorescenza. Immobilizzazione sui rilievi agarosio è un simple e modo efficace di formazione immagine celle su un copri senza danneggiare la loro integrità. Il tampone dovrebbe contenere un terreno di coltura a bassa fluorescenza.
L'imaging cellulare può essere effettuata sia in widefield, riflessione interna totale in fluorescenza (TIRF) oppure utilizzando HILO (fortemente inclinato e laminato Sheet Optical) microscopia. Nella configurazione HILO, il raggio laser penetra più profondamente nel campione che in TIRF, ma non illuminare l'intero campione da per widefield, consentendo un maggior rapporto di 23 segnale-rumore. A seconda della risoluzione potenza del laser e l'ora utilizzate, biomolecole internalizzati possono essere contati (mediante analisi stepwise photobleaching, Figura 3), localizzati o tracciati 24-28. Internalizzazione dei costrutti doppiamente etichettati con un paio di FRET fluorofori consente la quantificazione di FRET sia a singola cellula o livelli di singola molecola (Figura 6).
Diversi parametri possono essere variatia seconda dell'uscita desiderata e il sistema biologico studiato. In primo luogo, la quantità di materiale internalizzato per cella può essere regolato modificando la concentrazione di biomolecole etichettati aggiunto al cellule prima elettroporazione (Figura 2). Intensità di campo elettroporazione influenzerà anche sia l'efficienza di carico e la vitalità cellulare; come previsto, mentre il rendimento di carico aumenta con l'aumentare intensità di campo, la vitalità delle cellule elettroporate diminuisce (Figura 4A). Entrambi i parametri possono essere quantificate registrando la percentuale di carico e di divisione delle cellule dopo l'elettroporazione. Questo test vitalità accoppiata con imaging di fluorescenza verifica anche l'osservazione di biomolecole internalizzate nelle cellule viventi e lasciare continua osservazione per diverse generazioni (Figura 4B).
In sintesi, questo protocollo permette l'internalizzazione delle contrassegnati in modo fluorescente di DNA e proteine in molecoleE. coli o S. CEREVISIAE 26. Molecole individuali marcate con fluorofori organici possono essere monitorati con alta risoluzione spazio-temporale per tempi un ordine di grandezza superiore a PQ. Infine, questo metodo è compatibile con widefield, TIRF e rilevazione confocale, nonché sistemi di eccitazione a impulsi, come ALEX (alternata laser di eccitazione 28,29).
Protocollo
Preparazione 1. Cella
- Preparazione di batteri electrocompetent laboratorio artigianale
- Preparare una precoltura 5-10 ml notte da una singola colonia del ceppo E.coli di interesse in un mezzo a bassa fluorescenza come M9 o EZ Rich Definito Medium.
- Al mattino, inoculare una nuova cultura di 400 ml con la precoltura durante la notte in modo che OD 600 nm inizia alle 0.02. Aggiungere ai 400 ml di bassa fluorescenza media 2,5 ml di 1 M MgSO 4 e 2,5 ml di 1 M MgCl 2.
- Grow a 37 ° C e 250 rpm fino OD 600 nm raggiunge 0,4 e 0,6.
- Arrestare la crescita raffreddando cultura in un bagno di ghiaccio-acqua per 10-15 min.
Nota: Da ora in poi, effettuare tutte le fasi a 4 ° C (su ghiaccio). - Centrifugare cultura 15 min a 1000 x g. Eliminare il surnatante e risospendere il pellet cellulare in 250 ml refrigerati e sterile distillata H 2 O.
- Ripetere la centrifugazione e risospensione passi tWICE, diminuendo il volume di acqua da 100 ml e poi 50 ml.
- Centrifugare cultura 10 min a 1000 x g. Eliminare il surnatante e risospendere il pellet cellulare in 25 ml di freddo e sterile H 2 O distillata + 10% glicerolo.
- Ripetere la centrifugazione e risospensione passi tre volte, riducendo il volume di soluzione di glicerolo 10% a 10 ml, 5 ml, e infine a 500 microlitri.
- Aliquota le cellule in aliquote di 20 microlitri ciascuna, Flash-congelare in azoto liquido e conservare a -80 ° C.
- Cellule batteriche electrocompetent commerciali
- Diluire un'aliquota cellulare commerciale 1: 1 con acqua distillata fredda sterile. Preparare delle aliquote 20 microlitri e conservare a -80 ° C.
- Preparazione lievito electrocompetent
Nota: Electrocompetent S. cellule cerevisiae sono preparati prima di ogni esperimento elettroporazione e non possono essere conservati a -80 ° C, come per E. coli.- Per iniziare, inoculare 5Media 0 ml YPD con una singola colonia del ceppo desiderato di interesse.
- Incubare a 30 ° C e 250 rpm fino alla OD 600 nm raggiunge 0,6 e 0,8.
- Cellule centrifugare a 1000 xg per 5 minuti a 4 ° C.
- Risospendere il pellet in 25 ml refrigerati e sterile distillata H 2 O.
- Ripetere le fasi di lavaggio due volte risospendere in 25 ml di acqua e risospendere due volte in 2 ml di una soluzione raffreddata di sorbitolo al 1 M.
- Risospendere le cellule in 250 ml di 1 M sorbitolo e dividere le celle in 50 microlitri aliquote.
2. Agarose preparazione pad
- Per rimuovere le particelle fluorescenti sfondo, bruciare un coprioggetto in un forno a 500 ° C per 1 ora. "Clean bruciato" vetrini possono essere conservati per settimane a temperatura ambiente rivestito in foglio di alluminio.
Nota: Altri metodi di pulizia comuni quali la pulitura al plasma o soluzione Piranha potrebbero essere utilizzati purché sfondofluorescenza delle diapositive puliti rimane quasi nullo. - Preparare una soluzione di agarosio a bassa fluorescenza per fusione in un forno a microonde un agarosio 2% - soluzione di acqua distillata (70 ° C). Aggiungere immediatamente 500 ml di una chiara soluzione di agarosio 2% a 500 ml di 2X terreno di coltura a basso fluorescenza e mescolare delicatamente.
- Prima si raffredda e si induriscono, prontamente pipettare questo agarosio - soluzione media su un vetrino coprioggetto microscopio (n 1,5 spessore) per formare un tampone di circa 2 centimetri di diametro e un'altezza di pochi millimetri. Evitare di bolle e pop con punte per pipette, se necessario.
- Appiattire il pad con un secondo coprioggetto "bruciato" (No spessore 1.5, vedi Figura 1).
Nota: Questo coverslip superiore aiuta a formare un pad omogeneo piatta e protegge da polvere e asciugatura, mentre le cellule sono in preparazione. Terreno minimo come M9 o ricchi mezzo come EZ Rich Media Definito sono state testate per la loro bassa fluorescenza.
3. Electroporatione
- Incubazione
- Aggiungere fino a 5 ml di molecole marcate memorizzati in un buffer di basso contenuto di sale ( Nota: La concentrazione di molecole fluorescente nelle soluzioni madre e quindi la quantità di molecole marcate aggiunti alla cella prima elettroporazione è direttamente correlata con l'efficienza di carico (Figura 3, e Discussione). Poiché alcune proteine sono meno compatibili con condizione di basso sale, la concentrazione di sale nel buffer di memorizzazione può essere aumentato, ma il volume di molecole marcate aggiunti al cellule prima elettroporazione deve poi essere diminuito.
- Trasferire la miscela di cellule e biomolecole marcate nella cuvetta elettroporazione pre-raffreddata (0,1 e 0,2 cm di distanza per batteri e lieviti, rispettivamente). Battere delicatamente la cuvetta in panchina per eliminare eventuali bolle possibili dalla soluzione.
- Posizionare il cuvette nel elettroporatore e applicare un impulso elettrico ad alta tensione per la soluzione (0,9-1,8 kV / cm, vedi la discussione per maggiori dettagli sulla scelta di tensione). Tale impulso di forma pori transitori nelle membrane cellulari consentono biomolecole etichettati per diffondere nelle cellule.
- Controllare che la costante di tempo visualizzato sul elettroporatore è da 4 a 6 ms. Costanti di tempo inferiori sono spesso causa di concentrazione di sale troppo alta e / o la presenza di bolle nella cuvetta, e porteranno a molto bassa o nulla carico delle cellule.
- Recupero
- Immediatamente dopo l'elettroporazione, aggiungere 500 ml di terreno ricco come SOC, Piano EZ Rich Definito, YPD o qualsiasi mezzo ricco di cellule.
- Incubare il campione a 37 ° C per i batteri e 29 ° C per il lievito per 2 a 10 min. Per misure di vitalità, in cui l'utente vuole valutare la percentuale di cellule in crescita e dividendo dopo elettroporazione, utilizzare un tempo di recupero più lungo (fino a 1 ora) come we osservare tali tempi di latenza prima della prima divisione cellulare.
- Fasi di lavaggio
- Lavare le cellule per rimuovere eventuali biomolecole non internalizzato riducendo la velocità le cellule per 1 min a 3300 xg e 4 ° C. Eliminare il surnatante e risospendere le cellule in 500 microlitri PBS.
Nota: Per ogni campione, preparare un controllo negativo di cellule incubate con la stessa quantità di biomolecole etichettati ma non elettroporate e lavati esattamente allo stesso modo del campione principale. - Ripetere i passaggi precedenti per 3 volte.
- Nel caso di proteine internalizzazione, ottimizzare la procedura di lavaggio a seconda delle proprietà e il comportamento della proteina marcata di interesse. Le seguenti operazioni sono esempio di possibili ottimizzazioni:
- Eseguire i primi 3 cicli di lavaggio utilizzando PBS contenente NaCl 100 mM e 0,005% Triton X100 per rimuovere le proteine non internalizzato che potrebbero attaccare alla membrana cellulare esterna 22, 26.
- Filtrare le cellule elettroporate con un filtro di diametro dei pori di 0,22 micron montato all'interno di una provetta da 1,5 ml microcentrifuga pipettando le cellule elettroporate nel filtro. Spin giù per 3 minuti a 800 xg e 4 ° C. Gettare il flow-through. Aggiungere 500 microlitri nuovo PBS sulle celle e girare loro ancora una volta come prima e ripetere la procedura, una volta 22.
- Aggiungere una piccola quantità di proteasi K (10 ng in 500 microlitri di PBS) durante il primo ciclo di lavaggio per consentire la digestione di qualsiasi proteina non-internalizzato.
- Eseguire i primi 3 cicli di lavaggio utilizzando PBS contenente NaCl 100 mM e 0,005% Triton X100 per rimuovere le proteine non internalizzato che potrebbero attaccare alla membrana cellulare esterna 22, 26.
- Spin giù le cellule per 1 minuto a 3300 xg e 4 ° C. Eliminare il surnatante e risospendere le cellule in 150 ml di PBS.
- Distribuire la soluzione di cellule caricate sul pad agarosio rimuovendo il coprioggetto superiore e diffusione 10 ml di sospensione cellulare per gocciolina gocciolina. Sostituire un coprioggetto inutilizzato pulito bruciato (No spessore 1,5, corrispondente al microscopio specifica oggettiva) su tegli parte superiore del pad e premere delicatamente sul vetrino.
- Proteggere le cellule elettroporate dalla luce memorizzando le pastiglie in una scatola opaca mentre l'imaging diversi campioni.
- Lavare le cellule per rimuovere eventuali biomolecole non internalizzato riducendo la velocità le cellule per 1 min a 3300 xg e 4 ° C. Eliminare il surnatante e risospendere le cellule in 500 microlitri PBS.
Acquisizione dati 4. Microscopio
Nota: Single-cell e singola molecola microscopia a fluorescenza in microrganismi viventi possono essere eseguite su qualsiasi microscopio a fluorescenza appropriate (su misura o commerciale).
- Impostazioni
- Widefield o HILO illuminazione
- Campioni di immagine con qualsiasi microscopio TIRF / singola molecola.
Nota: Come esempio, si usa in laboratorio un microscopio invertito personalizzato con un TIRF set-up. Travi da un 532 nm e un diodo laser 637 nm vengono combinati e collimati prima focalizzazione sul piano focale posteriore dell'obiettivo. Fluorescenza dal campione viene raccolto attraverso lo stesso obiettivo, separato dalla luce di eccitazione utilizzando una lunga-pass e un filtro notch, e suddiviso in recanali d e verde utilizzando uno specchio dicroico. I due canali vengono esposte sulla metà separate del chip di un (EM-CCD) fotocamera elettroni moltiplicando charge-coupled device. I video vengono registrati utilizzando la modalità cinetica. Immagini luce bianca sono ottenuti utilizzando una lampada a luce bianca e un condensatore collegato al microscopio come fonte di illuminazione. - Per l'osservazione generale singola molecola, impostare la modalità di illuminazione del microscopio per TIRF o HILO 23 (vedi la discussione per ulteriori dettagli circa TIRF contro l'imaging HILO). Per impostare una modalità HILO su un microscopio TIRF, diminuire leggermente l'angolo di incidenza della luce di eccitazione di spostare l'attenzione leggermente superiore alla superficie coprioggetto (immagine della cella interiore piuttosto che la sua membrana inferiore a contatto con il coprioggetto, vedi 4.5.4 ).
- Per l'analisi a livello di cella, lunga molecola singola traccia esperimenti o step-saggio di analisi photobleaching, impostare la modalità di illuminazione del microscopio di un ensuri modalità widefieldng l'osservazione continua del volume cellulare intero e quindi di tutte le molecole marcate internalizzate.
- Campioni di immagine con qualsiasi microscopio TIRF / singola molecola.
- In genere, i poteri d'eccitazione intorno a 0,5-3 mW (~ 50-400 W / cm 2).
Nota: potenze laser inferiori sono utili per raggiungere l'osservazione a lunga vita di fluorescenza e il monitoraggio (più di 1 minuto), mentre potenze laser superiori potrebbero essere necessarie per una maggiore risoluzione spazio-temporale o analisi photobleaching graduale. - Utilizzare tempi di esposizione vanno da 15 ms per il monitoraggio esperimenti a 100 ms per l'osservazione più generale e quantificazione intensità. Nota: Altri frame rate e le modalità possono essere utilizzate come illuminazione stroboscopica, in particolare per lo studio di specie a rapido diffondenti 30.
- In microscopio TIRF, registrare il canale di fluorescenza su un elettrone moltiplicando CCD (EMCCD) fotocamera con un ingrandimento risultante in una lunghezza di pixel ~ 100 nm / pixel. La configurazione TIRF è descrivere in maggiori dettagli in riferimento 26.
- Widefield o HILO illuminazione
- Acquisizione dati
- Spegnere o bloccare l'illuminazione laser fino all'inizio dell'esperimento. Accendere la EMCCD guadagno fotocamera per evitare di danneggiare la fotocamera a causa di sovraesposizione.
- Posizionare il pad panino agarosio capovolta sul palco microscopio, con il lato della cella coperto rivolto verso il basso, al fine di portare la cella vicino all'obiettivo. Impostare l'attenzione per le cellule in luce trasmessa modalità microscopio 28. Registrare l'immagine di ogni cella di vista sotto l'imaging luce bianca al fine di individuare la cella contorni prima di spegnere la luce bianca.
- Proteggere il campione dalla luce ambientale laboratorio.
- Per la modalità di eccitazione HILO, regolare l'angolo del fascio di eccitazione a ciascun massimo rapporto segnale-rumore illuminando solo la sezione del campione vicino alla superficie coprioggetto.
- Per raggiungere l'illuminazione HILO, focalizzare il raggio laser nel piano focale posteriore di un 100x NA 1.4 obiettivo 28 (nume superioreaperture mochimico come il 1.45 e 1.49 sono adatti). Spostando la lente di focalizzazione perpendicolare al fascio, l'attenzione si sposta oltre al centro dell'obiettivo in modo che il fascio esce l'obiettivo con un angolo.
- Regolare la posizione dell'obiettivo per massimizzare il rapporto segnale-rumore, intensità di fluorescenza intracellulare rispetto extracellulare segnale di fondo.
- Accendere il guadagno della telecamera e iniziare l'acquisizione dei dati prima di accendere il laser.
- Durante la registrazione di dati, acquisire una immagine bianco-luce di ogni FOV prima o dopo la registrazione dei dati di fluorescenza; questo aiuta ad identificare i bordi delle celle nei canali di fluorescenza.
- Per la stima della redditività
- Utilizzare un ricco medio bassa fluorescenza nel pad agarosio per consentire alle cellule di crescere dopo l'elettroporazione.
- Equilibrare il microscopio alla temperatura ottimale per il microrganismo studiato (37 ° C per E. coli, 29 ° C per S. cerevisiae) Con un sistema di riscaldamento obiettivo.
- Record sia luce e fluorescenza immagini in bianco ogni 30 minuti, avendo cura di rimanere esattamente lo stesso campo visivo durante l'intera registrazione di dati. Un ritardo di ≈1 ora è tipicamente osservata prima cellule iniziano a dividersi.
- Contare il numero di biomolecole interiorizzate per cella
- Impostare la potenza del laser a valori elevati (2-3 MW) e tempo di esposizione lungo (100 ms).
- Impostare l'illuminazione modalità widefield per illuminare l'intera cella.
- Film record come descritto al punto 4.2 avendo cura di registrare fotogrammi aggiuntivi (50-100 fotogrammi) dopo il completamento del photobleaching fluorescenza.
Analisi 5. I dati
- Analisi generale
- Analizzare le immagini registrate e film, sia in luce bianca e fluorescenza eccitazioni, utilizzando un software di imaging, come ad esempio il software ImageJ gratuito.
- In ImageJ, aprire le immagini oi filmati registrati sul microscopio (formato TIF) in File> Apri> La posizione del file.
- Per confrontare qualitativamente intensità di fluorescenza sullo schermo del computer, assicurarsi che tutte le immagini di fluorescenza vengono visualizzate con le stesse impostazioni di luminosità e contrasto in Image> Adjust> Luminosità / Contrasto. Regolare manualmente o automaticamente le impostazioni di una immagine selezionata, premere il tasto "Set" e selezionare l'opzione "Propagare a tutte le altre immagini".
- Impostare il tipo di informazioni da estrarre: Analizza> Set misurazioni, e scegliere (almeno) "Area", "deviazione standard", "Min & Max valore di grigio" e "significano valore di grigio".
- Per confrontare intensità di fluorescenza delle cellule, selezionare l'area di interesse s utilizzando il pulsante di selezione a mano libera di ImageJ ed estrarre le intensità cellulari in Analizza> Misura. La tabella risultante contiene i valori di misura epuò essere salvato e / o copiati ad altri software. Il valore "medio" corrisponde alla intensità media per pixel nell'area selezionata e può essere confrontato direttamente tra celle o tra una cella e lo sfondo.
- In un campione di cellule elettroporate, una cella viene considerato caricato se la sua intensità media per pixel è maggiore l'intensità media per pixel del controllo negativo più 3 volte la sua deviazione standard (Av (I caricato cella)> Av (I -EP) + 3 * StdDev (I -EP)).
- Costruire immagini e filmati fluorescenza overlay falsi colori per valutare la qualità e il caricamento dei campioni.
- In ImageJ, immagini sovrapposte, come l'immagine di luce bianca e l'immagine di fluorescenza corrispondente allo stesso FOV in Immagine> Colori> Unisci canali. Selezionare un colore per ogni immagine (C4 (grigio) per la luce bianca, C1 (rosso) per il canale rosso, C2 (verde per canale verde ... ecc.).
- Controllaresull'immagine sovrapposizione che la fluorescenza si trova entro i confini delle cellule (immagine luce bianca) e che fluorescenza di fondo è basso ed omogeneo (nessun punti luminosi di fuori dei confini delle celle).
- Prima di analizzare un gran numero di cellule, controllare qualitativamente le immagini corrispondenti al campione negativo sono simili alle immagini cella vuota e visualizzare intensità molto inferiori rispetto alle cellule elettroporate.
- Per gli esperimenti di vitalità, contare manualmente le percentuali di divisione, non di divisione, ma visibilmente intatti (identici) e (morti), le cellule danneggiate dallo stesso campo di vista in crescita nel tempo (vedere 4.2.6).
- Valutare la fattibilità di almeno 200 cellule per campione (elettroporate, controllo negativo e celle vuote) al fine di raccogliere un numero sufficiente di statistiche.
- Analizzare le immagini registrate e film, sia in luce bianca e fluorescenza eccitazioni, utilizzando un software di imaging, come ad esempio il software ImageJ gratuito.
- Analisi basata Cell-
- Contare il numero di biomolecole interiorizzate per cella mediante analisi photobleaching graduale
- Cellule Segmento di selecting l'area di interesse utilizzando il pulsante di selezione a mano libera di ImageJ e disegnare una forma che circonda appunto la cella (equivalente a membrana cellulare).
- Estrarre le intensità di cellule nel tempo in Image> Stack> Profilo Plot Z-asse. Il grafico risultante rappresenta l'intensità media per pixel per l'area all'interno del perimetro delle cellule contro ogni fotogramma filmato risultante in una curva photobleaching per quella specifica cella. Esso contiene una diminuzione esponenziale iniziale della intensità cella raggiunge un asintoto inferiore (fluorescenza di fondo). I valori di misura e possono essere salvati e / o copiati altri software cliccando su "Salva" o "Copia".
- Copiare e incollare i valori sbiancanti in una colonna foglio di calcolo (I crudo).
- Calcolare l'autofluorescenza media per pixel che rimane dopo photobleaching (I auto) la media dei valori grezzi I ottenuti negli ultimi 50-100 fotogrammi (più basso Asymptote).
- Subtract l'autofluorescenza media per pixel che rimane dopo photobleaching per quella cella dalla curva photobleaching iniziale: I candeggio = I raw - I automatica.
- Utilizzare basale-sottratto photobleaching timetraces (I candeggio contro frame) che mostra meno di 10 punti quantizzati per valutare la dimensione del passo medio (intensità fluoroforo unitaria) a causa della sbiancamento di un unico fluoroforo 26.
- Valutare il numero di molecole internalizzate per cella dividendo l'intensità cellula basale-sottratto iniziale (I candeggio a t = 0) dall'intensità fluoroforo unitaria.
- Efficienza FRET Single-cell
- Misurare l'intensità media cellule per pixel in entrambi i canali di emissione del donatore e accettore (su eccitazione donatore) ed entro il limite di cella per ciascun canale come spiegare in 5.1.1.4.
- Misurare l'intensità media dei pixel per lo sfondo in ogni chAnnel da una zona vuota della diapositiva.
- Sottrarre questa intensità da sfondo l'intensità media per pixel. Utilizzare queste intensità di fluorescenza di fondo, sottratto per calcolare FRET per ogni cella calcolando l'intensità accettore background-sottratto diviso per il totale (+ accettore donatore) intensità background-sottratto al momento di eccitazione del donatore: I accettore / (I accettore + I donatori)
- Contare il numero di biomolecole interiorizzate per cella mediante analisi photobleaching graduale
- Analisi singola molecola
- Monitoraggio singola molecola e analisi di diffusione
Nota: Il protocollo per il monitoraggio diffusione molecole fluorescenti nelle cellule viventi e di valutare il loro coefficiente di diffusione apparente è stato descritto 26, 28.- In breve, montare le immagini di singole fluorofori in ogni fotogramma da una gaussiana ellittica 2D. Collegamento localizzato molecole di una traccia, se sono apparsi in fotogrammi consecutivi all'interno di una finestra di 5-7 pixel (0,48-0,67 micron). Utilizzare un memorparametro y di 1 fotogramma al conto per la scomparsa transitoria di un fluoroforo causa di lampeggiante o la localizzazione perdere.
- In vivo analisi smFRET
- Individuare manualmente molecole localizzate all'interno delle cellule da film passando attraverso un film in ImageJ ed identificare molecole immobili (o abbastanza immobile) nella FRET (accettore) del canale.
- Per estrarre le intensità nel canale accettore e donatore corrispondente ad una molecola immobile, selezionare l'area intorno molecola in ciascun canale utilizzando il pulsante di selezione "ovale" di ImageJ (cerchio attorno ad ogni singola fluoroforo, utilizzando un raggio ~3-pixel) e estrarre i intensità molecola in Analizza> Misura. La tabella risultante contiene i valori di misura e può essere salvato e / o copiati ad altri software.
- Calcolare i valori di fondo per canale per l'intensità media dei pixel da un cerchio delle stesse dimensioni in un'area vuota della diapositiva su tutti i fotogrammi analizzati.
- Utilizzare valori di fluorescenza di fondo sottratto nei canali donatore e accettore (su eccitazione donatore) per la fluorescenza e ti agiti tempo tracce, come nella cella singola Fret caso (vedi 5.2.1.7).
Nota: analisi e algoritmi automatizzati e robusto sono stati descritti in Bibliografia 26-28, 31.
- Monitoraggio singola molecola e analisi di diffusione
Risultati
Preparazione del campione
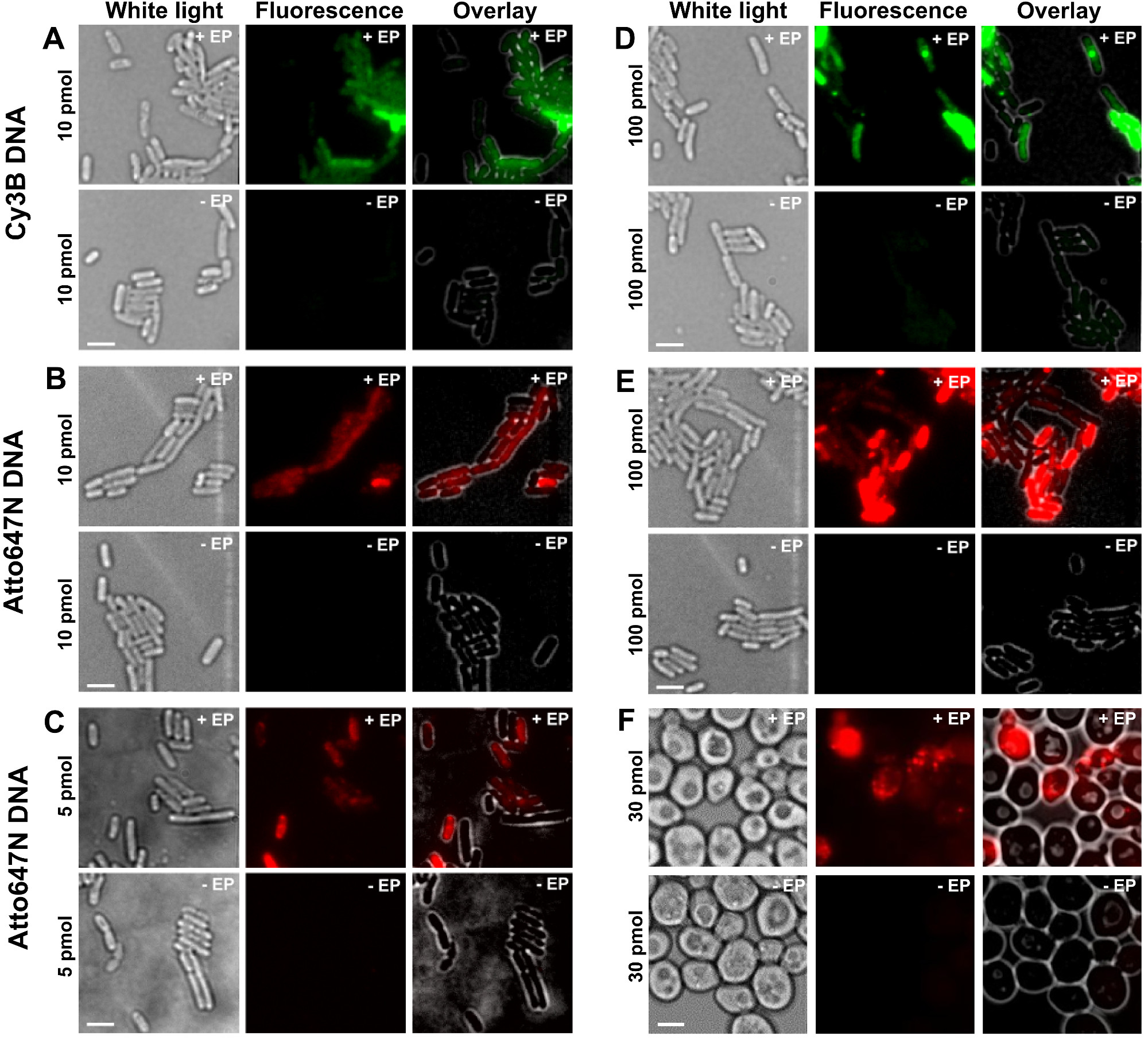
Le varie fasi del protocollo sono presentati come schemi della figura 1. Come esempio, abbiamo rappresentato il caricamento di batteri con doppiamente marcato (donatore e accettore coloranti) frammenti di DNA. Risultati rappresentativi per internalizzazione DNA sono mostrati nella Figura 2. Per ogni campione elettroporate, i dati per le celle vuote e le cellule non elettroporate sono stati registrati (Figura 2). "Le celle vuote" corrispondono a electrocompetent cellule non incubate con biomolecole fluorescenti né elettroporate; loro intensità nel canale fluorescente riflette il livello autofluorescenza in condizioni identiche sperimentali (potenza del laser, risoluzione tempo, temperatura, etc.). "Le cellule non-elettroporate" (chiamato anche -EP, cioè, meno EP) corrispondono a un controllo negativo in cui electrocompetent cellule sono state incubate con la biomolec fluorescentimoduli ma non elettroporate. Queste cellule non elettroporate dovrebbero mostrare un livello di fluorescenza simile al autofluorescenza delle celle vuote e significativamente inferiore all'intensità di fluorescenza, visualizzata da cellule elettroporate caricati. Questo conferma la rimozione di eventuali biomolecole etichettati non internalizzati che potrebbe hanno aderito alla membrana cellulare esterna.

Figura 2: I risultati rappresentativi per l'internalizzazione di dsDNA marcato con diversi fluorofori a differenti concentrazioni di batteri (AE) e lievito (F) Da sinistra a destra:. Bianco-luce, fluorescenza e sovrapporre le immagini. - / + EP denota incubazione senza / con elettroporazione. Bar Scala: 3 micron. A. CY3B dsDNA, 10 pmol, E. coli. B. ATTO647N dsDNA, 10 pmol, E. coli. C. Alexa647 dsDNA, 5 pmol, E. coli. D. CY3B dsDNA, 100 pmol, E. coli. E. ds ATTO647NDNA, 100 pmol, E. coli. F. ATTO647N dsDNA, 30 pmol, lievito. Questo dato è stato modificato dal riferimento 26. Clicca qui per vedere una versione più grande di questa figura.
{kind=link}
Contare il numero di biomolecole interiorizzate per cella
La procedura per stimare il numero di biomolecole marcate interiorizzate per cella utilizzando l'analisi photobleaching è presentato in figura 3 e Movie integrativa 2, insieme a risultati rappresentativi ottenuti con diverse concentrazioni di DNA marcato. Cella di carico aumenta l'efficienza con la quantità iniziale di incubato DNA marcato, permettendo all'utente di ottimizzare il numero di molecole marcate per cella da un livello "single-molecola" (<10, Film complementare 2B) ad un livello "insieme" (> 10 , supplementare Movie 2A). Un modo robusto di stimare la percentuale di cellule caricate è to contare il numero di cellule elettroporate visualizzazione un'intensità cella sopra l'intensità cellulare medio di cellule elettroporate non più 3 volte la deviazione standard (cioè, Av (I-EP) + 3 * StdDev (I-EP) dove Av = media, I = intensità per pixel, Std. Dev. = deviazione standard e -EP = non elettroporate) come mostrato in Figura 3.

Figura 3:. Contando il numero di molecole internalizzate mediante analisi photobleaching (A) singola cella photobleaching analisi. Esempi di timetraces intensità di fluorescenza (blu: i dati grezzi; rosso: fit; tramezze: WL e immagini di fluorescenza di E. coli caricato con ATTO647N marcato dsDNA prima e dopo sbiancamento). Top: evento di sbiancamento single-step. Middle: cella contenente ± 3 molecole mostrano sbiancamentoe lampeggiante. In basso: cella contenente> 10 passi corrispondenti ad almeno 10 molecole (B) Istogramma di un singolo passo per intensità di altezza da un algoritmo passo-montaggio automatico da 57 celle contenenti meno di 6 passi distinguibili.. Fit singola gaussiana è centrata a 11 ± 3 au, corrispondente ad un'intensità fluoroforo unitario di 8100 fotoni al secondo. L'asterisco indica la raccolta tutte le altezze gradino superiore o uguale a 50 au (C) Istogramma di molecole internalizzate per cella elettroporate con diverse quantità di ATTO647N dsDNA, calcolato dividendo la fluorescenza iniziale dall'intensità fluoroforo unitaria bin. Dall'alto in basso: celle vuote (cioè, non incubate con molecole fluorescenti e non elettroporate), non elettroporate (ma incubate con molecole fluorescenti, di nome -EP), e le cellule elettroporate incubate con 10 e 100 pmol dsDNA (nome + EP). Le celle vuote e non elettroporate corrispondonoad autofluorescenza, le cellule, mentre elettroporate mostrano un'ampia distribuzione di molecole interiorizzato, con una maggiore percentuale di cellule altamente caricate a 100 pmol (≥ 4 molecole, vedere bin asterisco-marcato). Efficienza internalizzazione (frazione di cellule con Int.> Significa 3x + Std. Dev. Di campione non -EP) per i 10 e 100 pmol campioni era di 94% e 90%, rispettivamente. 121 ± 106 molecole per 10 pmol dsDNA, e 176 ± 187 molecole per 100 pmol dsDNA: numero di molecole internalizzate per cella media. Impostazioni: 100 ms di esposizione, illuminazione widefield. Bar Scala: 1 micron. Questo dato è stato modificato dal riferimento 26. Clicca qui per vedere una versione più grande di questa figura.
{kind=link}
Carico delle cellule e la vitalità
Oltre a modificare la quantità di biomolecole etichettati aggiunto alle cellule prima elettroporazione, lautente può regolare la quantità di molecole internalizzate scegliendo diverse intensità di campo durante elettroporazione (Figura 4, Movie supplementare 1). Intensità di campo più elevate portano ad una maggiore efficienza di internalizzazione, ma portano a una leggera diminuzione della vitalità cellulare. Per proteine interiorizzazione, l'uso di una fase di filtrazione può aiutare a rimuovere-non interiorizzate proteine marcate (vedi 3.3.3.1.1). In tali casi, la filtrazione delle cellule assicura che le proteine fluorescenti osservati sono effettivamente internalizzati all'interno del citoplasma batterico; abbiamo notare, tuttavia, che la filtrazione ha anche un impatto negativo sulla vitalità cellulare (per maggiori dettagli, vedere REF 22).

Figura 4: Influenza della tensione elettroporazione su carico e vitalità cellulare (A) Bar grafico che rappresenta l'effetto di intensità di campo elettroporazione sul caricamento effic.iency (barre rosse) e la vitalità delle cellule (barre verdi). 84 ± 8% della cella non elettroporate (0 kV / cm) divide dopo 1 h su un pad agarosio a 37 ° C. Alle stesse condizioni, il 78 ± 3% e 49 ± 3% delle cellule elettroporate a 0,9 kV / cm e 1,8 kV / cm rispettivamente dividono dopo 1 ora. Per l'efficienza di carico, 73 ± 8% delle cellule vengono caricati a 0,9 kV / cm, mentre il 92 ± 6% delle cellule vengono caricati a 1,8 kV / cm. Le barre di errore rappresentano la deviazione standard calcolata da tre misurazioni indipendenti; più di 200 cellule sono state analizzate per ciascun campione e ogni ripetizione. In generale aumenta l'efficienza di carico con tensione elettroporazione per il leggero scapito della vitalità cellulare. (B) misure di fluorescenza a base di cellule su diverse generazioni dimostrano che l'intensità complessiva di fluorescenza viene ripartito equamente tra le due cellule figlie. Cella 1 e 2 si riferisce al numero di cellulare l'immagine luce bianca (a sinistra) e immagine di fluorescenza a t = 0. Scalabar: 1 micron). Questo dato è stato modificato dal riferimento 26.
Protein internalizzazione
Risultati rappresentativi per proteine internalizzazione sono nelle figure 5A & B. È particolarmente importante rimuovere la maggior quantità di residuo libera (non reagito) colorante dal campione proteico possibile prima elettroporazione. Negli esempi delle figure 5A e B, la CY3B marcato campione Klenow Fragment (CY3B-KF, dove KF è il frammento Klenow della E. coli DNA polimerasi I, 66 kDa) contiene solo 1% colorante libero; tale contributo tintura al carico complessiva cella sia trascurabile. Il confronto tra il campione di interesse elettroporate con entrambe le cellule non elettroporate (incubate con la stessa quantità di proteina marcata) e cellule elettroporate con la quantità equivalente di colorante libero costituiscono due controlli necessari per garantire che le molecole fluorescenti osservatisono infatti interiorizzato proteine marcate.

Figura 5: Proteine internalizzazione in batteri vivi (A) Rappresentante di fluorescenza campo di sovrapposizione di punti di vista.. Le cellule elettroporate a tensione di 1,4 kV con 50 pmol RNAP ω subunità da una soluzione proteica magazzino che conteneva solo l'1% libera CY3B tintura. Non elettroporate (non -EP) e celle vuote sono definiti come in precedenza. Dye gratuito è stato interiorizzato alla stessa concentrazione del campione elettroporate la RNAP ω. Imaging in modalità campo largo, 532-nm di eccitazione a 1 mW, 50 ms di esposizione. (B) Distribuzione di intensità di cellule-media non corretti per i campioni in (A), data in proporzione del numero di cellule totale. Più di 400 cellule per campione sono stati segmentati. Questo dato è stato modificato dal riferimento 22. (C) internalizzazione di UNLABeled T7 RNA polimerasi (RNAP T7, 98 kDa) in electrocompetent DH5α portando il pRSET-EmGFP plasmide codificante smeraldo GFP (EmGFP) sotto il controllo di un promotore T7. A sinistra: Schema di dosaggio. Middle: sovrapposizione di fluorescenza. A destra: istogrammi di base di cellule intensità di fluorescenza del campione non elettroporate (in alto) e le cellule incubate e elettroporate con T7 RNAP (in basso); circa l'11% delle cellule elettroporate mostrano ad alta intensità di fluorescenza (frazione di cellule con Int.> significa + 3x Std. Dev. di campione non -EP) indicando espressione di EmGFP. Gli asterischi indicano bidoni in grado di raccogliere tutte le intensità superiore o uguale 1.100 au bar Scala: 3 micron. Questo dato è stato modificato dal riferimento 26.
Figura 5C presenta un'altra applicazione di proteine elettroporazione. Qui, la proteina elettroporate è non marcato, ma la sua internalizzazione innesca una risposta fluorescente osservabile. Questo esperimento verifica la preindicationi e la funzionalità delle proteine elettroporate nel citoplasma cellulare. Senza etichetta T7 RNA polimerasi (98 kDa) è stato interiorizzato in E. coli DH5α ceppo contenente un plasmide codificante per la proteina fluorescente EmGFP sotto il controllo di un promotore T7 26. Poiché il gene per la T7 RNA polimerasi è assente in DH5α, espressione EmGFP nei nostri esperimenti richiede che funzionale T7 RNA polimerasi viene introdotto nelle cellule mediante elettroporazione (Figura 5C). Dopo elettroporazione con 1 pmol T7 RNAP,> 11% delle cellule (blu bar, Figura 5C) esposto fluorescenza superiore al controllo negativo (incubate con la stessa quantità di T7 RNAP, ma non elettroporate). Questo risultato stabilisce che una parte delle molecole T7 RNAP internalizzati mediante elettroporazione mantengono la loro integrità in vivo e può eseguire i comandi previsti nel citoplasma cellulare.
In vivo FRET alla singola molecolae livelli unicellulari
Infine, l'interiorizzazione e l'analisi delle specie doppiamente etichettati in batteri che vivono è presentato in figura 6 e Movie supplementare 3. fusioni proteine fluorescenti non sono l'ideale per gli studi in vivo smFRET, la capacità di fornire biomolecole doppiamente marcati nelle cellule viventi utilizzando elettroporazione è una dei grandi vantaggi di questo metodo. Figura 6A presenta cella singola FRET analisi di batteri caricati con diversi standard di DNA FRET (utilizzando CY3B e fluorofori Atto647N come donatore-accettore paio FRET). Le cellule sono elettroporate con 20 pmol di tre standard dsDNA FRET brevi doppiamente marcati con efficienze FRET apparenti (E *) di 0,17, 0,48, 0,86 e (precedentemente determinati 26) in vitro. Tutti DNA entrano nelle cellule in modo efficiente (Figura 6A, a sinistra) e il picco principale di ogni singola cellula e distribuzione * concorda bene con i risultati in vitro (Figura 6A, A destra). Nei campioni intermedio e alto-tasto, le popolazioni di cellule con basso E * del previsto sono osservati, presumibilmente a causa di una combinazione di accettore candeggio e inattività fotofisici, carico cella variabile (quindi, a rapporto variabile segnale-rumore) e la degradazione del DNA .

Figura 6:.. Risultati rappresentativi per cella singola e singola molecola FRET osservazione nei batteri che vivono Ensemble e smFRET studi singoli batteri (A) Analisi di cellule caricate con 20 pmol ognuno dei tre standard FRET DNA espositrici bassa (~ 0,17), intermedia (~ 0,48), e alta (~ 0,86) FRET (come misurato con misurazioni singola molecola in vitro; vedi REF 26). A sinistra: la luce bianca e rossa immagini verdi / (FRET) fluorescenza overlay (Scala bar: 3 micron). Esempi di valori FRET dalle cellule differenti sono indicate (bianco). Impianto ht (dall'alto in basso):. FRET cell-based non corretta (E *) istogrammi per donatore solo (verde scuro), basso (verde chiaro), intermedio (giallo), e alto (rosso) FRET standard di DNA (B-D) In vivo smFRET. Le cellule sono carichi di DNA 0.25 pmol intermedio-FRET (pannello B), 0.25 pmol high-FRET DNA (pannello C), e 5 pmol doppiamente-etichettati KF (pannello D). Colonna sinistra: verde / rosso di fluorescenza sovrapposizione di singolo fotogramma prima e dopo accettore photobleaching. Colonna centrale: tracce di tempo corrispondenti alla molecola nel cerchio giallo. Efficienza FRET, intensità di emissione dei donatori, e le intensità di emissione accettore sono visualizzati in blu, verde e rosso, rispettivamente. Colonna destra: istogrammi tasto della donatore solo molecole (verde) e molecole donatore-accettore (giallo, rosso, e grigio) da 20 tracce di tempo per ogni campione. Bar Scala: 3 micron per A, 1 micron per B-D. Questo dato è stato modificato dal riferimento 26.208fig6large.jpg "target =" _ blank "> Clicca qui per vedere una versione più grande di questa figura.
Per osservare smFRET in vivo per DNA o proteine di campioni, gli importi bassi (0,25 pmol) di standard di DNA intermedio e alto-FRET (Figura 6B, C) o 5 pmol di fluorofori doppio marcato KF (Alexa647 / CY3B come FRET coppia, Figura 6D) sono elettroporate in E. coli. Tali concentrazioni hanno portato a molte cellule caricate con poche (n = 1-10) molecole marcate, che consente la localizzazione diretta, il monitoraggio, e ti agiti monitoraggio per singole molecole. Alcune molecole diffondono liberamente, mentre altri appaiono immobile o diffusi lentamente (Movie supplementare 3). Timetraces di biomolecole immobilizzate doppiamente marcati (Figura 6, al centro) durano da 1 a 30 s e mostrare le caratteristiche di smFRET: cambiamenti anticorrelated in fluorescenza donatore e accettore su accettatore sbiancamento (per esempio, t ~ 16 secondi; la figura 6B, middle ), seguita da singolasbianca -step donatore (per esempio, t ~, 19 sec; Figura 6B). Distribuzioni FRET generati da tali timetraces (Figura 6, a destra) di risultato in un valore medio che si trova in eccellente accordo con pubblicati studi in vitro 26,31,32. Questi risultati stabiliscono la possibilità per gli studi smFRET quantitative DNA e proteine interiorizzate, e suggeriscono che le proteine mantengono la loro integrità e la struttura su elettroporazione e l'interiorizzazione (come sostenuto da esperimenti RNAP T7 internalizzazione).
Film supplementare 1:. La vitalità cellulare Sinistra: immagini luce bianca. Immagini di fluorescenza: Destra. Animated GIF animato che mostra la divisione dopo elettroporazione (1,8 kV / cm) di batteri caricate con 10 pmol Atto647 DNA marcato. L'apparente calo complessivo di fluorescenza è dovuta alla diluizione di DNA marcato sulla divisione cellulare ed anche parte al photobleaching che si verifica durante ciascunmisurazione.
Movie integrativa 2: studi photobleaching cell-based A.. Esempio rappresentativo di una cella molto carico (che contiene> 100 molecole di DNA Atto647N marcato). In alto a sinistra, l'immagine luce bianca della cella di interesse (rettangolo rosso). In alto a destra, di film delle cellule caricate mostrano il loro decadimento di fluorescenza per diversi minuti. In basso, traccia temporale di decadimento generale intensità di fluorescenza delle cellule di interesse. Fluorofori organici possono mostrare photobleaching vite 2 ordini di grandezza superiore a quella PQ (qui, ~ 41 sec per Atto647N). B. Esempio rappresentativo di una cella caricato con meno di 10 molecole marcate (3 in questo caso). Top, stesso pannello A., tracce momento l'intensità complessiva di fluorescenza della cella di interesse che mostra solo sbiancamento step e / o lampeggiante corrispondente a fluorofori organici singoli. L'altezza media di queste fasi corrisponde all'intensità unitario singola molecola (qui ~12 au) utilizzata per stimare il numero iniziale di molecole internalizzate per cella. Film sotto continuo rosso eccitazione laser a 300? W di potenza e 100 msec per frame.
Film supplementare 3: I n VIVO singola molecola FRET Top:. Le cellule caricate con 0,25 pmol alta FRET DNA (come nella Figura 6C) continuamente monitorato a 50 ms per frame sotto illuminazione nTIRF con 1 mW verde (532 nm) del laser. Ogni frame è un / rosso (FRET) di fluorescenza verde sovrapposizione di ogni canale. Diffondere e rosso immobile (intatto) e verde (un'unica etichetta attiva) molecole di DNA possono essere osservati. Trace tempo corrispondente alla molecola nel cerchio giallo: inferiore. Efficienza FRET, intensità di emissione dei donatori, e le intensità di emissione accettore sono visualizzati in blu, verde e rosso, rispettivamente. Evento Anti-correlato sbiancamento accettore (transizioni rosso a verde) corrisponde alla firma di singola molecola FRET.
Discussione
Molti parametri possono essere variati durante elettroporazione cellulare e acquisizione dati a seconda del sistema biologico di interesse e la precisa natura dell'esperimento (a livello di cella o l'analisi singola molecola). Ad esempio, quando electroporating DNA in batteri, 0,25-5 pmol di frammenti marcati dsDNA porta ad una bassa efficienza interiorizzazione, permettendo il rilevamento diretto singola molecola (es., Senza la necessità di photobleaching anticipo). Sopra 5 pmol dsDNA, le cellule tendono ad essere pesantemente caricato, un regime più adatto per l'analisi singola cellula. Tutti DNA etichettati dovrebbero essere precedentemente gel purificato per eliminare ogni traccia di colorante libero (fluoroforo non reagito) dalla soluzione di DNA magazzino. Inoltre, i potenziali problemi con la degradazione del DNA, in particolare per gli esperimenti smFRET, possono essere affrontate utilizzando DNA con acidi nucleici innaturali, o motivi che proteggono exonuclease accessibili termini come loop tornanti.
Un'altra adjustablparametro e in elettroporazione è l'intensità del campo applicato durante elettroporazione. Intensità bassa di campo (~ 1 kV / cm) porterà ad una bassa efficienza di carico appropriata per gli studi singola molecola. Intensità di campo più elevate (fino a 1,8 kV / cm) aumenterà l'efficienza di carico; Tuttavia, vi è una correlazione inversa tra intensità di campo e vitalità cellulare dopo elettroporazione (vedere Figura 4). Per riferimento, una intensità di campo normale usati per batteri e lieviti elettroporazione è ~ 1,5 kV / cm. La costante di tempo, che rappresenta la lunghezza di questo decadimento, è un parametro conveniente seguire, poiché la costante di tempo gocce non appena si verifica alcun fenomeno arco nella cuvetta. In Impostazioni normali, la costante di tempo deve essere superiore a 4 ms; valori più bassi porterà a bassa efficienza di carico o cellule danneggiate anche non caricati. La maggior parte elettroporatori offrono altri gradi di libertà (come "troncamento impulsi" o "pulse forma") che può essere modificato per sintonizzare siacarico delle cellule e la vitalità. Abbiamo applicato questo metodo per entrambi i batteri e lieviti, tuttavia procedure analoghe dovrebbero anche consentire l'internalizzazione di biomolecole marcati nelle cellule di mammifero utilizzando appropriate impostazioni elettroporatore dal loro membrana è in realtà meno complessa (singolo doppio strato lipidico) e dal elettroporazione è già stato utilizzato con tali cellule 21.
Quando internalizzazione proteine marcate, tutto colorante libero deve essere rimosso dal marcato soluzione proteica magazzino prima elettroporazione. Molecole di colorante libero, a causa delle loro dimensioni più piccole, possono essere internalizzati preferenzialmente sulle proteine di interesse, e sono difficili da distinguere durante l'analisi dei dati (nonostante la loro diffusione più veloce previsto). Come guida, per un campione di proteina marcata organicamente sia adatto per elettroporazione, la quantità di colorante libero residuo deve essere inferiore al 2% (rilevato utilizzando scansione fluorescenza di una SDS-PAGE) 22. Questo processo è particolarmente importante,come alcune molecole potrebbero attaccarsi alle membrane esterne di batteri o lieviti elettroporate. A questo proposito, il campione di controllo negativo dovrebbe visualizzare l'intensità di fluorescenza per cella nettamente inferiore cellule elettroporate, idealmente a partire da livello autofluorescenza di celle vuote (cellule che non sono stati incubati con eventuali biomolecole fluorescente né elettroporate, Figura 2).
Come con dsDNA, l'efficienza internalizzazione di proteine marcate è legato alla quantità di biomolecole aggiunti alle cellule prima di elettroporazione. Tuttavia, altri parametri, quali le dimensioni e la carica, giocano un ruolo nella internalizzazione. Piccole proteine presentano efficienze elevate internalizzazione, mentre le proteine più grandi (fino a 98 kDa) possono essere internalizzati con successo ma con minore efficienza (Figura 5) 26. Il punto isolelectric della proteina, potenziali interazioni con la membrana cellulare e gli altri parametri fisico ancheinfluenza cella di carico durante elettroporazione. Come risultato, gli utenti devono ottimizzare esperimenti per il proprio sistema, sapendo che un'alta concentrazione iniziale di proteina marcata (> 50 pM) darà le migliori possibilità di successo per il caricamento. Elettroporazione offre anche un nuovo strumento per turbare e analizzare la funzione cellulare introducendo proteine e altre biomolecole in cellule (o etichettati o senza etichetta). Gli esperimenti T7 RNA polimerasi (Figura 5C) presenti un esempio di un esperimento in cui possiamo introdurre una biomolecola che può cambiare l'espressione genica in vivo utilizzando elettroporazione.
Quando si esegue esperimenti fluorescenza singola molecola, illuminazione TIR è solitamente favorita rispetto altre modalità di illuminazione in quanto offre il miglior rapporto segnale-rumore di soli eccitanti fluorofori all'interno di una sezione sottile sopra la superficie coprioggetto (~ 100 nm). Tuttavia di imaging etichettati biomolecole diffondono all'interno microrganismi viventi potrebbero riquire illuminazione più profondo (fino a 0,8 micron per E. coli). Illuminazione più profondo si ottiene in modalità HILO, mantenendo un alto rapporto segnale-rumore. D'altra parte, l'imaging ampio campo è particolarmente importante per l'analisi fotoscolorimento graduale, in cui l'utente sta valutando il numero di molecole internalizzati dalle photobleaching un'intera cellula caricato con elevato potere laser e dividendo l'intensità iniziale fluorescenza cellulare dall'intensità unitaria prodotta da una singola molecola (passo singolo photobleaching, Figura 3). Immagini Widefield è richiesta anche per il monitoraggio molecola a lungo termine, al fine di localizzare le molecole diffondenti di interesse, anche se le loro traiettorie coprono l'intero volume delle cellule.
In questo protocollo, presentiamo come elettroporazione, una tecnica standard per i biologi e biochimici per la consegna acidi nucleici nelle cellule, costituisce un metodo semplice per la consegna di biomolecole fluorescenti in vari tipi di cellule. Thè romanzo, la tecnica high-throughput offre uno strumento unico per osservare molecole marcate nel loro ambiente nativo. In aggiunta biomolecole marcate con fluorofori che coprono una vasta gamma di lunghezze d'onda, elettroporazione può trasportare molecole modificate con molti gruppi chimici, come i nucleotidi innaturali e aminoacidi, chelanti di metalli, reticolanti, e gruppi di ingabbiamento. Se il sistema biologico che interessa non è essenziale per lo sviluppo delle cellule, il gene codificante per la proteina bersaglio può anche essere eliminato (o smontati), assicurando che le proteine osservati dopo interiorizzazione rappresentano tutti (o quasi) della piscina proteina intracellulare . In sostanza, elettroporazione può "trapianto" la flessibilità di bioconiugati in vitro in cellule viventi e quindi beneficiare sforzi in biologia sintetica, la biologia dei sistemi, e in vivo rilevazione singola molecola.
Divulgazioni
The authors have nothing to disclose.
Riconoscimenti
We thank Stephan Uphoff for discussions.
R.C. was supported by Linacre College, Oxford University. A.P. was supported by the German Academic Exchange Service (DAAD), the German National Academic Foundation and EPSRC. M.S. was supported by the Wellcome Trust. A.N.K. was supported by a UK BBSRC grant (BB/H01795X/1), and a European Research Council Starter grant (261227).
Materiali
| Name | Company | Catalog Number | Comments |
| ElectroMax DH5-alpha Comptent cells | Invitrogen | 11319-019 | or any other commercial or lab-mage electrocompetant bacteria or yeast. |
| EZ Rich Defined Madia | Teknova | M2105 | low fluorescence rich media |
| MicroPulser Electroporation Apparatus | Biorad | 165-2100 | or any classical electroporator for microorganism transformation |
| Certified Molecular Biology agarose | Biorad | 161-3100 | low fluorescence agarose for agarose pad |
| Microscope coverslips No 1.5 thickness | Menzel | BB024060SC | remove background particles by heating slides in furnace at 500 °C for 1h |
| Single-molecule fluorescence microscope | Home-built | described in REFs | |
| Localization software | Custom-written, available online | MATLAB and C++ software package that can be adapted for localization analysis. | |
| Tracking software | Available online | MATLAB implementation by Blair and Dufresne. |
Riferimenti
- Tsien, R. Y. The green fluorescent protein. Annu Rev Biochem. 67, 509-544 (1998).
- Leake, M. C., et al. Stoichiometry and turnover in single, functioning membrane protein complexes. Nature. 443, 355-358 (2006).
- Taniguchi, Y., Kawakami, M. Application of HaloTag protein to covalent immobilization of recombinant proteins for single molecule force spectroscopy. Langmuir. 26, 10433-10436 (2010).
- Xie, X. S., Choi, P. J., Li, G. W., Lee, N. K., Lia, G. Single-molecule approach to molecular biology in living bacterial cells. Annual review of biophysics. 37, 417-444 (2008).
- Lee, J. H., et al. Highly multiplexed subcellular RNA sequencing in situ. Science. 343, 1360-1363 (2014).
- Miesenbock, G., De Angelis, D. A., Rothman, J. E. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 394, 192-195 (1998).
- Sauer, M. Localization microscopy coming of age: from concepts to biological impact. J Cell Sci. 126, 3505-3513 (2013).
- Dempsey, G. T., Vaughan, J. C., Hao Chen, K., Zhuang, X. Evaluation of fluorophores for optimal performance in localizationbased super-resolution imaging. Nat Meth. 8, 1027-1041 (2011).
- Shaner, N. C., Steinbach, P. A., Tsien, R. Y. A guide to choosing fluorescent proteins. Nat Meth. 2, 905-909 (2005).
- Landgraf, D., Okumus, B., Chien, P., Baker, T. A., Paulsson, J. Segregation of molecules at cell division reveals native protein localization. Nat. Methods. 9, 480-482 (2012).
- Jaitin, D. A., et al. Massively Parallel Single-Cell RNA-Seq for Marker-Free Decomposition of Tissues into Cell Types. Science. 343, 776-779 (2014).
- Aldridge, S., et al. AHT-ChIP-seq: a completely automated robotic protocol for high-throughput chromatin immunoprecipitation. Genome Biol. 14, R124 (2013).
- Keppler, A., et al. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat Biotechnol. 21, 86-89 (2003).
- Wombacher, R., et al. Live-cell super-resolution imaging with trimethoprim conjugates. Nat. Methods. 7, 717-719 (2010).
- Zhang, Z., et al. A new strategy for the site-specific modification of proteins in vivo. Biochemistry. 42, 6735-6746 (2003).
- McNeil, P. L., Murphy, R. F., Lanni, F., Taylor, D. L. A method for incorporating macromolecules into adherent cells. J Cell Biol. 98, 1556-1564 (1984).
- Clarke, M. S., McNeil, P. L. Syringe loading introduces macromolecules into living mammalian cell cytosol. J Cell Sci. 102, 533-541 (1992).
- Sakon, J. J., Weninger, K. R. Detecting the conformation of individual proteins in live cells. Nat. Methods. 7, 203-205 (2010).
- Taylor, L. S. Electromagnetic syringe. IEEE Trans. Biomed. Eng. 25, 303-304 (1978).
- Dower, W. J., Miller, J. F., Ragsdale, C. W. High efficiency transformation of E. coli by high voltage electroporation. Nucleic Acids Res. 16, 6127-6145 (1988).
- Neumann, E., Schaefer-Ridder, M., Wang, Y., Hofschneider, P. H. Gene transfer into mouse lyoma cells by electroporation in high electric fields. EMBO J. 1, 841-845 (1982).
- Sustarsic, M., et al. Optimized delivery of fluorescently labeled proteins in live bacteria using electroporation. Histochem Cell Biol. , (2014).
- Tokunaga, M., Imamoto, N., Sakata-Sogawa, K. Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat. Methods. 5, 159-161 (2008).
- Sinha, A., et al. A cascade of DNA-binding proteins for sexual commitment and development in Plasmodium. Nature. 000, 1-5 (2014).
- English, B. P., et al. Single-molecule investigations of the stringent response machinery in living bacterial cells. Proc Natl Acad Sci U S A. 108, E365-E373 (2011).
- Crawford, R., et al. Long-lived intracellular single-molecule fluorescence using electroporated molecules. Biophys J. 105, 2439-2450 (2013).
- Uphoff, S., Reyes-Lamothe, R., Garza de Leon, F., Sherratt, D. J., Kapanidis, A. N. Single-molecule DNA repair in live bacteria. Proc Natl Acad Sci U S A. 110, 8063-8068 (2013).
- Uphoff, S., Sherratt, D. J., Kapanidis, A. N. Visualizing Protein-DNA Interactions in Live Bacterial Cells Using Photoactivated Single-molecule Tracking. J Vis Exp. , (2014).
- Hohlbein, J., Gryte, K., Heilemann, M., Kapanidis, A. N. Surfing on a new wave of single-molecule fluorescence methods. Phys Biol. 7, 031001 (2010).
- Xie, X. S., Yu, J., Yang, W. Y. Perspective - Living cells as test tubes. Science. 312, 228-230 (2006).
- Santoso, Y., et al. Conformational transitions in DNA polymerase I revealed by single-molecule FRET. Proc Natl Acad Sci U S A. 107, 715-720 (2010).
- Hohlbein, J., et al. Conformational landscapes of DNA polymerase I and mutator derivatives establish fidelity checkpoints for nucleotide insertion. Nature communications. 4, 2131 (2013).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati