
Method Article
Identificação de contatos intensificadores-promotores em corpos embrionários por captura quantitativa de conformação de cromossomo (4C)
Neste Artigo
Resumo
Relatamos a aplicação da captura de conformação de cromossomos quantitativos seguida de sequenciamento de alto throughput em corpos embrionários gerados a partir de células-tronco embrionárias. Esta técnica permite identificar e quantificar os contatos entre melhoradores putativos e regiões promotoras de um determinado gene durante a diferenciação de células-tronco embrionárias.
Resumo
Durante o desenvolvimento dos mamíferos, os destinos celulares são determinados através do estabelecimento de redes regulatórias que definem a especificidade, o tempo e os padrões espaciais da expressão genética. Corpos embrióides (EBs) derivados de células-tronco pluripotentes têm sido um modelo popular para estudar a diferenciação das três principais camadas de germes e definir circuitos regulatórios durante a especificação do destino celular. Embora seja sabido que os melhoradores específicos do tecido desempenham um papel importante nessas redes interagindo com os promotores, atribuindo-os aos seus genes-alvo relevantes ainda permanece desafiador. Para que isso seja possível, são necessárias abordagens quantitativas para estudar os contatos intensificadores-promotores e suas dinâmicas durante o desenvolvimento. Aqui, adaptamos um método 4C para definir os melhoradores e seus contatos com promotores cognatos no modelo de diferenciação EB. O método usa frequentemente enzimas de restrição de corte, sônica e um protocolo de PCR mediado por ligadura aninhada compatível com kits de preparação de bibliotecas de DNA comerciais. Posteriormente, as bibliotecas 4C são submetidas a sequenciamento de alto throughput e analisadas bioinforformaticamente, permitindo a detecção e quantificação de todas as sequências que tenham contatos com um promotor escolhido. Os dados de seqüenciamento resultantes também podem ser usados para obter informações sobre a dinâmica dos contatos do intensificador-promotor durante a diferenciação. A técnica descrita para o modelo de diferenciação EB é fácil de implementar.
Introdução
Em camundongos, a massa celular interna (ICM) de embriões de 3,5 dias de idade contém células-tronco pluripotentes embrionárias. O ICM desenvolve-se ainda no epiblasto no dia 4.5, gerando células de ectoderme, mesoderme e endoderme, as três principais camadas germinativas do embrião. Embora as células pluripotentes no ICM existam apenas transitoriamente in vivo, elas podem ser capturadas na cultura pelo estabelecimento de células-tronco embrionárias de camundongos (mESCs)1,2,3. Os mESCs permanecem em um estado indiferenciado e proliferam indefinidamente, mas após estímulos intrínsecos e extrínsecos também são capazes de sair do estado de pluripotência e gerar células das três camadas de germes de desenvolvimento2,4. Curiosamente, quando cultivados em suspensão em pequenas gotículas, os mESCs formam agregados tridimensionais (ou seja, EBs) que se diferenciam em todas as três camadas de germes5. O ensaio de formação eb é uma ferramenta importante para estudar o processo de especificação da linhagem precoce.
Durante a especificação da linhagem, as células de cada camada de germe adquirem um programa específico de expressão genética4. A expressão espacial precisa dos genes é regulada por diversos elementos cis-regulatórios, incluindo promotores centrais, intensificadores, silenciadores e isoladores6,7,8,9. Intensificadores, segmentos de DNA regulatório tipicamente abrangendo algumas centenas de pares de bases, coordenam a expressão genética específica do tecido8. Os intensificadores são ativados ou silenciados por meio da vinculação de fatores de transcrição e cofatores que regulam a estrutura local da cromatina8,10. Técnicas comumente utilizadas para identificar melhoradores putativos são a imunoprecipitação de cromatina em todo o genoma, seguida de sequenciamento (ChIP-seq) e o ensaio para cromatina acessível à transposase usando técnicas de sequenciamento (ATAC-seq). Assim, os intensificadores ativos são caracterizados por marcas específicas de histona ativa e pelo aumento da acessibilidade do DNA local11,12,13,14. Além disso, acredita-se que os melhoradores de desenvolvimento requerem interação física com seu promotor cognato8,9. De fato, foi demonstrado que as variantes e exclusões de aprimoramentos que interrompem os contatos do intensificador-promotor podem levar a malformações de desenvolvimento15. Portanto, há necessidade de novas técnicas que forneçam informações adicionais para a identificação de melhoradores funcionais que controlem a expressão genética do desenvolvimento.
Desde o desenvolvimento da técnica de captura de conformação de cromossomo (3C)16,o mapeamento dos contatos cromossômicos tem sido intensamente utilizado para avaliar a distância física entre elementos regulatórios. É importante ressaltar que foram desenvolvidas recentemente variantes de alto throughput de técnicas 3C, fornecendo diferentes estratégias de fixação, digestão, ligadura e recuperação de contatos entre fragmentos de cromatina17. Entre eles, in situ Hi-C tornou-se uma técnica popular que permite o sequenciamento de produtos de ligadura 3C genoma-wide18. No entanto, os altos custos de sequenciamento necessários para alcançar uma resolução adequada para a análise de contatos intensificadores-promotores torna essa técnica impraticável para o estudo de loci específicos. Assim, foram desenvolvidos métodos alternativos para analisar loci direcionados em resolução mais alta19,20,21,22. Um desses métodos, ou seja, 4C, conhecido como uma estratégia um contra todos, permite a detecção de todas as sequências que entram em contato com um site selecionado como ponto de vista. No entanto, uma desvantagem da técnica 4C padrão é a PCR inversa necessária, que amplifica fragmentos de tamanho diferente, favorecendo pequenos produtos e quantificação de viés após sequenciamento de alta taxa de throughput. Recentemente, umI-4C, uma nova variante da técnica 4C usando identificadores moleculares exclusivos (UMI) foi desenvolvida para perfis de contato cromossômico quantitativos e direcionados que contornam esse problema23. Esta abordagem utiliza cortadores freqüentes, sonagem e um protocolo de PCR mediado por ligadura aninhada, envolvendo assim a amplificação de fragmentos de DNA com distribuição de comprimento relativamente uniforme. Essa homogeneidade reduz vieses no processo de amplificação das preferências de PCR para sequências mais curtas e permite uma recuperação eficiente e contagem precisa de moléculas/fragmentos espacialmente conectados.
Aqui descrevemos um protocolo que adapta a técnica UMI-4C para identificar e quantificar contatos de cromatina entre promotores e intensificadores de fatores instrutivos de transcrição de linhagem durante a diferenciação do EB.
Protocolo
1. Geração do corpo embrionário a partir de células-tronco embrionárias do camundongo
- Prepare o meio de cultura sem soro mESC: DMEM/F12 e meio neurobasal misturado a uma razão de 1:1. O meio de cultura é complementado com solução de aminoácidos não essenciais MEM (1x), piruvato de sódio (1 mM), L-glutamina (2 mM), penicilina-estreptomicina (100 U/mL), beta-mercapto Suplementos de etanol (50 μM), N2 e B27 (1x), PD0325901 (1 μM), CHIR99021 (3 μM) e fator inibidor de leucemia (LIF) (1.000 U/mL).
- Preparar meio de diferenciação EB: DMEM suplementado com soro bovino fetal de 10% (FBS), aminoácidos não essenciais mem (1x), piruvato de sódio (1 mM), L-glutamina (2 mM), penicilina-estreptomicina (100 U/mL), beta-mercaptoetanol (50 μM).
- MESCs de cultura em pratos plásticos de 10 cm pré-revestidos com gelatina de 0,1% (w/v) em meio sem soro de cultura mESC.
- Quando os mESCs atingirem 60% de confluência, remova o meio de cultura e lave suavemente 1x com 2 mL de PBS esterilizado.
- Remova o PBS completamente e adicione 2 mL de meio de descolamento celular. Incubar o prato de cultura a 37 °C por 5 min.
- Inativar a reação adicionando 8 mL de meio de diferenciação EB no prato.
- Dissociar as colônias mESC, pipetando para cima e para baixo 15-20 vezes para obter uma suspensão unicelular.
- Centrifugar as células a 300 x g à temperatura ambiente (RT) por 5 min e remover cuidadosamente o sobrenadante.
- Contagem de células (por exemplo, usando um hemocímetro).
- Resuspender a pelota celular com o meio de diferenciação EB e ajustar a concentração para 2 x 104 células/mL.
- Inverta a tampa de um prato de cultura de 15 cm e use uma pipeta multicanal de 200 μL para depositar gotas de 20 μL de células resuspensas (~400 células/gota) na tampa.
- Inverta a tampa cuidadosamente na câmara inferior e incuba rasteiro com quedas de suspensão a 37 °C com 5% de CO2 e 95% de umidade por 3 dias.
- Reúna os EBs lavando a tampa suavemente com 10 mL de PBS e transfira a suspensão contendo EB para um tubo de plástico de 50 mL.
- Coloque o tubo em RT por 30 min, de modo que os EBs afundem até o fundo pela gravidade. Remova cuidadosamente o sobrenadante.
- Recoloque os EBs suavemente com 10 mL de meio de diferenciação EB fresco e transfira para uma placa de Petri bacteriológica de 10 cm.
- Verifique a formação de EB 3-6 dias depois usando um microscópio invertido. Os EBs gerados devem ser redondos e homogêneos em tamanho.
- Incubar as culturas a 37 °C com 5% de CO2 e 95% de umidade. Os EBs continuarão a se diferenciar nas três camadas de germes e poderão ser coletados em vários pontos de tempo para análise.
2. Dissociação de EBs
- Recolher os EBs de dois a três pratos de 10 cm em um tubo de plástico de 50 mL. Centrifugar os EBs a 300 x g em RT por 5 min, em seguida, remover cuidadosamente o sobrenadante.
- Resuspender os EBs com 10 mL de PBS. EBs centrífugas a 300 x g em RT por 3 min e remova o sobrenadante.
- Adicionar 2 mL de trypsin-EDTA (0,25%) para a pelota e incubar o tubo a 37 °C por 15 min. Pipeta para cima e para baixo a cada 3 minutos para obter uma suspensão unicelular.
- Adicione 8 mL de meio de diferenciação EB para parar a reação de tripsina. Verifique a dissociação do EB sob o microscópio e conte as células.
3. Fixação
- Ressuspensão das células em meio de cultura EB fresco a 1 x 106 células/mL. Para um tubo de 50 mL, use um máximo de 4,5 x 107 células em 45 mL de meio.
- Adicione o paraformaldeído de um estoque de 37% (não mais de 6 meses) a uma concentração final de 1%.
ATENÇÃO: Siga as normas de saúde e segurança adequadas durante o manuseio do paraformaldeído por ser um produto químico perigoso. - Incubar por 10 min em RT sob rotação.
- Sacie o formaldeído adicionando glicina a uma concentração final de 0,125 M.
- Incubar por 5 min em RT em rotação.
- Transfira as células fixas para gelo e mantenha frio a 4 °C a partir de agora.
- Pelota as células a 300 x g por 5 min em uma centrífuga refrigerada.
- Descarte o sobrenadante e resuspenda a pelota em PBS frio (1 mL para 5 x 106 células), em seguida, transfira para tubos de bloqueio de 1,5 mL.
- Pelotaas as células a 300 x g por 5 min a 4 °C, descarte o sobrenadante e congele as pelotas em nitrogênio líquido. Armazene a -80 °C ou siga com o protocolo abaixo.
4. Lse celular e enzima de restrição digerir
- Ressuspensão suave da pelota celular em 0,25 mL de tampão de lyse gelada recém-preparado (10 mM Tris-HCl pH = 8,0, 10 mM NaCl, 0,2% Inibidores de Igepal CA630 e 1x protease) por 2-5 x 106 células. Para preparar 5 mL de tampão de lysis, consulte Tabela 1.
- Incubar as células por 15 min no gelo.
- Centrífuga a 1.000 x g por 5 min a 4 °C. Descarte o sobrenadante e mantenha a pelota, que contém os núcleos.
- Lave os núcleos pelleted com 500 μL de tampão de lyse fria.
- Ressuspena suavemente a pelota em um tubo de 1,5 mL com 50 μL de 0,5% de SDS em 1x tampão 2, em seguida, incubar o tubo em um bloco de aquecimento a 62 °C por 10 min.
- Remova os tubos do bloco de aquecimento e adicione 170 μL de tampão de digestão contendo 25 μL de 10% de triton X-100 para saciar o SDS. Misture bem por pipetting, evitando espuma excessiva.
- Incubar a 37 °C por 15 min.
- Adicione 25 μL de tampão de digestão, misture invertendo e tome 8 μL como um controle não digerido. Mantenha a amostra de controle não digerida a -20 °C. Adicione a enzima de restrição 100 U MboI (4 μL de um estoque de 25 U/μL) aos núcleos restantes e digerir a cromatina por 2 h a 37 °C em rotação. Adicione outra alíquota de 100 U de MboI e incubar por mais 2 h.
- Adicione mais 100 U de MboI e incubar sob rotação durante a noite a 37 °C.
- No dia seguinte, adicione mais 100 U de MboI e incubar por 3 h a 37 °C em rotação.
- Tome 8 μL como uma amostra de controle digerida. De-crosslink digerido amostras de controle e as amostras de controle não digeridas da etapa 4.8 adicionando 80 μL de tampão TE (10 mM Tris pH = 8, 1 mM EDTA) e 10 μL de proteinase K (10 mg/mL). Incubar a 65 °C por 1h.
- Execute as alíquotas de 20 μL em um gel de 0,6% para verificar a eficiência da digestão. As digestãos bem sucedidas mostram principalmente fragmentos na faixa de 3,0-0,5 kb.
5. Ligadura de proximidade e reversão de crosslink
- Incubar as amostras digeridas pelo MboI a 65 °C em um bloco de aquecimento por 20 min para inativar o MboI e, em seguida, esfriar para RT.
- Centrifugar os tubos por 5 min a 1.000 x g em RT, remover o sobrenadante e dissolver a pelota em 200 μL de tampão ligase fresco.
- Adicione 1.000 μL da mistura mestre de ligadura a cada amostra. Para preparar 1.000 μL da mistura master de ligadura, consulte Tabela 2.
- Misture invertendo e incubando em RT durante a noite com rotação lenta (9 rpm).
- Elimine o RNA e os resíduos de proteína adicionando 100 μL de proteinase K (10 mg/mL) e 10 μL de RNase A (10 mg/mL). Incubar amostras a 55 °C por 45-60 min.
- Continue incubando amostras a 65 °C por mais 4 h.
6. Scisamento de DNA e seleção de tamanho
- Tubos frios para RT.
- Centrífuga por 5 min a 1.000 x g a 4 °C.
- Divida a amostra em três alíquotas de 400 μL em tubos de 2 mL e adicione 2 μL de glicogênio (20 mg/m), 40 μL de acetato de sódio (3 M, pH = 5,2) e volumes de 2,5x (1 mL) de 100% de etanol para cada tubo. Misture invertendo e incubando a -80 °C por 45-60 min.
- Centrifugação a 16.000 x g a 4 °C por 25 min. Mantenha os tubos no gelo depois de girar e remova cuidadosamente o sobrenadante encanando.
- Lave as pelotas de DNA resuspendendo em 800 μL de 70% de etanol. Centrífuga a 16.000 x g a 4 °C por 5 min.
- Remova o sobrenadante e realize a lavagem mais uma vez com 800 μL de 70% de etanol.
- Dissolver a pelota em 130 μL de tampão tris de 1x (10 mM Tris-HCl, pH = 8) e incubar a 37 °C por 15 min para dissolver totalmente o DNA. Se necessário, use pipetting para resuspender qualquer precipitado.
- Medir o rendimento do DNA; 2,5-5 μg de cromatina pode ser esperado para 1 x 106 células. Verifique a ligadura executando ± 200 ng do produto 3C em um gel de agarose de 0,6%. Ligaduras bem sucedidas mostram principalmente fragmentos de DNA > 3 kb. Armazene as amostras a -20 °C.
- Diluir uma amostra em um tubo de 0,65 mL adequado para sonação a 10 ng/μL em 100 μL de 1x tris volume tampão (1 μg por tubo). A quantidade padrão utilizada para a preparação da biblioteca é de 3 μg, por isso realize a sônica em três tubos separados, se necessário.
- Cisalhamento de DNA para um tamanho de 150-700 bp (média = 400-500 bp) usando os seguintes parâmetros no sônico: Ciclos: 6-8 de 20 s em -60 s off. Isso deve tornar o DNA adequado para a preparação da biblioteca de sequenciamento de alto throughput usando sequenciadores de Illumina.
- Transfira o DNA para um novo tubo de bloqueio normal. Pool múltiplas sônicas da mesma amostra.
- Aqueça uma garrafa de contas de purificação de DNA no RT. De agora em diante, use pontas baixas de ligação.
- Adicione 1,8x volumes de contas ao tubo de DNA e resuspenda suavemente.
- Incubar no RT por 5 min.
- Colete as contas com um rack magnético. Lave as contas 2x com 1 mL de etanol recém-preparado, mantendo os tubos no rack magnético.
NOTA: Remova todo o etanol, incluindo gotículas residuais. - Seque as contas a ar brevemente (2-3 min) em RT.
NOTA: Não seque contas maiores que 5 min. Isso diminuirá o rendimento do DNA. - Resuspender as contas com 90 μL de tampão tris de 1x (10 mM Tris-HCl, pH = 8) para eluir o DNA.
- Meça o rendimento do DNA e analise uma alíquota de 5 μL em um gel de 1,5%. Deve haver muito pouca perda em relação ao rendimento da prisão.
7. Preparação da biblioteca para sequenciamento
- Adicione 15 μL de mix mestre do kit de preparação da biblioteca. Para reparar as extremidades do DNA cortado, combine 10 μL de tampão de reação de reparo final de 10x e 5 μL de mistura de enzimas de reparo final.
- Incubar em RT por 30 min.
- Adicione 1,1x volume de contas de purificação de DNA e resuspenda suavemente.
- Incubar no RT por 5 min.
- Colete as contas com um rack magnético. Lave as contas duas vezes com 1 mL de etanol recém-preparado, mantendo os tubos no rack magnético. Remova o etanol.
- Seque a ar das contas por 2-3 min em RT. Resuspenda as contas com 42 μL de tampão tris 1x (10 mM Tris-HCl, pH = 8) para eluir o DNA.
- Adicione 8 μL de mistura mestre de rejeitos dA a cada amostra. Para preparar a mistura mestre de rejeito dA, combine 5 μL de tampão de reação de rejeito de 10x dA e 3 μL de fragmento de Klenow exo menos.
- Incubar a 37 °C por 30 min.
- Adicione 2 μL de fosfatase alcalina intestinal (CIP) ao DNA desfosforilato.
- Incubar por 30 min a 37 °C, depois 60 min a 50 °C.
- Adicione 1,1x volumes de contas de purificação de DNA e resuspenda suavemente.
- Incubar no RT por 5 min.
- Colete as contas com um rack magnético. Lave as contas 2x com 1 mL de etanol recém-preparado, mantendo os tubos no rack magnético.
- Seque brevemente as contas (2-3 min) em RT. Resuspenda as contas com 35 μL de tampão tris 1x (10 mM Tris-HCl, pH = 8) para eluir o DNA.
- Realize a reação de ligadura do adaptador. Utilize concentrações reduzidas de adaptador/ligase como mencionado na Tabela 3.
- Incubar a 20 °C por 15 min.
- Adicione 3 μL da mistura de uracil DNA glicosilase e DNA glicosilase lyase endonuclease VII (por exemplo, USER), misture por pipetting e incubar a 37 °C por 15 min.
- Aumente o volume para 100 μL com água, ferva 5 min a 96 °C e mantenha as amostras no gelo.
- Adicione 1,1x volumes de contas de purificação de DNA e resuspenda suavemente.
- Incubar no RT por 5 min.
- Colete as contas com um rack magnético. Lave as contas 2x com 1 mL de etanol recém-preparado, mantendo os tubos no rack magnético.
- Seque a ar das contas por 2-3 min em RT. Resuspenda as contas com 50 μL de tampão tris 1x (10 mM Tris-HCl, pH = 8) para eluir o DNA.
8. 4C cromatina biblioteca de interação amplificação e purificação
- Amplie a biblioteca 4C usando 10 μL de biblioteca para realizar o primeiro PCR. A configuração e o programa pcr podem ser encontrados na Tabela 4.
- Executar PCR aninhado. A configuração e o programa PCR aninhados podem ser encontrados na Tabela 5.
- Pool produtos PCR para cada biblioteca e purificar com uma esfera de purificação de DNA de 1,1x.
- Meça o rendimento do DNA e analise uma alíquota de 5 μL em um gel de 1,5%.
- Ajuste a concentração da biblioteca e sequencie a biblioteca. Se indexadas, as bibliotecas podem ser agrupadas antes do sequenciamento.
Resultados
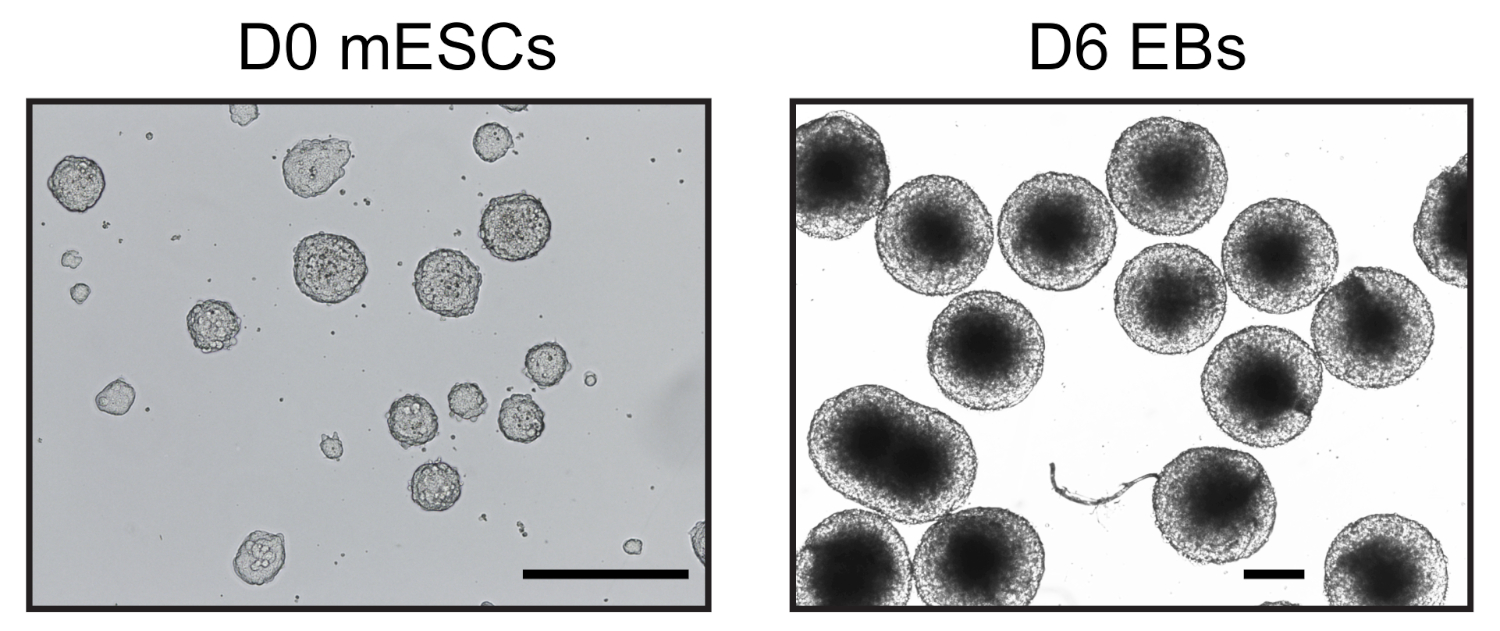
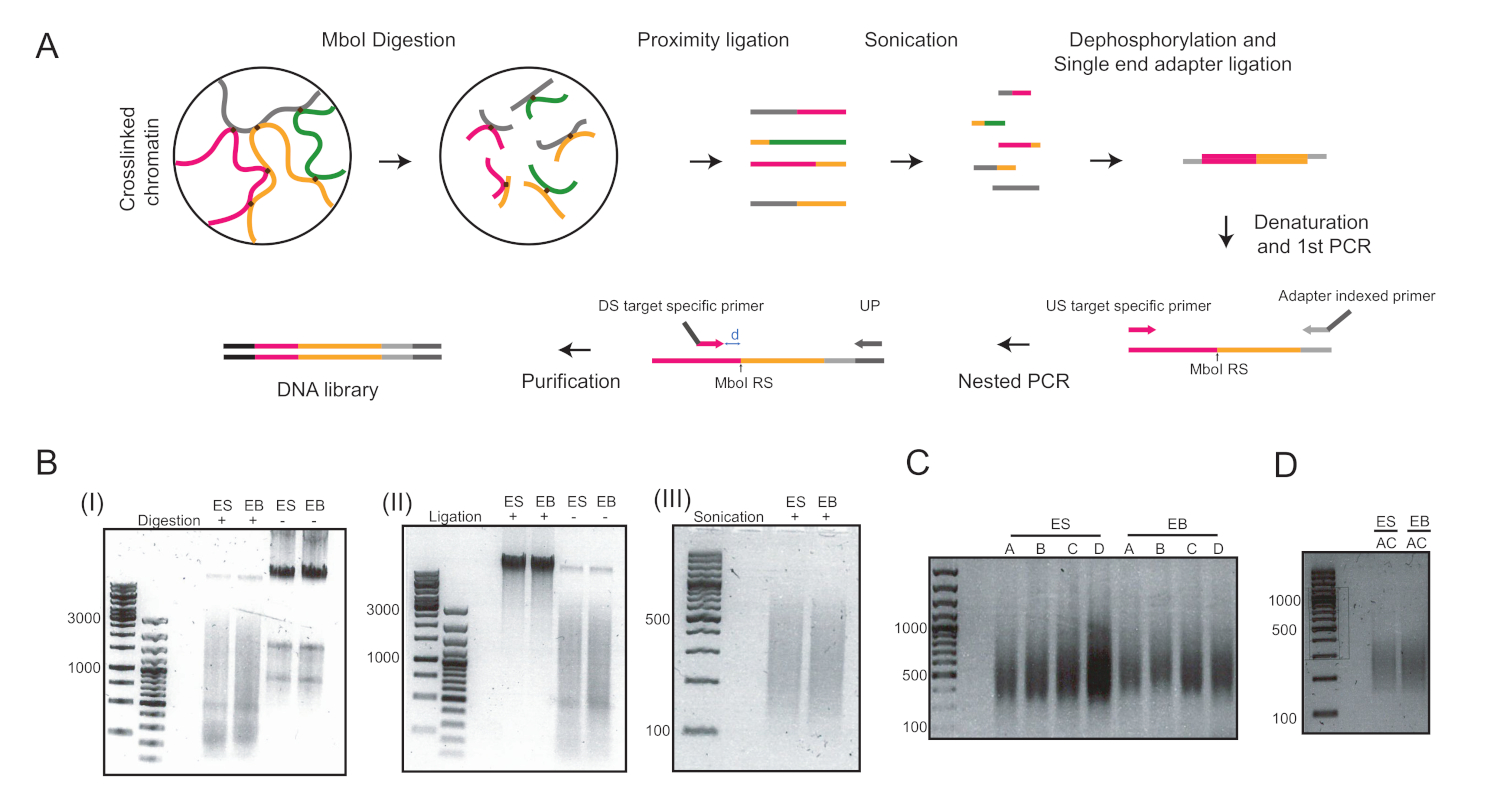
Seis dias após a indução da diferenciação da ESC nas gotas de enforcamento, obtivemos uma população homogênea de EBs que foram utilizadas para análises posteriores (Figura 1). Adaptamos o método UMI-4C23 para quantificar a interação específica com cromatina em promotores de genes específicos de linhagem nos EBs24. Uma visão geral esquemmática do protocolo com géis representativos de controle de qualidade em diferentes etapas é mostrada na Figura 2A. O primeiro controle de qualidade foi realizado para determinar a eficiência da digestão da enzima de restrição MboI. A digestão eficiente mostrou um tamanho de fragmento inferior a 3 kbp(Figura 2B). Note-se que a digestão da cromatina mESC e EB foi difícil e, por vezes, a cromatina residual não digerida persistiu. O segundo controle de qualidade foi realizado após a ligadura para verificar se a maioria dos fragmentos eram agora > 3 kbp(Figura 2B). Em seguida, fragmentos de cromatina obtidos após a sonagem foram analisados por eletroforese em gel. Eram esperados tamanhos de fragmentos de 400-500 bp (Figura 2B).
Após desfosforilação e ligadura de adaptador single-end, duas rodadas de PCR foram realizadas para amplificar as metas de interesse. Uma abordagem aninhada foi usada para projetar um conjunto de dois primers para cada lócus. Isso ajudou a melhorar a especificidade. Cada alvo foi amplificado separadamente com dois pares de primer diferentes para otimizar as condições de PCR (ou seja, pares de primer A e B para os pares de lócus e primer Pou5f1 C e D para o lócus T, respectivamente) e resultou em uma mancha de DNA em torno de 400 bp(Figura 2C). Alternativamente, multiplex PCR foi realizado para amplificar os alvos A e C simultaneamente (Figura 2D) e resultou em um tamanho de fragmento semelhante após a purificação(Figura 2D). Os primers usados para a preparação da biblioteca 4C (loci de Pou5f1 e T) podem ser encontrados na Tabela 6.
Para análise de dados, as leituras de sequenciamento bruto foram primeiro alinhadas com o genoma do rato mm10, todas duplicadas e leituras de baixa qualidade (< 20) foram removidas. Para cada isca, as informações sobre cada fragmento de restrição foram obtidas com base no número de fragmentos de leitura, e um perfil de contato bruto foi obtido. Em seguida, a região de interesse foi definida como todos os fragmentos de restrição com 2 kbp e 250 kbp de distância para a isca. O tamanho de cada fragmento de restrição foi aumentado pela agregação dos fragmentos de restrição adjacentes sequencialmente para suavizar os perfis até que um limiar de 5% do número total de contatos brutos fosse atingido na região de interesse. Para garantir que as réplicas fossem integradas e as condições fossem comparadas, incluímos encostas e interceptações aleatórias no nível do fragmento de restrição. O perfil médio por condição e a variação da dobra entre eles foram traçados como mostrado na Figura 3. Durante a diferenciação do EB, os contatos entre os intensificadores e o promotor do gene de pluripotência Pou5f1 diminuíram, enquanto os contatos intensificadores-promotores da linhagem mesendoderm fator de transcrição instrutiva T aumentaram(Figura 3),fornecendo insights funcionais sobre esses intensificadores de desenvolvimento.

Figura 1: Imagens representativas de mESC e corpos embriodados derivados. Dia 0 mESC cultivado em condições livres de soro (esquerda) e dia homogêneo 6 EBs (direita) observados por um microscópio invertido. Barra de escala = 500 μm. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 2: fluxo de trabalho 4C e imagens representativas das principais etapas do protocolo. (A) Fluxo de trabalho esquemático do quantitativo 4C. RS = site de restrição; EUA = rio acima; DS = rio abaixo; UP = primer universal; D = a distância entre RS e DS deve ser idealmente de 5-15 bp.(B) Exemplos de cromatina digerida por MboI (I), cromatina ligada nos núcleos (II) e cromatina sônica (III). Os números à esquerda indicam os tamanhos de DNA determinados pela escada de DNA para cada amostra. (C) Exemplos de amplificação pcr nos dois loci: Pou5f1 (primers A e B) e T (primer C e D). (D) Exemplos de amplificação multiplex PCR em Pou5f1 e T loci utilizando primers A e C. ES = células-tronco embrionárias; EB = corpos embrionários. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 3: Exemplos de perfis 4C. Perfis quantitativos 4C para iscas localizadas nos promotores de genes Pou5f1 e T ensaios em mESCs e Dia 6 EBS. O painel superior mostra parcelas de contatos médios gerados a partir de duas réplicas biológicas independentes; o painel inferior mostra a mudança média da dobra de contato dos EBs do dia 6 em relação aos mESCs (média das duas réplicas). Caixas azuis claras indicam a localização de melhoradores com mudanças dinâmicas durante a diferenciação. Figura adaptada de Tian et al.24. Clique aqui para ver uma versão maior desta figura.
{kind=link}
| Para 5mL | |
| 1M Tris-HCl, pH8.0 | 50 μL |
| 5M Nacl | 10 μL |
| 10% Igepal CA630 | 100 μL |
| 50x Inibidores completos de protease da Roche | 100 μL |
| Água de MilliQ | 4,74 mL |
Tabela 1: Tampão de lysis.
| Para 1000μL | |
| Água de MilliQ | 869 μL |
| 10X NEB T4 DNA Ligase Buffer | 120 μL |
| 20mg/mL Soro bovino Albumina | 6 μL |
| Ligase de DNA U/μL T4 2000 | 5 μL |
Tabela 2: Preparação do mix mestre de ligadura.
| Por 15 μL | |
| Buffer de reação de ligadura rápida 5X | 10 μL |
| Adaptador NEBNext | 3 μL |
| Ligase de DNA Rápido T4 | 2 μL |
Tabela 3: Reação de ligadura do adaptador.
| Configuração pcr | |
| Adaptor ligado biblioteca-sobre-deads | 10 μL |
| Água de grau PCR | 20,25 μL |
| Primer específico de 10 μM | 3,75 μL |
| Primer de índice NEB de 10 μM | 3,75 μL |
| Tampão herculase II 5X | 10 μL |
| DNTPs de 10 Mm | 1,25 μL |
| Polimerase Herculase II | 1 μL |
| Volume total | 50 μL |
| Programa PCR | |
| Passo 1: 98 °C - 2 min | |
| Passo 2: 98 °C - 20s | |
| Passo 3: 65 °C - 30s | |
| Passo 4: 72 °C - 45s | |
| Passo 5: vá para o passo 2 para fazer um total de 15-18 ciclos | |
| Passo 6: 72 °C - 3 min | |
| Passo 7: 4 °C – segurar |
Tabela 4: Amplificação da biblioteca de interação cromatina 4C, primeiro PCR.
| Configuração PCR aninhada | |
| Fragmento de DNA do primeiro PCR | 10 μL |
| Água de grau PCR | 20,25 μL |
| Primer específico de 10 μM+P5 Illumina primer | 3,75 μL |
| 10 μM P7 Illumina primer | 3,75 μL |
| Tampão herculase II 5X | 10 μL |
| DNTPs de 10 Mm | 1,25 μL |
| Polimerase Herculase II | 1 μL |
| Volume total | 50 μL |
| Programa PCR aninhada | |
| Passo 1: 98 °C - 2 min | |
| Passo 2: 98 °C - 20s | |
| Passo 3: 65 °C - 30s | |
| Passo 4: 72 °C - 45s | |
| Passo 5: vá para o passo 2 para fazer um total de 15-18 ciclos | |
| Passo 6: 72 °C - 3 min | |
| Passo 7: 4 °C – segurar |
Tabela 5: Amplificação da biblioteca de interação cromatina 4C, PCR aninhado.
| Nome | Seqüência (5'-3') |
| DS-Oct4-A | AATGATACGGCGACCGAGATCTACACTCTTTCCCTACACGACG CTCTCTCTTCTTGCAAAAAAAGCACCAGGCCAG |
| EUA-Oct4-A | TCTTGCAAAAGATAAAAAGCACCAGGCC |
| DS-Oct4-B | AATGATACGGCGACCGAGATCTACACTCTTTCCCTACACGACG CTCTCTCTGTGGGGGTCAGGGGGCgCGGGGGGGGGCT |
| EUA-Oct4-B | ACCAGGGGGGGGGGGGGTGTGTGGGGGGGGGGGGGGGGGGGG |
| DS-T-C | AATGATACGGCGACCGAGATCTACACTCTTTCCCTACACGACG CTCTCTCTCCTGGCCTGCACATTCGCCAAAGGAGC |
| EUA-T-C | GATTACACCTGGCCTGCACATTCGCCAA |
| DS-T-D | AATGATACGGCGACCGAGATCTACACTCTTTCCCTACACGACG CTCTCTCTGGTTTGgAGAGGTCAAGGAGACCCGGGAG |
| EUA-T-D | GCTGAGGCTTGGAGAGGTCAAGGAGACC |
| UP-4C | CAAGCAGAAGACGGCATACGA |
| Adap-i1 | CAAGCAGAAGACGGCATACGAGATCGTGATGACTGGAGTTCAGA CGTGTGCTCTTCCGATC |
| Adap-i2 | CAAGCAGAAGACGGCATACTACTACTACACGGTGACTGGAGTTCAGA CGTGTGCTCTTCCGATC |
| Adap-i3 | CAAGCAGAAGACGGCATACGAGATGCCAAGTGACTGGAGTTCAGA CGTGTGCTCTTCCGATC |
| Adap-i4 | CAAGCAGAAGACGGCATACGAGATTGGTGGTGACTGGAGTTCAGA CGTGTGCTCTTCCGATC |
Tabela 6: Primers usados para a preparação da biblioteca 4C.
Discussão
O método de cultura de queda suspensa não precisa de fatores de crescimento adicionais ou citocinas e gera reprodutivelmente populações homogêneas de EBs a partir de um número predeterminado de mESCs5. Aqui descrevemos um protocolo de 4C quantitativo adaptado da abordagem UMI-4C para quantificar o contato melhorador-promotor de fatores de transcrição específicos de linhagem no modelo de diferenciação eB. Identificamos regiões de cromatina que entram em contato com promotores de genes Pou5f1 e T de forma dinâmica durante a diferenciação do EB. O Pou5f1 foi desregulado durante a diferenciação do EB e a freqüência de contato entre o promotor Pou5f1 e seu potencializador distal diminuiu. Por outro lado, t foi regulado durante a diferenciação de EB e identificamos três melhoradores para os quais as frequências de contato com seu promotor são diminuídas (Figura 3). Para confirmar a identificação, um ensaio de imunoprecipitação de cromatina (ChIP) da marca de histona ativa H3K27ac pode ser realizado24, uma vez que esta marca histona tem se mostrado associada à ativação do intensificador e os intensificadores perdem essa marca durante sua inativação11.
Uma técnica 4C padrão tem sido amplamente utilizada para pesquisar o perfil de contato com cromatina de locais genômicos específicos25. No entanto, essa abordagem é difícil de interpretar quantitativamente mesmo após extensa normalização26,27,28 devido aos vieses introduzidos pela heterogeneidade do tamanho do fragmento de PCR e a impossibilidade de distinguir duplicatas pcr. Nosso método quantitativo 4C é em grande parte idêntico à técnica UMI-4C que permite a quantificação de moléculas únicas usando sonificação e um passo pcr mediado por ligadura aninhada para contornar a limitação da abordagem clássica 4C23. No entanto, ao contrário do UMI-4C que usa identificadores moleculares únicos, nosso protocolo quantitativo 4C permite a quantificação de moléculas únicas com base na quebra de DNA específica produzida pela etapa de sonificação. Torna nosso protocolo compatível com kits comerciais de preparação de bibliotecas de DNA, obviede a necessidade de primers com identificadores moleculares exclusivos.
Nosso protocolo envolve vários passos-chave que devem ser considerados. Como no método 4C clássico28, fatores críticos do nosso protocolo são a eficiência da digestão e a ligadura durante a preparação das moléculas 3C. A baixa eficiência de digestão/ligadura pode diminuir drasticamente a complexidade da interação com um fragmento de interesse, resultando em uma resolução reduzida. Como descrito anteriormente23, outra etapa crítica do protocolo é o design dos primers para a amplificação da biblioteca. Os segundo primers de reação pcr devem ser localizados 5-15 nt a partir do local de restrição interrogado. Em uma leitura de seqüenciamento de 75 nt, isso permite pelo menos 40 nt esquerda do comprimento de captura para mapeamento. O primer usado na primeira reação pcr deve ser projetado a montante do segundo primer sem sobreposição e ambos devem ser específicos o suficiente para garantir uma amplificação eficiente do DNA. Para multiplexação, os primers devem ser projetados independentemente, visando uma temperatura de fusão (Tm) de 60-65 °C. Além disso, quanto a outras técnicas 3C, a resolução do método quantitativo 4C é determinada pela enzima de restrição utilizada no protocolo25. Este protocolo usa uma enzima de restrição com um site de reconhecimento de 4 bps, MboI. A resolução máxima com esta enzima é de cerca de 500 bp, mas esta é altamente dependente de lócus e raramente alcançada. Outra limitação é que as interações que ocorrem entre elementos localizados no mesmo fragmento de restrição não são detectáveis. Além disso, as interações que ocorrem a uma distância de um local de restrição não podem ser distinguidas do fundo não digerido. O uso de uma etapa de preenchimento antes da ligadura pode permitir a detecção dessas interações.
Quantitative 4C é ideal para interrogar contatos de cromatina de loci alvo. No entanto, a etapa específica de amplificação do PCR limita o número de loci que podem ser investigados simultaneamente. Uma maneira de aumentar o número de loci direcionados é multiplexar as etapas do PCR para amplificar simultaneamente vários alvos, mas isso requer compatibilidade dos primers usados e testes de cada par de primer antes da implementação. Se forem desejadas mudanças globais na arquitetura de cromatina nos promotores, abordagens em todo o genoma como Hi-C, PC Hi-C ou HiChIP seriam mais apropriadas29,,30,,31.
Divulgações
Os autores não têm nada para revelar.
Agradecimentos
Gostaríamos de agradecer a F. Le Dily, R. Stadhouders e membros do laboratório Graf por seus conselhos e discussões. G.S. foi apoiado por uma bolsa marie sklodowska-curie (H2020-MSCA-IF-2016, miRStem), T.V.T por uma bolsa de pós-doutorado Juan de la Cierva (MINECO, FJCI-2014-22946). Este trabalho foi apoiado pelo Conselho Europeu de Pesquisa no âmbito do7º Programa-Quadro FP7 (ERC Synergy Grant 4D-Genome, convênio de subvenção 609989 à T.G.), pelo Ministério da Economia, Indústria e Competitividade da Espanha (MEIC) à parceria da EMBL, Centro de Excelencia Severo Ochoa 2013-2017 e Ao Programa CERCA Generalitat de Catalunya.
Materiais
| Name | Company | Catalog Number | Comments |
| 0.1% EmbryoMax gelatin | EMD Millipore | ES-006-B | Cell culture |
| 0.25% Trypsin-EDTA | 25200072 | ||
| AMPure XP | Beckman Coulter | 10136224 | 4C/DNA purification |
| B27 supplement | Gibco | 17504044 | Cell culture |
| Beta-mercaptoethanol | Gibco | 31350010 | Cell culture |
| Bioruptor Pico | Diagencode | B01060010 | 4C/sonication |
| BSA | NEB | B9000S | 4C |
| CHIR99021 | Selleck Chemicals | S1263 | Cell culture |
| CIP | NEB | M0212 | 4C |
| cOmplete Protease Inhibitor Cocktail | Roche | 4693116001 | 4C |
| DMEM/F12 medium | Gibco | 11320033 | Cell culture |
| dNTP | NEB | N0447S | 4C |
| ESGRO Leukaemia Inhibitory Factor (LIF) | EMD Millipore | ESG1107 | Cell culture |
| Formaldehyde solution (37%) | Sigma | 252549-25ML | 4C |
| Glycin | Sigma | GE17-1323-01 | 4C |
| Glycogen | ThermoFischer | R0551 | 4C |
| Herculase II Fusion DNA polymerase | Agilent | 600675 | 4C |
| IGEPAL CA-630 | Sigma | I3021-50ML | 4C |
| Knockout DMEM | 10829018 | ||
| L-glutamine | Gibco | 25030081 | Cell culture |
| MboI | NEB | R0147M | 4C |
| MEM non-essential amino acids | Gibco | 11140050 | Cell culture |
| N2 supplement | Gibco | A1370701 | Cell culture |
| NEBNext DNA Library prep | NEB | E6040 | 4C |
| NEBuffer 2.1 | NEB | B7202S | 4C/digestion |
| Neurobasal medium | Gibco | 21103049 | Cell culture |
| PD0325901 | Selleck Chemicals | S1036 | Cell culture |
| Penicillin Streptomycin | Gibco | 15140122 | Cell culture |
| Proteinase K | NEB | P8107S | 4C |
| Qubit 4 Fluorometer | ThermoFischer | Q33238 | 4C |
| Qubit dsDNA HS Assay Kit | ThermoFischer | Q32851 | 4C |
| RNase A | ThermoFischer | EN0531 | 4C |
| Sodium pyruvate solution | Gibco | 11360070 | Cell culture |
| StemPro Accutase Cell Dissociation Reagent | Gibco | A1110501 | Cell culture |
| T4 DNA Ligase Reaction Buffer | NEB | B0202S | 4C |
| T4 DNA Ligase Reaction Buffer | NEB | M0202M | 4C |
Referências
- Evans, M. J., Kaufman, M. H. Establishment in culture of pluripotential cells from mouse embryos. Nature. 292 (5819), 154-156 (1981).
- Martello, G., Smith, A. The nature of embryonic stem cells. Annual Review Cell and Developmental Biology. 30, 647-675 (2014).
- Martin, G. R. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proceedings of the National Academy of Science U. S. A. 78 (12), 7634-7638 (1981).
- Loh, K. M., Lim, B., Ang, L. T. Ex uno plures: molecular designs for embryonic pluripotency. Physiological Reviews. 95 (1), 245-295 (2015).
- Sheridan, S. D., Surampudi, V., Rao, R. R. Analysis of embryoid bodies derived from human induced pluripotent stem cells as a means to assess pluripotency. Stem Cells International. 2012, 738910 (2012).
- Gaszner, M., Felsenfeld, G. Insulators: exploiting transcriptional and epigenetic mechanisms. Nature Reviews in Genetics. 7 (9), 703-713 (2006).
- Lenhard, B., Sandelin, A., Carninci, P. Metazoan promoters: emerging characteristics and insights into transcriptional regulation. Nature Reviews in Genetics. 13 (4), 233-245 (2012).
- Long, H. K., Prescott, S. L., Wysocka, J. Ever-Changing Landscapes: Transcriptional Enhancers in Development and Evolution. Cell. 167 (5), 1170-1187 (2016).
- Schoenfelder, S., Fraser, P. Long-range enhancer-promoter contacts in gene expression control. Nature Reviews in Genetics. 20 (8), 437-455 (2019).
- Spitz, F., Furlong, E. E. Transcription factors: from enhancer binding to developmental control. Nature Reviews in Genetics. 13 (9), 613-626 (2012).
- Creyghton, M. P., et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proceedings of the National Academy of Sciences U. S. A. 107 (50), 21931-21936 (2010).
- Heintzman, N. D., et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 459 (7243), 108-112 (2009).
- Klemm, S. L., Shipony, Z., Greenleaf, W. J. Chromatin accessibility and the regulatory epigenome. Nature Reviews in Genetics. 20 (4), 207-220 (2019).
- Rada-Iglesias, A., et al. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 470 (7333), 279-283 (2011).
- Lettice, L. A., et al. Development of five digits is controlled by a bipartite long-range cis-regulator. Development. 141 (8), 1715-1725 (2014).
- Dekker, J., Rippe, K., Dekker, M., Kleckner, N. Capturing chromosome conformation. Science. 295 (5558), 1306-1311 (2002).
- de Wit, E., de Laat, W. A decade of 3C technologies: insights into nuclear organization. Genes and Development. 26 (1), 11-24 (2012).
- Lieberman-Aiden, E., et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 326 (5950), 289-293 (2009).
- Simonis, M., et al. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nature Genetics. 38 (11), 1348-1354 (2006).
- Splinter, E., de Wit, E., van de Werken, H. J., Klous, P., de Laat, W. Determining long-range chromatin interactions for selected genomic sites using 4C-seq technology: from fixation to computation. Methods. 58 (3), 221-230 (2012).
- Stadhouders, R., et al. Multiplexed chromosome conformation capture sequencing for rapid genome-scale high-resolution detection of long-range chromatin interactions. Nature Protocols. 8 (3), 509-524 (2013).
- van de Werken, H. J., et al. Robust 4C-seq data analysis to screen for regulatory DNA interactions. Nature Methods. 9 (10), 969-972 (2012).
- Schwartzman, O., et al. UMI-4C for quantitative and targeted chromosomal contact profiling. Nature Methods. 13 (8), 685-691 (2016).
- Tian, T. V., et al. Whsc1 links pluripotency exit with mesendoderm specification. Nature Cell Biology. 21 (7), 824-834 (2019).
- Chen, H., et al. Dynamic interplay between enhancer-promoter topology and gene activity. Nature Genetics. 50 (9), 1296-1303 (2018).
- Apostolou, E., et al. Genome-wide chromatin interactions of the Nanog locus in pluripotency, differentiation, and reprogramming. Cell Stem Cell. 12 (6), 699-712 (2013).
- de Wit, E., et al. The pluripotent genome in three dimensions is shaped around pluripotency factors. Nature. 501 (7466), 227-231 (2013).
- Krijger, P. H. L., Geeven, G., Bianchi, V., Hilvering, C. R. E., de Laat, W. 4C-seq from beginning to end: A detailed protocol for sample preparation and data analysis. Methods. , (2019).
- Mumbach, M. R., et al. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nature Methods. 13 (11), 919-922 (2016).
- Rao, S. S., et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 159 (7), 1665-1680 (2014).
- Schoenfelder, S., Javierre, B. M., Furlan-Magaril, M., Wingett, S. W., Fraser, P. Promoter Capture Hi-C: High-resolution, Genome-wide Profiling of Promoter Interactions. Journal of Visualized Experiments. (136), e57320 (2018).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados