
Method Article
Идентификация контактов Enhancer-Promoter в эмбриоидных телах по количественному захвату конформации хромосомы (4C)
В этой статье
Резюме
Мы сообщаем о применении количественного улавливания конформации хромосом с последующим секвенированием с высокой пропускной способностью эмбриональных тел, генерируемых из эмбриональных стволовых клеток. Этот метод позволяет определить и количественно контакты между преподобными усилителями и промоториками регионов данного гена во время дифференциации эмбриональных стволовых клеток.
Аннотация
Во время развития млекопитающих, судьбы клеток определяются путем создания регуляторных сетей, которые определяют специфику, сроки и пространственные модели экспрессии генов. Эмбрибридальные тела (EBs), полученные из плюрипотентных стволовых клеток, были популярной моделью для изучения дифференциации трех зародышевых слоев и определения регуляторных схем во время спецификации судьбы клеток. Хотя хорошо известно, что конкретные ткани усилители играют важную роль в этих сетях, взаимодействуя с промоутерами, назначение их их соответствующих генов цели по-прежнему остается сложной задачей. Для того чтобы это стало возможным, необходимы количественные подходы для изучения контактов с усилителем-промоутером и их динамикой в процессе разработки. Здесь мы адаптировали метод 4C для определения усилителей и их контактов с коньяк-промоутерами в модели дифференциации EB. Метод использует часто резки ограничения ферментов, sonication, и вложенных-лиги опосредованного ПЦР протокол совместим с коммерческими наборами подготовки библиотеки ДНК. Впоследствии библиотеки 4C подвергаются секвенированию и анализу биоинформации с высокой пропускной способностью и анализируются биоинформатически, что позволяет выявлять и количественно анализировать все последовательности, которые имеют контакты с выбранным промоутером. Полученные данные по секвенированию также могут быть использованы для получения информации о динамике контактов с усилителем-промоутером во время дифференциации. Метод, описанный для модели дифференциации EB, прост в реализации.
Введение
У мышей внутренняя клеточная масса (ICM) 3,5-дневных эмбрионов содержит эмбриональные плюрипотентные стволовые клетки. ICM далее развивается в эпибласт на 4,5 день, генерации эктодерм, мезодерм, и эндодерм овых клеток, основных трех слоев зародыша в эмбрионе. Хотя плюрипотентные клетки в ICM существуют только преходяще in vivo, они могут быть захвачены в культуре путем создания эмбриональных стволовых клеток мыши (mESCs)1,2,3. MESCs остаются в недифференцированном состоянии и размножаются бесконечно, но на внутренних и выхнанных стимулов они также способны выхода из состояния плюрипотентности и генерации клеток из трех слоев зародышевых развития2,4. Интересно, что при культуре в подвеске в небольших капель, mESCs образуют трехмерные агрегаты (т.е. EBs), которые дифференцируются во все три слоя зародыша5. Исследование формирования EB является важным инструментом для изучения процесса спецификации ранней линии.
Во время спецификации линии, клетки каждого слоя зародыша приобретают специфическую программу экспрессии гена4. Точное пространственно-временное выражение генов регулируется различными цис-регуляторными элементами, включая основные промоторы, усилители, глушители и изоляторы6,,77,8,9. Enhancers, регулятивные сегменты ДНК, как правило, охватывающих несколько сотен пар базы, координировать ткани конкретных экспрессии генов8. Усилители активируются или заглушаются при связывании транскрипционных факторов и кофакторов, которые регулируют местную структуру хроматина8,10. Широко используемые методы для выявления предполагаемого усилителей генома всей хроматина иммунопрециция следуют секвенирования (ChIP-seq) и анализ транспозаз-доступных хроматин с использованием последовательности (ATAC-seq) методы. Таким образом, активные усилители характеризуются специфическими активными гистонными знаками и повышенной локальной доступностью ДНК11,,12,,13,,14. Кроме того, развитие усилители, как полагают, требуют физического взаимодействия с их коньяк промоутер8,9. Действительно, было показано, что усилитель варианты и удаления, которые нарушают усилитель-промоутер контакты могут привести к развитию пороков15. Поэтому существует необходимость в новых методах, которые предоставляют дополнительную информацию для идентификации функциональных усилителей, которые контролируют экспрессию генов развития.
С момента развития метода захвата хромосомного захвата (3С)16,картирование хромосомных контактов интенсивно используется для оценки физического расстояния между регулятивными элементами. Важно отметить, что в последнее время были разработаны высокопроизводительные варианты 3C-методов, обеспечивающие различные стратегии фиксации, пищеварения, перевязки и восстановления контактов между фрагментами хроматина17. Среди них, на месте Hi-C стала популярной техникой, позволяющей секвенировать 3C перевязочные продукты генома в ширину18. Тем не менее, высокие затраты на секвенирование, необходимые для достижения разрешения, подходящего для анализа контактов с усилителем-промоутером, делают этот метод нецелесообразным для изучения конкретных локусов. Поэтому были разработаны альтернативные методы для анализа целевых локутов при более высоком разрешении19,,20,,21,,22. Один из этих методов, а именно 4C, известный как один против всех стратегий, позволяет обнаруживать все последовательности, которые контактируют с выбранным в качестве точки зрения сайтом. Однако недостатком стандартной техники 4C является обратный ПЦР, который усиливает фрагменты разного размера, отдав предпочтение небольшим продуктам и предусмотрителям количественной оценки после секвенирования высокой пропускной способности. Недавно, UMI-4C, новый вариант техники 4C с использованием уникальных молекулярных идентификаторов (UMI) был разработан для количественного и целевого хромосомного профилирования контактов, что обходит эту проблему23. Этот подход использует частые резцы, sonication, и вложенно-ligation-опосредованного протокола PCR, таким образом включая усиление фрагментов дна с относительно равномерным распределением длины. Эта однородность уменьшает предубеждения в процессе усиления преференций ПЦР для более коротких последовательностей и позволяет эффективно восстанавливать и точно подсчитывать пространственно соединенные молекулы/фрагменты.
Здесь мы описываем протокол, который адаптирует метод UMI-4C для выявления и количественной оценки контактов хроматина между промоутерами и усилителями поучительных транскрипционных факторов линии во время дифференциации EB.
протокол
1. Эмбриодидное тело из эмбриональных стволовых клеток мыши
- Подготовка mESC безсыворотки среды культуры: DMEM / F12 и нейробазальной среды смешанных в соотношении 1:1. Культурная среда дополнена раствором несущественных аминокислот MEM (1x), пируватом натрия (1 мМ), L-глютамином (2 мМ), пенициллином-стрептомицином (100 U/mL), бета-меркапто Добавки N2 и B27 (1x), PD0325901 (1 мкм), CHIR99021 (3 МКм) и ингибиторный фактор лейкемии (LIF) (1000 U/mL).
- Подготовка СРЕДы дифференциации EB: DMEM дополнен 10% сыворотки крупного рогатого скота плода (FBS), MEM несущественные аминокислоты (1x), пируват натрия (1 мМ), L-глутамамин (2 мМ), пенициллин-стрептомицин (100 U/mL), бета-меркаптоэтанол (50 Мм).
- Культура mESCs на 10 см пластиковые блюда предварительно с покрытием 0,1% (w/v) желатин в mESC культуры сыворотки свободной среде.
- Когда mESCs достичь 60% вспыхивания, удалить среду культуры и мыть осторожно 1x с 2 мл стерилизованных PBS.
- Удалить PBS полностью и добавить 2 мл среды отслоения клеток. Инкубировать культурное блюдо при 37 градусах по Цельсию в течение 5 мин.
- Инактивируйте реакцию, добавляя 8 мл среды дифференциации EB в блюдо.
- Диссоциативные колонии mESC путем pipetting вверх и вниз 15-20 раз, чтобы получить одноклеточную подвеску.
- Центрифуги клетки на 300 х г при комнатной температуре (RT) в течение 5 мин и тщательно удалить супернатант.
- Подсчитывайте клетки (например, с помощью гемоситометра).
- Отрежь клеточные гранулы со средой дифференциации EB и отрегулируйте концентрацию до 2 х 104 ячеек/мл.
- Переворачивать крышку 15-сантиметрового блюда культуры и использовать 200 л многоканальной пипетки, чтобы нанести 20 капель l resuspended клеток (400 клеток/капли) на крышке.
- Тщательно переворачивайте крышку на нижнюю камеру и инкубируйте блюдо висячими каплями при температуре 37 градусов с 5% CO2 и 95% влажности в течение 3 дней.
- Соберите EBs, аккуратно промыв крышку 10 мл PBS и перенесите содержащую EB подвеску на пластиковую трубку 50 мл.
- Поместите трубку на RT в течение 30 минут, так что EBs опустятся на дно под действием силы тяжести. Аккуратно удалите супернатант.
- Отрежь EBs осторожно с 10 мл свежей среды дифференциации EB и передать 10 см бактериологического блюда Петри.
- Проверьте формирование EB 3-6 дней спустя с помощью перевернутого микроскопа. Генерируемые EBs должны быть круглыми и однородными по размеру.
- Инкубировать культуры при 37 градусах Цельсия с 5% CO2 и 95% влажности. EBs будут продолжать дифференцировать в 3 слоя зародыша и могут быть собраны на различных временных точках для анализа.
2. Диссоциация ЕБ
- Соберите EBs от двух до трех 10 см посуды в 50 мл пластиковой трубки. Центрифуге EBs на 300 x g на RT на 5 минут, затем тщательно удалить супернатант.
- Приостанавливай EBs с 10 мл PBS. Центрифуга EBs на 300 х г на RT в течение 3 мин и удалить супернатант.
- Добавить 2 мл трипсина-EDTA (0.25%) к гранулы и инкубировать трубку при 37 градусов по Цельсию в течение 15 мин. Пипетка вверх и вниз каждые 3 мин, чтобы получить одноклеточную подвеску.
- Добавьте 8 мл среды дифференциации EB, чтобы остановить реакцию трипсина. Проверьте диссоциацию EB под микроскопом и подсчитайте клетки.
3. Фиксация
- Resuspend клетки в свежей среде культуры EB на 1 х 106 ячеек/мл. Для трубки 50 мл используйте максимум 4,5 х 107 ячеек в 45 мл средней.
- Добавьте параформальдегид из 37% бульона (не старше 6 месяцев) до 1% конечной концентрации.
ВНИМАНИЕ: Следуйте соответствующим правилам здоровья и безопасности при обращении с параформальдегидом, поскольку это опасное химическое вещество. - Инкубировать в течение 10 минут на RT под вращением.
- Утолить формальдегид, добавив глицин до конечной концентрации 0,125 М.
- Инкубировать в течение 5 мин на RT под вращением.
- Перенесите фиксированные клетки на лед и держите холодным при 4 градусах по Цельсию с этого момента.
- Пеллет клетки на 300 х г в течение 5 минут в охлажденной центрифуге.
- Откажитесь от супернатанта и resuspend гранулы в холодной PBS (1 мл для 5 х 106 ячеек), а затем передать до 1,5 мл безопасной блокировки труб.
- Пеллет клетки на 300 х г в течение 5 минут при 4 градусах Цельсия, отбросить супернатант, и оснастки заморозить гранулы в жидком азоте. Храните при -80 градусах по Цельсию или продолжайте с протоколом ниже.
4. Клеточный лисис и ферментное дайджест ограничения
- Аккуратно приостановите клеточные гранулы в 0,25 мл свежеприготовленного ледяного лисичного буфера (10 мм Tris-HCl pH 8,0, 10 мм NaCl, 0,2% Ингибиторы Igepal CA630 и 1x ингибиторы протеазы) на 2-5 х 106 элементов. Для подготовки 5 мл буфера лисиса см.
- Инкубировать клетки в течение 15 минут на льду.
- Центрифуга при 1000 х г в течение 5 мин при 4 градусах По цельсию. Откажитесь от супернатанта и держите гранулы, которые содержат ядра.
- Вымойте гранулированные ядра с 500 зл и буфером холодного лизиса.
- Аккуратно приостановите пеллету в трубке 1,5 мл с 50 qL 0,5% SDS в 1x буфере 2, затем инкубируйте трубку в нагревательном блоке при 62 градусах По Цельсия в течение 10 минут.
- Удалите трубки из нагревательного блока и добавьте 170 л буфера пищеварения, содержащего 25 зл и 10% тритона X-100, чтобы утолить SDS. Хорошо перемешать путем пипетки, избегая чрезмерного пенообразующего.
- Инкубировать при 37 градусах по Цельсию в течение 15 мин.
- Добавьте 25 кл.л. буфера пищеварения, смешайте путем инвертирования и примите 8 зл в качестве непереваренный контроль. Держите непереваренный контрольный образец при -20 градусов по Цельсию. Добавьте 100 U MboI фермент ограничения (4 л из 25 U / L бульона) в оставшиеся ядра и переварить хроматин в течение 2 ч при 37 градусов по Цельсию при вращении. Добавить еще один aliquot 100 U MboI и инкубировать еще 2 ч.
- Добавить еще 100 U MboI и инкубировать под вращение на ночь при 37 градусов по Цельсию.
- На следующий день добавьте еще 100 U MboI и инкубировать в течение 3 ч при 37 градусах Цельсия при вращении.
- Возьмите 8 зл в качестве переваренных контрольный образец. De-crosslink переварили контрольные образцы и непереваренные контрольные образцы со ступени 4.8, добавляя 80 qL буфера TE (10 мм Tris pH 8, 1 мМ EDTA) и 10 л протеиназа K (10 мг/мл). Инкубировать при 65 градусах по Цельсию на 1 ч.
- Выполнить 20 аликотов на 0,6% гель, чтобы проверить эффективность пищеварения. Успешные пищеварения показывают в основном фрагменты в диапазоне 3,0-0,5 кб.
5. Перевязка близости и разворот поперечной связи
- Инкубировать MboI-переваренные образцы при 65 градусах ПоЦельси в нагревательном блоке в течение 20 минут, чтобы инактивировать MboI, затем охладить RT.
- Centrifuge трубки в течение 5 минут на 1000 х г на RT, удалить супернатант, и растворить гранулы в 200 л свежего буфера лигазы.
- Добавьте в каждый образец смесь 1000 юаней мастера перевязки. Для подготовки 1000 л из перевязки мастер смеси, см Таблица 2.
- Смешайте путем инвертирования и инкубировать на RT ночь с медленным вращением (9 об / мин).
- Устраните остатки РНК и белка, добавив 100 л протеиназу K (10 мг/мл) и 10 л RNase A (10 мг/мл). Инкубировать образцы при 55 градусах по Цельсию в течение 45-60 мин.
- Продолжить инкубации образцов при 65 градусах По Цельсию еще 4 ч.
6. Стрижка ДНК и выбор размера
- Прохладные трубки для RT.
- Центрифуга в течение 5 мин при 1000 х г при 4 градусах По цельсию.
- Разделите образец на три 400 аликотов в 2 мл трубок и добавьте 2 л гликогена (20 мг/м), 40 л ацетата натрия (3 М, рН и 5,2) и 2,5 x объемы (1 мл) по 100% этанола в каждую трубку. Смешайте путем инвертирования и инкубировать при -80 градусов по Цельсию в течение 45-60 мин.
- Центрифуга при 16 000 х г при 4 градусах по Цельсию в течение 25 мин. Держите трубки на льду после вращения и тщательно удалите супернатант путем пипетки.
- Вымойте гранулы ДНК, приостановив в 800 зл 70% этанола. Центрифуга при 16 000 х г при 4 градусах по Цельсию в течение 5 мин.
- Удалить супернатант и выполнить мыть еще раз с 800 зл 70% этанола.
- Растворите гранулы в 130 кл. 1x tris буфера (10 мм Tris-HCl, рН no 8) и инкубировать при 37 градусов по Цельсию в течение 15 минут, чтобы полностью растворить ДНК. При необходимости используйте пайпеттинг, чтобы повторно приостановить любой осадок.
- Измерение урожайности ДНК; 2,5-5 мкг хроматина можно ожидать для 1 х 106 клеток. Проверьте перевязку, запустив 200 нг продукта 3C на 0,6% агарозного геля. Успешные перевязки в основном показывают фрагменты ДНК Храните образцы при -20 градусах Цельсия.
- Разбавить образец в трубке 0,65 мл, подходящей для звукования, до 10 нг/Л в 100 л 1x Tris объем буфера (1 мкг на трубку). Стандартное количество, используемое для подготовки библиотеки составляет 3 мкг, поэтому выполнять звукование в трех отдельных труб, если это необходимо.
- Стрижка ДНК размером 150-700 bp (в среднем 400-500 bp) с использованием следующих параметров на звуковой: Циклы: 6-8 из 20 с -60 с. Это должно сделать ДНК подходящей для высокой пропускной способности секвенирования подготовки библиотеки с использованием золотистов Illumina.
- Перенесите сложеную ДНК в обычную новую трубку с безопасным замком. Объедините несколько звуковых данных из одного и того же образца.
- Разогрейте бутылку бисера для очистки ДНК на RT. Отныне используйте низкие советы по связыванию.
- Добавьте 1,8-x объемов бисера в трубку ДНК и аккуратно притяните.
- Инкубировать на RT в течение 5 мин.
- Соберите бисер с магнитной стойкой. Вымойте бисер 2x с 1 мл свежеприготовленного 80% этанола, сохраняя трубки в магнитной стойке.
ПРИМЕЧАНИЕ: Удалите все этанола, в том числе остаточные капли. - Воздушно-сухие бусы кратко (2-3 мин) на RT.
ПРИМЕЧАНИЕ: Не сушите бусы дольше 5 мин. Это снизит урожайность ДНК. - Отрептуйте бусы с 90 qL 1x Tris буфер (10 mM Tris-HCl, рН No 8) для того чтобы elute дна.
- Измерьте выход ДНК и проанализируйте аликот на 1,5% на 1,5%. Там должно быть очень мало потерь по сравнению с презвукозационным выходом.
7. Подготовка библиотеки к секвенированию
- Добавьте 15 юаней мастер-микса из комплекта подготовки библиотеки. Для восстановления концов сложеной ДНК, объединить 10 зл 10 йл 10x конечного ремонта реакции буфера и 5 зл конечного ремонта фермента смеси.
- Инкубировать на RT в течение 30 мин.
- Добавьте 1,1-x объем бисера для очистки ДНК и аккуратно притяните.
- Инкубировать на RT в течение 5 мин.
- Соберите бисер с магнитной стойкой. Вымойте бисер дважды с 1 мл свежеприготовленного 80% этанола, сохраняя трубки в магнитной стойке. Удалите этанол.
- Воздушно-сухие бусы в течение 2-3 мин на RT. Отрежь бисер с 42 зл 1x Tris буфера (10 мм Tris-HCl, рН No 8), чтобы уэлитировать ДНК.
- Добавьте 8 зЛ мастер-миксования dA-хвостового к каждому образцу. Для подготовки мастер-микса dA-хвоста смешайте 5 зл 10x dA-хвостового буфера реакции и 3 ЗЛ фрагмента Klenow экзо минус.
- Инкубировать при 37 градусах по Цельсию в течение 30 мин.
- Добавьте 2 Л икры кишечной щелочной фосфатазы (CIP) в дефосфорилат ДНК.
- Инкубировать в течение 30 минут при 37 градусах по Цельсию, затем 60 мин при 50 градусах По кв. м.
- Добавьте 1,1x объемы бисера для очистки ДНК и аккуратно притяните.
- Инкубировать на RT в течение 5 мин.
- Соберите бисер с магнитной стойкой. Вымойте бисер 2x с 1 мл свежеприготовленного 80% этанола, сохраняя трубки в магнитной стойке.
- Воздушно-сухие бусинки кратко (2-3 мин) на RT. Resuspend бусы с 35 зл 1x Tris буфера (10 мм Tris-HCl, рН No 8), чтобы elute ДНК.
- Выполните реакцию перевязки адаптера. Используйте пониженные концентрации адаптера/лигаза, как упоминалось в таблице 3.
- Инкубировать при 20 градусах по Цельсию в течение 15 мин.
- Добавьте 3 зЛ смеси уракила ДНК гликосилазы и ДНК гликозилазы лизозилы VII (например, USER) фермента, смешивать путем пипеттинга и инкубировать при 37 градусах По Цельсию в течение 15 мин.
- Увеличьте громкость до 100 qL с водой, кипятите 5 мин при температуре 96 градусов по Цельсию, затем держите образцы на льду.
- Добавьте 1,1x объемы бисера для очистки ДНК и аккуратно притяните.
- Инкубировать на RT в течение 5 мин.
- Соберите бисер с магнитной стойкой. Вымойте бисер 2x с 1 мл свежеприготовленного 80% этанола, сохраняя трубки в магнитной стойке.
- Воздушно-сухие бусы в течение 2-3 мин на RT. Отрежь бисер с 50 зл и 1x Tris буфер (10 мм Tris-HCl, рН No 8), чтобы уэлитировать ДНК.
8. 4C хроматин взаимодействия библиотеки усиления и очистки
- Усиль библиотеку 4C, используя 10 qL библиотеки для выполнения первого ПЦР. Настройка и программа PCR можно найти в таблице 4.
- Выполните вложенные ПЦР. Вложенные настройки и программы ПЦР можно найти в таблице 5.
- Бассейн ПЦР продукты для каждой библиотеки и очистить с 1.1x ДНК очистки бисера.
- Измерьте выход ДНК и проанализируйте аликот в размере 5 л на геле 1,5%.
- Отрегулируйте концентрацию библиотеки и последовательность библиотеки. При индексации библиотеки могут быть объединены до секвенирования.
Результаты
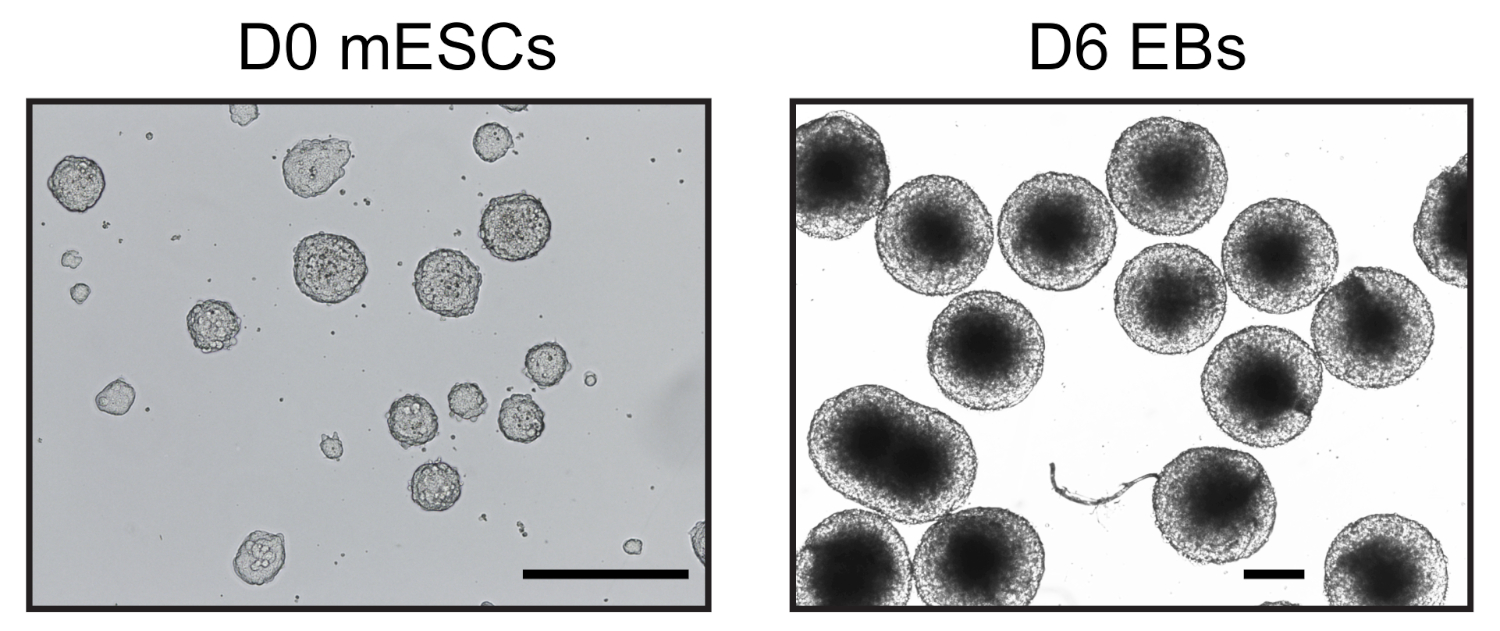
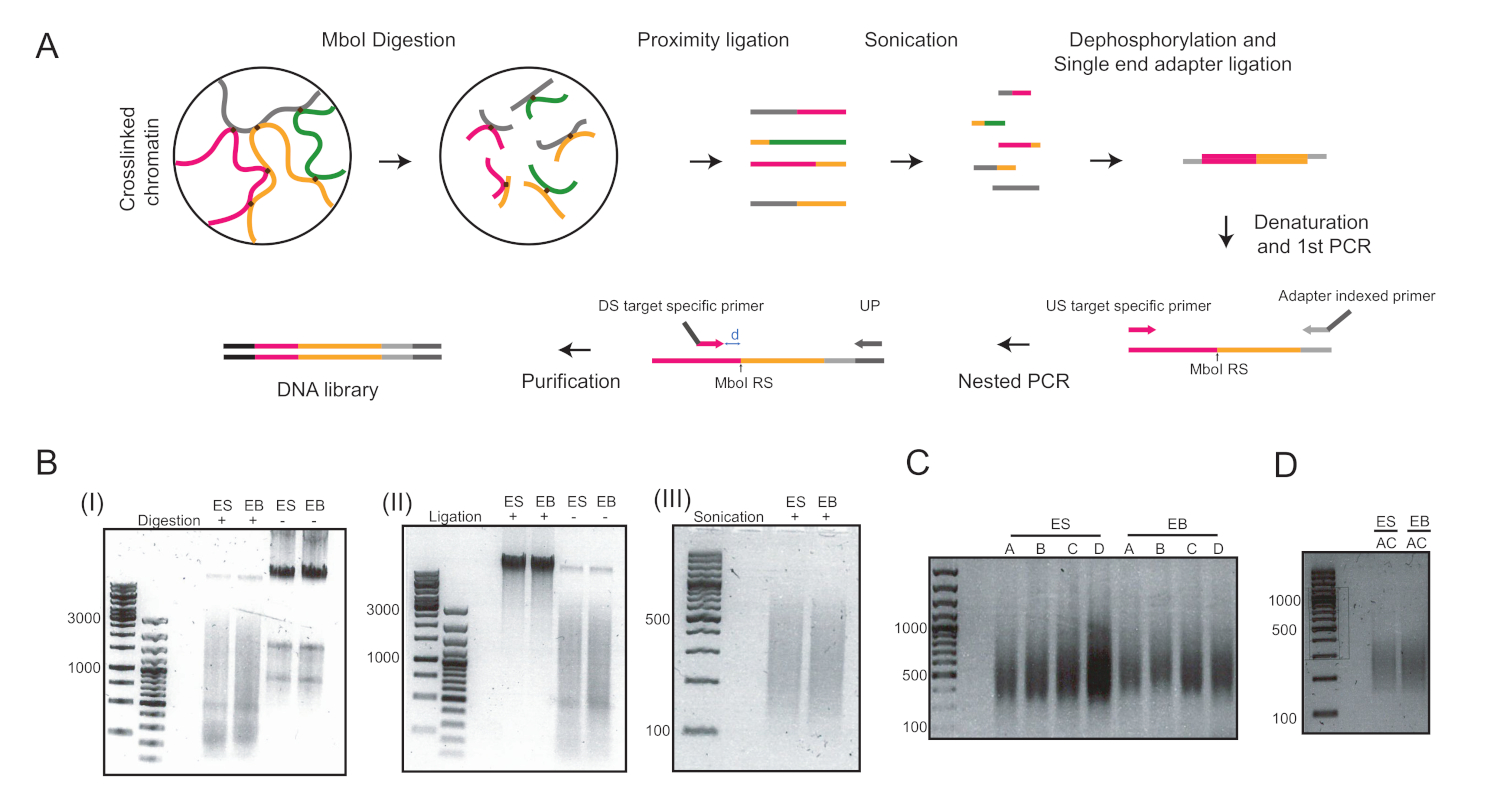
Через шесть дней после индукции дифференциации ESC в висячих каплях, мы получили однородную популяцию ЭБ, которые были использованы для дальнейшего анализа(рисунок 1). Мы адаптировали метод UMI-4C23 для количественной оценки специфического взаимодействия хроматина у промоторов родовых специфических генов в EBs24. Схематический обзор протокола с репрезентативными гелями контроля качества на разных этапах показан на рисунке 2A. Первый контроль качества был проведен для определения эффективности пищеварения фермента MboI ограничения. Эффективное пищеварение показало размер фрагмента менее 3 кбит/с(рисунок 2B). Следует отметить, что мЭСК и EB хроматин пищеварения было трудно, а иногда и остаточный непереваренный хроматин сохраняется. Второй контроль качества был проведен после перевязки, чтобы убедиться, что большинство фрагментов в настоящее время являются "gt; 3 кбит/с"(рисунок 2B). Затем фрагменты хроматина, полученные после звукования, были проанализированы гелевым электрофорисисом. Фрагмент размером 400-500 б.п. были ожидаемы(рисунок 2B).
После дефосфорилирования и перевязки одноконечных адаптеров были проведены два раунда ПЦР для усиления целей, представляющих интерес. Вложенный подход был использован для разработки набора из двух праймеров для каждого локуса. Это помогло улучшить специфичность. Каждая цель была усилена отдельно с двумя различными парами грунтовки для оптимизации условий ПЦР (т.е. праймер пар A и B для lou5f1 локус и праймер пары C и D для T локус, соответственно) и привело к МАЗок ДНК около 400 bp (Рисунок 2C). Кроме того, мультиплекс ПЦР был выполнен для одновременного усиления целей A и C(рисунок 2D)и привел к аналогичному размеру фрагмента после очистки(рисунок 2D). Праймеры, используемые для подготовки библиотеки 4C (лоджи Pou5f1 и T) можно найти в таблице 6.
Для анализа данных, необработанные считывания последовательности были впервые выровнены против генома мыши refence mm10, были дублированы, а низкое качество (Nolt; 20) считываний было удалено. Для каждой приманки информация о каждом фрагменте ограничения была получена путем вычисления количества прочитанных фрагментов, и был получен необработанный контактный профиль. Далее, область интереса была определена как все фрагменты ограничения с 2 кбит/с и 250 кбит/с расстояниедо к приманке. Размер каждого фрагмента ограничения увеличивался путем последовательного агрегирования смежных фрагментов ограничений для сглаживания профилей до достижения порога в 5% от общего числа необработанных контактов в интересующем регионе. Для обеспечения интеграции реплики и сопоставления условий мы включили как склоны, так и случайные перехваты на уровне фрагмента ограничения. Средний профиль на состояние и изменение складов между ними были построены, как показано на рисунке 3. Во время дифференциации EB, контакты между усилителями и промоутер омрачаемость ген pou5f1 уменьшился, в то время как усилитель-промоутер контакты мезендодера линии поучительный транскрипционный фактор T увеличился (Рисунок 3), обеспечивая функциональные идеи об этих усилителей развития.

Рисунок 1: Репрезентативные изображения mESC и производных эмбриональных тел. День 0 mESC культивируется в условиях без сыворотки (слева) и однородный день 6 EBs (справа) наблюдается перевернутый микроскоп. Шкала бар 500 мкм. Пожалуйста, нажмите здесь, чтобы просмотреть большую версию этой цифры.
{kind=link}

Рисунок 2: рабочий процесс 4C и репрезентативные изображения основных этапов протокола. (A) Схематический рабочий процесс количественного 4C. RS - сайт ограничения; США - вверх по течению; DS - вниз по течению; UP - универсальная грунтовка; D - расстояние между RS и DS в идеале должно быть 5-15 bp. (B) Примеры MboI-переваренный хроматин (I), в-якли ligated хроматин (II), и sonicated хроматин (III). Цифры слева указывают на размеры ДНК, определяемые лестницей ДНК, пробежкой для каждого образца. (C) Примеры усиления ПЦР на двух локуляциях: Pou5f1 (праймеры A и B) и T (праймер C и D). (D) Примеры мультиплексного усиления ПЦР на Pou5f1 и T loci с использованием грунтовок A и C. ES - эмбриональных стволовых клеток; EB - эмбриональные тела. Пожалуйста, нажмите здесь, чтобы просмотреть большую версию этой цифры.
{kind=link}

Рисунок 3: Примеры профилей 4C. Количественные профили 4C для приманок, расположенных на промоутерах генов Pou5f1 и T, ассссиемых в mESC и День 6 ЭБШ. Верхняя панель показывает участки средних контактов, генерируемых двумя независимыми биологическими репликациями; нижняя панель показывает среднее изменение контактного склада в 6-й день EBs по сравнению с mESCs (средний показатель двух репликатов). Светло-голубые коробки указывают расположение усилителей с динамическими изменениями во время дифференциации. Рисунок адаптированы из Тянь и др.24. Пожалуйста, нажмите здесь, чтобы просмотреть большую версию этой цифры.
{kind=link}
| Для 5mL | |
| 1M Tris-HCl, pH8.0 | 50 зл |
| 5M NaCl | 10 зл |
| 10% Igepal CA630 | 100 л |
| 50x Рош полный ингибиторы протеазы | 100 л |
| Миллиовода | 4,74 мл л. |
Таблица 1: Буфер лисиса.
| Для 1000 л | |
| Миллиовода | 869 Лл |
| 10X NEB T4 ДНК Лигаза Буфер | 120 л |
| 20 мг/мЛ Сыворотка Бойвина Альбумин | 6 зл |
| 2000 U/L T4 ДНК Лигаза | 5 зл |
Таблица 2: Подготовка мастера лигации смеси.
| Для 15 Зл | |
| 5X Быстрая лигационная реакция буфера | 10 зл |
| NeBNext Адаптор | 3 зл |
| Быстрый T4 ДНК лигаза | 2 л |
Таблица 3: Реакция перевязки адаптера.
| Настройка ПЦР | |
| Адаптер ligated библиотека-на-deads | 10 зл |
| Вода класса ПЦР | 20.25 Лл |
| 10 мкм Целевой конкретный грунтовка | 3,75 л л. |
| 10 мкм НЭБ Индекс грунтовка | 3,75 л л. |
| Буфер Геркуласа II 5X | 10 зл |
| 10 Мм dNTPs | 1,25 л л. |
| Геркуласа II полимераза | 1 зл |
| Общий объем | 50 зл |
| Программа ПЦР | |
| Шаг 1: 98 КК - 2 мин | |
| Шаг 2: 98 КК - 20s | |
| Шаг 3: 65 КК - 30s | |
| Шаг 4: 72 КК - 45s | |
| Шаг 5: перейдите на шаг 2, чтобы сделать в общей сложности 15-18 циклов | |
| Шаг 6: 72 КК - 3 мин | |
| Шаг 7: 4 КК - удерживайте |
Таблица 4: 4C хроматин взаимодействия библиотеки усиления, первый ПЦР.
| Вложенная настройка ПЦР | |
| Фрагмент ДНК из первого ПЦР | 10 зл |
| Вода класса ПЦР | 20.25 Лл |
| 10 км конкретных праймер | 3,75 л л. |
| 10 мкм P7 Illumina грунтовка | 3,75 л л. |
| Буфер Геркуласа II 5X | 10 зл |
| 10 Мм dNTPs | 1,25 л л. |
| Геркуласа II полимераза | 1 зл |
| Общий объем | 50 зл |
| Nested PCR программа | |
| Шаг 1: 98 КК - 2 мин | |
| Шаг 2: 98 КК - 20s | |
| Шаг 3: 65 КК - 30s | |
| Шаг 4: 72 КК - 45s | |
| Шаг 5: перейдите на шаг 2, чтобы сделать в общей сложности 15-18 циклов | |
| Шаг 6: 72 КК - 3 мин | |
| Шаг 7: 4 КК - удерживайте |
Таблица 5: 4C хроматин взаимодействия библиотеки усиления, вложенные ПЦР.
| Имя | Последовательность (5'-3') |
| DS-Oct4-A | ААТГАТАКЧАККАКСТАКАЦТАКАЦТАКТАКТАКТАКТАКТАКСТИКТТЦКЦТАКАКГАКГ CTCTTGATCTCTCTCTGCAAGATAACTAAGКАКККАГКАГКАГКАГКАГКАГКАГКАГКАГКАГКАГКАГКАГКАГКАГКАГКАГКАГКАГКАГКА |
| США-Октябрь4-A | ТККТГКЦААААААААААКАККАКАКАКК |
| DS-Oct4-B | ААТГАТАКЧАККАКСТАКАЦТАКАЦТАКТАКТАКТАКТАКТАКСТИКТТЦКЦТАКАКГАКГ CTCTTCCGATGTGTGATGGGGGTCAGCGGGGGGGGGGGGGCGC |
| США-Октябрь4-B | АККАГГГГГГГГГГГГГГГГГКККККККККТ |
| DS-T-C | ААТГАТАКЧАККАКСТАКАЦТАКАЦТАКТАКТАКТАКТАКТАКСТИКТТЦКЦТАКАКГАКГ CTCTTCCCTCCTGGGTCCCTGCACATTCGCCAAAGGAGC |
| US-T-C | ГАТТАКАККТГГТКТКакАТКККАКАА |
| DS-T-D | ААТГАТАКЧАККАКСТАКАЦТАКАЦТАКТАКТАКТАКТАКТАКСТИКТТЦКЦТАКАКГАКГ CTCTTGATCTGGCTCTTGGAGAGCAAGGAGACCCGGGAGGAGGAGGAGGAGGAGGAGGAGGAGGAGGAG |
| US-T-D | GCTGAGGCTTGGAGAGGTCAGAGGAGACC |
| UP-4C | КААГКАГААГААКККАТАЦГАКГАКГАКГАКГАКГАКГАКГАКГАКГА |
| Адап-i1 | КААГКААГААГААКГАЗГКААТАКАГАГАГАТАГАТГАТТАГАТТАКГАГГА CGTGTGTCTCTCTCTGATC |
| Адап-i2 | КААГКААГААГААКГАЗГКАТАГАГАГАТАКАТГГГГГГАТТАГГАГТЦАГА CGTGTGTCTCTCTCTGATC |
| Адап-i3 | КААГКААГААГААКГАЗГКАТАГАГАТГККТТААГГАТТАГАТТАГГАТТЦАГАГАГА CGTGTGTCTCTCTCTGATC |
| Адап-i4 | КААГКААГААГААКГАЗГАЗКАТАКГАГАТААТТГТЦАГАТТАКГАГТЦАГАГА CGTGTGTCTCTCTCTGATC |
Таблица 6: Праймеры, используемые для подготовки библиотеки 4C.
Обсуждение
Метод висевой культуры падения не требует дополнительных факторов роста или цитокинов и воспроизводит однородные популяции ЭБ из заранее определенного количества mESCs5. Здесь мы описываем протокол количественного 4C, адаптированный из подхода UMI-4C для количественной оценки контакта усилителя-промоутера конкретных транскрипционных факторов в модели дифференциации EB. Мы определили области хроматина, которые контактируют с промоутерами генов Pou5f1 и T динамическим образом во время дифференциации EB. Pou5f1 был downregulated во время дифференциации EB и частота контакта между промоутером Pou5f1 и его дистальным усилителем уменьшилась. И наоборот, T был upregulated во время дифференциации EB, и мы определили три усилители, для которых контактчастоты с их промоутер уменьшается(Рисунок 3). Для подтверждения идентификации, хроматин иммунопреципции (CHIP) анализ активного гистона знак H3K27ac может бытьвыполнена 24, так как это гистон знак ассоциируется с усилителем активации и усилители теряют эту отметку во время их инактивации11.
Стандартная техника 4C широко используется для обследования контактного профиля хроматина конкретных геномных сайтов25. Однако, этот подход трудно интерпретировать количественно даже после обширной нормализации26,27,28 из-за предубеждений, введенных неоднородности размера фрагмента ПЦР и невозможность различать дубликаты ПЦР. Наш количественный метод 4C во многом идентичен методу UMI-4C, который позволяет количественно определить одиночные молекулы с помощью звуковой и вложенно-опосредованного ПЦР шага, чтобы обойти ограничение классического подхода 4C23. Однако, в отличие от UMI-4C, который использует уникальные молекулярные идентификаторы, наш количественный протокол 4C позволяет количественно определить одиночные молекулы на основе специфического разрыва ДНК, производимого звуковым шагом. Это делает наш протокол совместимым с коммерческими комплектами подготовки библиотеки ДНК, устраняя необходимость праймеров с уникальными молекулярными идентификаторами.
Наш протокол включает в себя несколько ключевых шагов, которые должны быть рассмотрены. Как и в классическом методе 4C28,критическими факторами нашего протокола являются эффективность пищеварения и перевязка при подготовке молекул 3C. Низкая эффективность пищеварения/лиги может значительно снизить сложность взаимодействия с фрагментом интереса, что приводит к уменьшению разрешения. Как уже описано ранее23, еще одним важным шагом протокола является проектирование грунтов для усиления библиотеки. Второй ПЦР реакции праймеры должны быть расположены 5-15 nt от допроса места ограничения. В 75 nt секвенирования читать, это позволяет по крайней мере 40 nt слева от длины захвата для отображения. Праймер, используемый в первой реакции ПЦР, должен быть разработан вверх по течению второй грунтовки без перекрытия, и оба должны быть достаточно конкретными, чтобы обеспечить эффективное усиление ДНК. Для мультиплексирования грунтовки должны быть разработаны независимо, с целью температуры плавления (Tм)60-65 градусов по Цельсию. Кроме того, как и для других методов 3C, разрешение количественного метода 4C определяется ограничение мэнзим, используемый в протоколе25. Этот протокол использует фермент ограничения с 4 bp сайт распознавания, MboI. Максимальное разрешение с этим ферментом составляет около 500 bp, но это сильно зависит от локуса и редко достигается. Другим ограничением является то, что взаимодействия, возникающие между элементами, расположенными в одном и том же фрагменте ограничения, не обнаруживаются. Кроме того, взаимодействия, происходящие на расстоянии одного участка ограничения, нельзя отличить от непереваренного фона. Использование шага заполнения до перевязки может позволить обнаружить эти взаимодействия.
Количественный 4C идеально подходит для допроса хроматина контактов целевых локутов. Тем не менее, конкретный шаг усиления ПЦР ограничивает количество локутов, которые могут быть исследованы одновременно. Способ увеличить количество целевых локутов заключается в мультиплексном пЦР-шагах, чтобы одновременно усилить несколько целей, но для этого требуется совместимость используемых праймеров и тестирование каждой пары праймеров до реализации. Если глобальные изменения архитектуры хроматина у промоутеров желательны, то подходы, такие как Hi-C, PC Hi-C или HiChIP, были бы более подходящими29,,30,,31.
Раскрытие информации
Авторам нечего раскрывать.
Благодарности
Мы хотели бы поблагодарить Ф. Ле Дили, Р. Стахудеров и членов лаборатории «Граф» за их советы и обсуждения. Г.С. была поддержана стипендией Марии Склодовской-Кюри (H2020-MSCA-IF-2016, miRStem), T.V.T. постдокторской стипендией Хуана де ла Червы (MINECO, FJCI-2014-22946). Эта работа была поддержана Европейским исследовательским советом в рамках7-й Рамочной программы FP7 (ERC Synergy Grant 4D-Genome, грантовое соглашение 609989 т.г.), Министерствоэкономики, промышленности и конкурентоспособности Испании (MEIC) к партнерству EMBL, Centro de Excelencia Severo Ochoa 2013-2017 и CERCA Program Generalit de Cat.
Материалы
| Name | Company | Catalog Number | Comments |
| 0.1% EmbryoMax gelatin | EMD Millipore | ES-006-B | Cell culture |
| 0.25% Trypsin-EDTA | 25200072 | ||
| AMPure XP | Beckman Coulter | 10136224 | 4C/DNA purification |
| B27 supplement | Gibco | 17504044 | Cell culture |
| Beta-mercaptoethanol | Gibco | 31350010 | Cell culture |
| Bioruptor Pico | Diagencode | B01060010 | 4C/sonication |
| BSA | NEB | B9000S | 4C |
| CHIR99021 | Selleck Chemicals | S1263 | Cell culture |
| CIP | NEB | M0212 | 4C |
| cOmplete Protease Inhibitor Cocktail | Roche | 4693116001 | 4C |
| DMEM/F12 medium | Gibco | 11320033 | Cell culture |
| dNTP | NEB | N0447S | 4C |
| ESGRO Leukaemia Inhibitory Factor (LIF) | EMD Millipore | ESG1107 | Cell culture |
| Formaldehyde solution (37%) | Sigma | 252549-25ML | 4C |
| Glycin | Sigma | GE17-1323-01 | 4C |
| Glycogen | ThermoFischer | R0551 | 4C |
| Herculase II Fusion DNA polymerase | Agilent | 600675 | 4C |
| IGEPAL CA-630 | Sigma | I3021-50ML | 4C |
| Knockout DMEM | 10829018 | ||
| L-glutamine | Gibco | 25030081 | Cell culture |
| MboI | NEB | R0147M | 4C |
| MEM non-essential amino acids | Gibco | 11140050 | Cell culture |
| N2 supplement | Gibco | A1370701 | Cell culture |
| NEBNext DNA Library prep | NEB | E6040 | 4C |
| NEBuffer 2.1 | NEB | B7202S | 4C/digestion |
| Neurobasal medium | Gibco | 21103049 | Cell culture |
| PD0325901 | Selleck Chemicals | S1036 | Cell culture |
| Penicillin Streptomycin | Gibco | 15140122 | Cell culture |
| Proteinase K | NEB | P8107S | 4C |
| Qubit 4 Fluorometer | ThermoFischer | Q33238 | 4C |
| Qubit dsDNA HS Assay Kit | ThermoFischer | Q32851 | 4C |
| RNase A | ThermoFischer | EN0531 | 4C |
| Sodium pyruvate solution | Gibco | 11360070 | Cell culture |
| StemPro Accutase Cell Dissociation Reagent | Gibco | A1110501 | Cell culture |
| T4 DNA Ligase Reaction Buffer | NEB | B0202S | 4C |
| T4 DNA Ligase Reaction Buffer | NEB | M0202M | 4C |
Ссылки
- Evans, M. J., Kaufman, M. H. Establishment in culture of pluripotential cells from mouse embryos. Nature. 292 (5819), 154-156 (1981).
- Martello, G., Smith, A. The nature of embryonic stem cells. Annual Review Cell and Developmental Biology. 30, 647-675 (2014).
- Martin, G. R. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proceedings of the National Academy of Science U. S. A. 78 (12), 7634-7638 (1981).
- Loh, K. M., Lim, B., Ang, L. T. Ex uno plures: molecular designs for embryonic pluripotency. Physiological Reviews. 95 (1), 245-295 (2015).
- Sheridan, S. D., Surampudi, V., Rao, R. R. Analysis of embryoid bodies derived from human induced pluripotent stem cells as a means to assess pluripotency. Stem Cells International. 2012, 738910 (2012).
- Gaszner, M., Felsenfeld, G. Insulators: exploiting transcriptional and epigenetic mechanisms. Nature Reviews in Genetics. 7 (9), 703-713 (2006).
- Lenhard, B., Sandelin, A., Carninci, P. Metazoan promoters: emerging characteristics and insights into transcriptional regulation. Nature Reviews in Genetics. 13 (4), 233-245 (2012).
- Long, H. K., Prescott, S. L., Wysocka, J. Ever-Changing Landscapes: Transcriptional Enhancers in Development and Evolution. Cell. 167 (5), 1170-1187 (2016).
- Schoenfelder, S., Fraser, P. Long-range enhancer-promoter contacts in gene expression control. Nature Reviews in Genetics. 20 (8), 437-455 (2019).
- Spitz, F., Furlong, E. E. Transcription factors: from enhancer binding to developmental control. Nature Reviews in Genetics. 13 (9), 613-626 (2012).
- Creyghton, M. P., et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proceedings of the National Academy of Sciences U. S. A. 107 (50), 21931-21936 (2010).
- Heintzman, N. D., et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 459 (7243), 108-112 (2009).
- Klemm, S. L., Shipony, Z., Greenleaf, W. J. Chromatin accessibility and the regulatory epigenome. Nature Reviews in Genetics. 20 (4), 207-220 (2019).
- Rada-Iglesias, A., et al. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 470 (7333), 279-283 (2011).
- Lettice, L. A., et al. Development of five digits is controlled by a bipartite long-range cis-regulator. Development. 141 (8), 1715-1725 (2014).
- Dekker, J., Rippe, K., Dekker, M., Kleckner, N. Capturing chromosome conformation. Science. 295 (5558), 1306-1311 (2002).
- de Wit, E., de Laat, W. A decade of 3C technologies: insights into nuclear organization. Genes and Development. 26 (1), 11-24 (2012).
- Lieberman-Aiden, E., et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 326 (5950), 289-293 (2009).
- Simonis, M., et al. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nature Genetics. 38 (11), 1348-1354 (2006).
- Splinter, E., de Wit, E., van de Werken, H. J., Klous, P., de Laat, W. Determining long-range chromatin interactions for selected genomic sites using 4C-seq technology: from fixation to computation. Methods. 58 (3), 221-230 (2012).
- Stadhouders, R., et al. Multiplexed chromosome conformation capture sequencing for rapid genome-scale high-resolution detection of long-range chromatin interactions. Nature Protocols. 8 (3), 509-524 (2013).
- van de Werken, H. J., et al. Robust 4C-seq data analysis to screen for regulatory DNA interactions. Nature Methods. 9 (10), 969-972 (2012).
- Schwartzman, O., et al. UMI-4C for quantitative and targeted chromosomal contact profiling. Nature Methods. 13 (8), 685-691 (2016).
- Tian, T. V., et al. Whsc1 links pluripotency exit with mesendoderm specification. Nature Cell Biology. 21 (7), 824-834 (2019).
- Chen, H., et al. Dynamic interplay between enhancer-promoter topology and gene activity. Nature Genetics. 50 (9), 1296-1303 (2018).
- Apostolou, E., et al. Genome-wide chromatin interactions of the Nanog locus in pluripotency, differentiation, and reprogramming. Cell Stem Cell. 12 (6), 699-712 (2013).
- de Wit, E., et al. The pluripotent genome in three dimensions is shaped around pluripotency factors. Nature. 501 (7466), 227-231 (2013).
- Krijger, P. H. L., Geeven, G., Bianchi, V., Hilvering, C. R. E., de Laat, W. 4C-seq from beginning to end: A detailed protocol for sample preparation and data analysis. Methods. , (2019).
- Mumbach, M. R., et al. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nature Methods. 13 (11), 919-922 (2016).
- Rao, S. S., et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 159 (7), 1665-1680 (2014).
- Schoenfelder, S., Javierre, B. M., Furlan-Magaril, M., Wingett, S. W., Fraser, P. Promoter Capture Hi-C: High-resolution, Genome-wide Profiling of Promoter Interactions. Journal of Visualized Experiments. (136), e57320 (2018).
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеThis article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены