
Method Article
Mapeando as Relações Estrutura-Função de Fatores de Transcrição Oncogênica Desordenados usando análise transcriômica
Neste Artigo
Resumo
Domínios intrinsecamente desordenados são importantes para a função do fator de transcrição de fusão oncogênica. Para atingir terapeuticamente essas proteínas, é necessário uma compreensão mais detalhada dos mecanismos regulatórios utilizados por esses domínios. Aqui, usamos transcrição para mapear características estruturais importantes do domínio EWS intrinsecamente desordenado no sarcoma Ewing.
Resumo
Muitos cânceres são caracterizados por translocações cromossômicas que resultam na expressão de fatores de transcrição da fusão oncogênica. Tipicamente, essas proteínas contêm um domínio intrinsecamente desordenado (IDD) fundido com o domínio de vinculação de DNA (DBD) de outra proteína e orquestram mudanças transcricionais generalizadas para promover a malignidade. Essas fusões são muitas vezes a única aberração genômica recorrente nos cânceres que causam, tornando-os alvos terapêuticos atraentes. No entanto, direcionar fatores de transcrição oncogênica requer uma melhor compreensão do papel mecanicista que as IDDs de baixa complexidade desempenham em sua função. O domínio n-terminal do EWSR1 é um IDD envolvido em uma variedade de fatores de transcrição de fusão oncogênica, incluindo EWS/FLI, EWS/ATF e EWS/WT1. Aqui, usamos o sequenciamento de RNA para investigar as características estruturais do domínio EWS importante para a função transcricional do EWS/FLI no sarcoma Ewing. Primeiro esgotamento mediado por shRNA da fusão endógena das células de sarcoma de Ewing emparelhadas com expressão ectópica de uma variedade de construções mutantes EWS é realizada. Em seguida, o sequenciamento de RNA é usado para analisar os transcriptomes de células expressando esses construtos para caracterizar os déficits funcionais associados a mutações no domínio EWS. Ao integrar as análises transcriômicas com informações publicadas anteriormente sobre motivos de ligação de DNA EWS/FLI e localização genômica, bem como ensaios funcionais para a transformação da capacidade de transformação, conseguimos identificar características estruturais do EWS/FLI importantes para a oncogênese e definir um novo conjunto de genes-alvo EWS/FLI críticos para sarcoma Ewing. Este artigo demonstra o uso do sequenciamento de RNA como método para mapear a relação estrutura-função do domínio intrinsecamente desordenado de fatores de transcrição oncogênica.
Introdução
Um subconjunto de cânceres, incluindo muitas malignidades da infância e adolescência, são caracterizados por translocações cromossômicas que geram nova fusão oncogenes1,2,3,4,5,6. As proteínas de fusão resultantes frequentemente funcionam como fatores de transcrição oncogênica, orquestrando mudanças generalizadas na regulação transcricional para promover a tumorigênese7,8. Os cânceres com essas translocações geralmente possuem uma paisagem mutacional silenciosa, com poucas aberrações genômicas recorrentes além da fusão pathognomônica4,9. Como tal, direcionar diretamente a proteína de fusão é uma estratégia terapêutica atraente nessas doenças. No entanto, esses fatores de transcrição oncogênica consistem comumente em um domínio de baixa complexidade, intrinsecamente desordenado, transcriptionalmente ativado fundido com um domínio de vinculação de DNA (DBD)10,11,12,13,14. Tanto os domínios intrinsecamente desordenados (IDDs) quanto os DBDs dessas proteínas têm se mostrado difíceis de atingir com abordagens farmacológicas convencionais. O desenvolvimento de novas abordagens terapêuticas, portanto, requer uma compreensão molecular mais detalhada dos mecanismos empregados por essas fusões para regular aberrantemente a expressão genética.
A porção de IDD N-terminal de EWSR1 é comumente fundida a um DBD em câncer, incluindo EWS/FLI em sarcoma Ewing, EWS/WT1 em tumor de células pequenas e difusas pequenas células redondas, e EWS/ATF1 em sarcoma de células claras de partes macias10. O papel mecanicista do IDD EWS em cada uma dessas fusões é incompletamente compreendido. A família de fusões EWS/ETS, especificamente EWS/FLI, é a mais caracterizada funcionalmente até o momento. O EWS/FLI coordena alterações epigenéticas e transcricionais em todo o genoma que levam à ativação e repressão de milhares de genes7,11,15,16. Estudos têm demonstrado que o IDD é importante para o recrutamento de co-ativadores transcricionais (como p300, WDR5 e o complexo BAF), bem como co-repressores (como o complexo NuRD)11,15,17. A fusão do IDD EWS à porção terminal C do FLI1 confere nova especificidade de vinculação de DNA ao ETS DBD de FLI1, de tal forma que a oncoproteína de fusão (EWS/FLI) se liga a regiões repetitivas GGAA-microsatélites do genoma, além do consenso ETS motivo18,19,20. Combinado com a função de recrutamento do co-ativador, esta atividade emergente de ligação de DNA do EWS/FLI promove a formação de novo melhorador em GGAA-microsatélites distal para locais de início de transcrição (TSS) (microsatélites "melhorador" RNA polymerase II para promover transcrição em GGAA-microsatélites proximal para TSS (microsatélites "promotores")11,15,16,21.
Juntos, esses dados nos levaram a supor que elementos discretos dentro do domínio EWS contribuem para o recrutamento de co-reguladores distintos para diferentes tipos de sites de vinculação EWS/FLI. No entanto, discernir esses elementos dentro da porção EWS do EWS/FLI, e como eles funcionam, tem sido dificultado pela natureza altamente repetitiva e desordenada do domínio. Aqui utilizamos um sistema de resgate de knockdown publicado anteriormente em células de sarcoma Ewing para mapear funcionalmente esses elementos no IDD EWS. Neste sistema, o EWS/FLI é esgotado usando um shRNA direcionado ao 3'UTR do gene FLI1, e a expressão é resgatada com diferentes construções de CDNA mutantes EWS/FLI sem o 3'UTR7,17,22. Esses experimentos se concentraram em construções com várias exclusões para mapear a relação estrutura-função entre o IDD EWS e os importantes fenótipos oncogênicos, incluindo a ativação de uma construção de repórter GGAA-microsatélite, ensaios de formação de colônias e validação direcionada de genes ativados pelo EWS/FLI7,17,22 . No entanto, esses estudos não conseguiram encontrar sub-domínios discretos dentro do IDD EWS em EWS/FLI que são exclusivamente importantes para ativação ou repressão. Todos os construtos testados foram capazes de ativar e reprimir genes-alvo específicos, levando à formação eficiente de colônias, ou incapazes de regular qualquer um dos genes alvo EWS/FLI, levando à perda da formação decolônias 7,17,22.
Análises transcriômicas habilitadas pela adoção generalizada do sequenciamento da próxima geração são comumente utilizadas para comparar assinaturas de expressão genética em duas condições, frequentemente no contexto de triagem ou estudos descritivos. Em vez disso, queríamos aproveitar a capacidade de capturar dados de expressão em todo o genoma usando RNA-sequencincing (RNA-seq) para caracterizar as contribuições dos IDDs para a função do fator de transcrição. Neste caso, o RNA-seq é emparelhado com o sistema knockdown-rescue para explorar a relação estrutura-função do domínio EWS. Essa abordagem é aplicável a outros fatores de transcrição de fusão, incluindo outras fusões EWS ou fatores de transcrição do tipo selvagem com função mal compreendida, e tem múltiplas vantagens sobre os outros ensaios usados para estudos de mapeamento funcional, como ensaios de repórter ou qRT-PCR direcionado. Estes incluem testar determinantes estruturais de função no contexto de cromatina relevante, a capacidade de testar vários tipos de elementos de resposta em um ensaio (ou seja, ativado e reprimido, GGAA-microsatélite e não-microsatélite, etc.), e a capacidade resultante de detectar melhor a função parcial.
A implementação bem-sucedida dessa abordagem depende de um sistema baseado em células que captura os fenótipos de interesse (neste caso, células A673 com esgotamento EWS/FLI mediado por shRNA) e um painel de construções mutantes em um vetor de expressão apropriado para o sistema baseado em células (neste caso, pMSCV-hygro com vários mutantes EWS/FLI marcados por 3x-FLAG a serem entregues por transdução retroviral). A transdução viral de construções de esgotamento baseadas em CRISPR, construções de esgotamento baseadas em shRNA e construtos de expressão cDNA com seleção apropriada para gerar linhas de células estáveis é recomendada sobre transfecção transitória. A interpretação a jusante dos resultados é reforçada quando os dados transcriômicos podem ser emparelhados com outros dados relacionados à localização do fator de transcrição e outras leituras fenotípicas quando disponíveis.
Neste artigo, aplicamos esta abordagem para caracterizar a atividade do mutante DAF da EWS/FLI14. O mutante DAF tem 17 mutações de tyrosina para alanina nas regiões repetitivas do EWS IDD de EWS/FLI14. Este mutante EWS em particular havia sido relatado anteriormente e é incapaz de ativar a expressão genética do repórter quando fundido ao ATF1 DBD14. No entanto, dados preliminares do QRT-PCR sugeriram que este mutante foi capaz de ativar a transcrição do alvo EWS/FLI NR0B123. A abordagem transcriômica descrita aqui permitiu a detecção bem sucedida da função parcial do mutante DAF. Ao emparelhar esses dados transcriômicos com informações sobre os motivos de vinculação e reconhecimento EWS/FLI, mostramos ainda que o mutante DAF mantém a função em repetições de microsatélites GGAA. Esses resultados identificam o DAF como o primeiro mutante EWS/FLI parcialmente funcional e destacam a função em genes não microsatélites tão importantes para a oncogênese (como relatado23). Isso demonstra o poder desta abordagem de mapeamento de estrutura-função transcriptômica para fornecer insights sobre a função dos fatores de transcrição oncogênica.
Protocolo
1. Configurar painel in vitro de construções
NOTA: Esta etapa vai variar dependendo da proteína específica a ser analisada.
- Prepare alíquotas do vírus para esgotamento e construção de expressões conforme necessário.
- Semente um prato de cultura tecidual de 10 cm com células 3-5 x 106 HEK293-EBNA ou HEK293T para cada construção necessária para transdução viral. As células aderem durante a noite no DMEM (Modified Eagle Media, mídia de águia modificada) de Dulbecco, suplementadas com 10% de soro bovino fetal (FBS), penicilina/estreptomicina/glutamina (P/S/Q) e 0,3 mg/mL G418.
NOTA: As células HEK293-EBNA e HEK293T são recomendadas para a produção viral porque são fáceis de cultivar, têm alta eficiência de transfecção e expressam eficientemente proteínas recombinantes de plasmídeos episômicos. As células devem estar entre 50-70% confluentes no dia da transfecção. - Prepare uma mistura de transfecção para cada construção de transdução viral. Combine 2 mL de mídia de soro reduzido com 90 μL de reagente de transfecção.
NOTA: Recomenda-se a mídia de soro reduzido pré-aquecimento. - Adicione 10 μg cada um de um plasmídeo de embalagem viral (por exemplo, gag-pol), plasmid de envelope viral (por exemplo, VSV-G) e um de esgotamento baseado em CRISPR, esgotamento baseado em shRNA ou construção de expressão cDNA (por exemplo, pMKO ou pMSCV) para a mistura de transfecção. Misture bem com a pipetação suave.
- Deixe a mistura de transfecção ficar por 20 minutos à temperatura ambiente. Remova a mídia de crescimento HEK293-EBNA dos pratos da cultura tecidual e adicione 3 mL DMEM suplementado com 10% de FBS, P/S/Q e piruvato de sódio de 10 mM. A cada prato, adicione 2 mL de mistura de transfecção dropwise. Deixe as células sentarem-se em mídia de transfecção durante a noite em uma incubadora a 37 °C e 5% de CO2.
- Na manhã seguinte, adicione 20 mL de mídia DMEM com 10% de FBS, suplementação P/S/Q e piruvato de sódio de 10 mM. Incubar as células nele a 37 °C e 5% de CO2 durante a noite.
- Na manhã seguinte, substitua a mídia por mídia de coleta viral de 5 mL (VCM) (DMEM complementada com FBS inativado de calor de 10%, P/S/Q e HEPES de 20 mM).
- Após 4h, colete VCM das placas e armazene em um tubo cônico de 50 mL no gelo a 4 °C. Substitua por 5 mL de VCM fresco.
- Após 4h, colete VCM de placas no mesmo tubo cônico de 50 mL e armazene no gelo a 4 °C. Substitua por 8 mL de VCM fresco para coleta durante a noite.
- Pela manhã, recolhe vcm de placas e armazene no tubo cônico de 50 mL no gelo a 4 °C. Substitua por 5 mL de VCM fresco.
- Após 4h, recolhe VCM das placas e armazene no tubo cônico de 50 mL no gelo a 4°C. Substitua por 5 mL de VCM fresco. Depois de 4h, colete VCM das placas e adicione ao tubo cônico de 50 mL.
- Coleções de alíquotas de tubo de 50 mL em criotubos (2 mL por alíquota) após filtragem através de um filtro de 0,45 μm. Armazene alíquotas virais a -80 °C até usar.
NOTA: O protocolo pode ser pausado aqui, e as alíquotas virais podem ser armazenadas até estarem prontas para uso.
- Semente um prato de cultura tecidual de 10 cm com células 3-5 x 106 HEK293-EBNA ou HEK293T para cada construção necessária para transdução viral. As células aderem durante a noite no DMEM (Modified Eagle Media, mídia de águia modificada) de Dulbecco, suplementadas com 10% de soro bovino fetal (FBS), penicilina/estreptomicina/glutamina (P/S/Q) e 0,3 mg/mL G418.
- Células de sementes na densidade apropriada em um prato de cultura tecidual de 10 cm. Meta 50% confluência. Deixe as células aderirem durante a noite colocando-se na incubadora a 37 °C contendo 5% de CO2.
NOTA: Para células A673 esta é 5 x 106 células em 10 mL de mídia DMEM com 10% de FBS, suplementação P/S/Q e piruvato de sódio de 10 mM. Essas condições podem variar dependendo da taxa de crescimento das células utilizadas. - Esgotado fator endógeno de interesse. Se as células não precisarem ter a proteína endógena de interesse esgotada, pule para a etapa 1.4.
- Descongelar alíquota viral para transdução de shRNA ou crispr construto visando a proteína de interesse. Descongele as alíquotas congeladas rapidamente em um banho de água de 37 °C.
- Adicione 2,5 μL de polibrene de 8 mg/mL a cada alíquota viral e misture por tubulação suave. Remova a mídia das placas das células e adicione suavemente alíquota viral à placa de 10 cm por tubulação ao longo da lateral da placa. Balance a placa para espalhar os 2 mL de alíquota viral.
- Incubar a 37 °C na incubadora de cultura tecidual por 2h. Balance a placa a cada 30 minutos para evitar que quaisquer áreas da placa sequem.
- Adicione 5 mL de mídia DMEM com 10% de FBS, suplementação P/S/Q e piruvato de sódio de 10 mM, com 5 μL de polibrene de 8 mg/mL. Deixe as células incubarem durante a noite.
- Pela manhã, remova a mídia das células e as células de passagem para a mídia complementadas com um reagente de seleção. Ao passar células, semeá-las de forma a permitir que elas cresçam por 48-72 h e atinjam 50% de confluência.
NOTA: Para células A673 com pSRP-iEF-2, as células são semeadas em uma divisão de 1:5 e selecionadas por 72 h com 2 μg/mL de puramicina.
- Construções de expressão cDNA transdutor.
- Verifique as células para confirmar 50-70% de confluência.
- Descongelar alíquotas virais para transdução de cDNA construções(s) de interesse. Descongele as alíquotas congeladas rapidamente em um banho de água de 37 °C. Adicione 2,5 μL de polibrene de 8 mg/mL a cada alíquota viral e misture suavemente.
- Remova a mídia das células banhadas e adicione suavemente alíquota viral à placa de 10 cm por tubulação ao longo da lateral da placa. Balance a placa para espalhar os 2 mL de alíquota viral.
- Incubar a 37 °C na incubadora de cultura tecidual por 2h. Balance a placa a cada 30 minutos para evitar que quaisquer áreas da placa sequem.
- Adicione 5 mL de mídia DMEM com 10% de FBS, suplementação P/S/Q e piruvato de sódio de 10 mM, com 5 μL de polibrene de 8 mg/mL. Deixe as células incubarem durante a noite.
- Pela manhã, remova a mídia das células e células de passagem em mídia de dupla seleção. Crescer e passar células conforme necessário durante 7-10 dias para permitir a dupla seleção e expressão da construção cDNA.
NOTA: Esta divisão desta passagem pode exigir otimização para diferentes linhas celulares. Para células A673 com pSRP-iEF-2 e uma construção pMSCV-hygro, as células são passadas sem se dividir em 2 μg/mL puramicina e 100 μg/mL de higromicina.
2. Coletar células, validar a expressão de construtos e configurar ensaios fenotípicos correlativos
- Após 7-10 dias de dupla seleção, colete células em um tubo cônico de 15 mL. Conte células coletadas com hemótmetro. Aliquot coletou células para sequenciamento de RNA e para validar a expressão de construções de CDNA.
NOTA: Configure quaisquer ensaios fenotípicos correlativos exigidos pela questão da pesquisa sob investigação. Ensaios de formação de colônias são um exemplo de um ensaio fenotípico correlativo que são usados aqui.- Coletar entre 5 x 105 e 1 x 106 células para sequenciamento de RNA e 2 x 106 células para extração de proteínas. Células de pelotas por centrifugação a 1.000 x g a 4 °C por 5 min e remova o sobrenatante.
- Lave a pelota com PBS frio de 1 mL. Pelota por centrifugação a 1.000 x g a 4 °C por 5 min e remova o supernasce. As pelotas de congelamento de flash em nitrogênio líquido e armazenam a -80 °C.
- Configure quaisquer ensaios correlativos com as células restantes.
NOTA: O protocolo pode ser pausado aqui com amostras coletadas armazenadas no congelador de -80 °C.
- Validar o knockdown de proteína de interesse (se usado) e expressão do painel de construções.
- Descongelar pelotas de células para extração de proteínas no gelo. Células resuspend em gelo frio 500 μL tampão de extração nuclear (20 mM HEPES pH 7.9, 140 mM NaCl, 10% glicerol, 1,5 mM MgCl2, 1 mM EDTA, 1 mM DTT, 1% IGEPAL) com inibidor de protease. Deixe descansar por 5 minutos no gelo.
- Núcleos de pelotas por centrifugação a 1.000 x g a 4 °C por 5 min e remova o supernasce. Lave núcleos em 500 μL tampão de extração nuclear gelado (20 mM HEPES pH 7.9, 140 mM NaCl, 10% glicerol, 1,5 mM MgCl2, 1 mM EDTA, 1 mM DTT, 1% IGEPAL) com inibidor de protease.
- Núcleos de pelotas por centrifugação a 1.000 x g a 4 °C por 5 min e remova o supernasce. Nuclei de resuspend em 200 μL tampão RIPA frio com inibidor de protease (ajuste o volume do buffer RIPA de acordo com o tamanho da pelota.) Deixe-o sentado no gelo por 45-60 min com vigoroso vórtice a cada 15 minutos.
- Detritos de células de pelota por centrifugação a 16.000 x g a 4 °C por 45-60 min. Mantenha o supernatante e transfira para um tubo frio fresco
- Prepare amostras para eletroforese SDS-PAGE fervendo 5-10 μg de proteína com tampão de carga de 1x por 5 min. Execute um gel SDS-PAGE conforme necessário para a proteína de interesse.
- Transfira para uma membrana nitrocelulose ou PVDF conforme necessário para a proteína de interesse. Bloqueie e borrie com os anticorpos primários e secundários apropriados para confirmar o knockdown da proteína endógena (se usada) e expressão ectópica da construção cDNA.
NOTA: O protocolo pode ser pausado aqui.
- Extrair RNA. Avalie a qualidade e a quantidade do RNA.
- Descongelar pelotas de célula no gelo. Extrair RNA total usando um kit de extração baseado em coluna de spin de sílica de acordo com as instruções do fabricante.
- Resumidamente, lise as células usando o tampão de lise do kit. Aplique o lysate a uma coluna de spin de sílica com um giro breve a >13000 rpm por 30-60 segundos ou remova o gDNA aplicando o lysate a uma coluna de remoção gDNA com um breve giro a >13000 rpm por 30-60 segundos.
- Realize uma digestão de DNA na coluna se o lysato for diretamente aplicado a uma coluna de spin de sílica. Se usar uma coluna de remoção gDNA, aplique o eluato a uma coluna de giro de sílica com um giro breve a >13000 rpm para 30-60 s.
- Lave o RNA na coluna de acordo com as instruções do fabricante. Elute RNA em 30 μL de tampão de elução.
- Avalie a qualidade e a quantidade do RNA usando um fluorômetro ou qualquer outro instrumento comparável. Certifique-se de que a razão 260/280 esteja próxima de 2 e que haja pelo menos 2,5 μg de RNA para submeter para sequenciamento.
NOTA: À medida que as réplicas são coletadas, cada réplica deve ser processada com o mesmo protocolo de extração de RNA. - Use uma pequena alíquota de RNA para confirmar o knockdown estável da proteína de interesse, se necessário, por qRT-PCR. Armazene a amostra restante do RNA a -80 °C.
- Coletar réplicas biológicas repetindo as etapas 1-2 até que 3-4 conjuntos completos de RNA tenham sido coletados. Certifique-se de que cada réplica exibe expressão adequada de construções de CDNA e knockdown estável da proteína endógena (se utilizada).
3. Sequenciamento de próxima geração
- Envie o RNA extraído para ser sequenciado usando uma plataforma de sequenciamento de próxima geração com uma meta de 50 milhões de 150 leituras finais emparelhadas de par de 150 bases (bp). Siga as instruções da instalação que processa as amostras. Selecione para RNAs poli-adenylated e sequenciamento específico de fios.
4. Pipeline de contagem de alinhamento e transcrição
NOTA: Este protocolo pressupõe que, após o envio e processamento da amostra, um conjunto de arquivos FASTQ emparelhados sejam devolvidos para cada amostra. Esses arquivos são frequentemente compactados com um sufixo de "fastq.gz". Uma análise mais aprofundada desses arquivos FASTQ exigirá acesso a uma instalação de computação de alto desempenho (HPC) executando um sistema operacional Linux.
- Transferir arquivos
- Abra um terminal para o ambiente HPC com PuTTY. Faça um diretório para a análise chamada "projeto".
- Navegue até o diretório "path_to/project" e faça um novo diretório para os arquivos de fastq .gz cru compactados chamados "fastq". Também faça um diretório chamado "aparado". Isso é mostrado na Figura S1A-C.
- Transfira os arquivos de fastq bruto compactado.gz do armazenamento local para o diretório "path_to/project/fastq/" usando o WinSCP ou um programa semelhante. Verifique se há um arquivo "R1" e um "R2" para cada amostra, conforme mostrado na Figura S1B.
- Opcional: Se necessário, instale TrimGalore. Defina o diretório contendo o arquivo trim_galore executável na variável ambiente PATH no Linux.
NOTA: Leituras de baixa qualidade e adaptadores são aparados com TrimGalore. TrimGalore está disponível em https://github.com/FelixKrueger/TrimGalore. - Opcional: Navegue até o diretório para pacotes de software baixados (ou seja, "path_to/software"). Baixe o mais recente pacote TrimGalore usando o comando "curl -fsSL https://github.com/FelixKrueger/TrimGalore/archive/[versão].tar.gz -o trim_galore-[versão].tar.gz."
- Opcional: Desempacote o arquivo .gz piche. Use o comando "tar -xvzf trim_galore-[version_number].tar.gz".
- Opcional: Torne trimgalore executável. Use o comando "chmod a+x path_to/software/TrimGalore-[versão]/trim_galore". Certifique-se de que este novo diretório está no PATH. Use o comando "export PATH=path_to/software/TrimGalore-[versão]:$PATH".
- Navegue até path_to/project/fastq/. Use TrimGalore para cortar as leituras de baixa qualidade dos arquivos fastq.gz usando o comando mostrado na Figura S1C.
NOTA: Bandeiras adicionais para este comando podem ser relevantes e podem ser encontradas aqui: https://github.com/FelixKrueger/TrimGalore/blob/master/Docs/
Trim_Galore_User_Guide.md - Verifique os arquivos fastq .gz aparados no diretório path_to/project/aparado. Certifique-se de que são chamados de sample1_R1_val_1.fq.gz e sample1_R2_val_2.fq.gz
- Alinhe arquivos FASTQ aparados com STAR e gere contagens de transcrição.
NOTA: STAR está disponível em https://github.com/alexdobin/STAR)- Opcional: Instale a versão STAR 2.6 ou posterior. Coloque a ESTRELA executável no caminho.
- Opcional: Navegue até o diretório para pacotes de software baixados (ou seja, "path_to/software").
- Opcional: Baixe o pacote STAR usando o comando "curl -SLO https://github.com/alexdobin/STAR/archive/[versão].tar.gz". Desempacotar o arquivo .gz piche.
- Opcional: Use o comando "tar -xzf [versão].tar.gz". Torne a STAR executável. Use o comando "chmod a+x path_to/software/STAR-[versão]/bin".
- Opcional: Certifique-se de que este novo diretório está no caminho. Use o comando "export PATH=path_to/software/STAR-[version_number]/bin/linux_x86_64_static:$PATH".
NOTA: O manual STAR está disponível em: (https://github.com/alexdobin/STAR/blob/master/doc/STARmanual.pdf). - Certifique-se de que há índice de genoma para usar com STAR. Coloque isso em um diretório separado do path_to/projeto/diretório. Se um índice foi gerado anteriormente para experimentos anteriores, use isso. Alternativamente, use um índice pré-gerado apropriado se disponível aqui: http://refgenomes.databio.org/. Caso contrário, construa um novo índice usando o comando "STAR-runMode genomeGenerate" usando as instruções no manual STAR.
NOTA: Para o restante deste protocolo, o caminho para o índice STAR será referido como "path_to/STAR_index". - Navegue até o path_to/projeto/diretório. Faça um novo diretório chamado "STAR_output" como mostrado na Figura S1D.
- Navegue até o path_to/projeto/aparado/diretório. Use o comando mostrado na Figura S1D para executar o STAR para alinhar os arquivos fastq .gz aparados.
NOTA: Esta etapa é a mais computacionalmente intensiva e recomenda-se realizar isso em um cluster HPC com vários segmentos (ou seja, >16) designados para a tarefa de alinhamento. Dependendo do número de amostras e recursos computacionais disponíveis, esta etapa pode levar muitas horas a dias. - Encontre a saída necessária para os próximos passos que contenham as contagens por transcrição no seguinte local: path_to/project/STAR_output/sampleN_ReadsPerGene.out.tab.
NOTA: Na coluna ReadsPerGene.out.tab 1 contém informações sobre o recurso que está sendo contado. A coluna 2 contém as contagens de leitura não restritas, a coluna 3 mantém as contagens de leitura suspensas e a coluna 4 contém as contagens de leitura encalhadas inversas. As quatro primeiras linhas deste arquivo terão informações sobre as leituras alinhadas que não se alinharam a um único gene. Este protocolo requer as contagens de leitura não transe. - Use RStudio (preferível) ou R no ambiente HPC para compilar os dados da linha 5 e abaixo para as colunas 1 e 2 para cada amostra. Defina o diretório de trabalho para "projeto" em R.
- Leia em cada arquivo ReadsPerGene.out.tab usando o comando na Figura S2A. Para a primeira coluna, leve apenas os caracteres antes do "." na coluna "Ensembl gene ID" para facilitar o processamento a jusante.
- Compile contagens de todas as amostras em um dataframe chamado "totcts" usando os comandos na Figura S2B. Salve esta nova tabela de dados de contagem bruta como uma guia delimitada .txt arquivo, ou seja, sample_counts.txt, se desejar, usando o comando "write.table".
NOTA: A ordem do ID do gene Ensembl é a mesma para cada arquivo ReadsPerGene.out.tab em todas as amostras.
5. Expressão diferencial e análise a jusante
- Normalize para efeitos em lote entre amostras com ComBat.
NOTA: Existem duas variáveis possíveis que explicam mudanças na expressão genética, sendo a primeira a construção utilizada (ou seja, a amostra) e a segunda fatores externos associados à passagem das células ao longo do tempo (ou seja, o lote). Recomenda-se um passo para normalizar amostras para a variação em lote com o ComBat do pacote R.- Instale se necessário e carregue as bibliotecas para sva, DESeq2, AnotationDBI.org. Hs.eg.db, pheatmap, RColorBrewer, genefilter, Cairo, ggplot2, ggbiplot, rgl e remodele2 como mostrado na Figura S2C. Para instalação, use o comando "install.packages" ou Bioconductor por documentação para cada pacote.
- Primeiro filtrar os dados apenas para aqueles genes que têm pelo menos uma contagem por leitura. Salve esta nova tabela para denotar filtragem como visto na Figura S2D.
NOTA: Frequentemente, muitos genes terão contagem muito baixa ou nenhuma leitura. - Prepare uma segunda tabela para normalização em lote chamada "vars", como mostrado na Figura S2E. Defina os nomes da linha para os nomes únicos de cada amostra. Defina os nomes das colunas como "amostra", "lote" e "construa".
- Atribua a todas as amostras um número único na coluna "amostra" de 1 a n, sendo n o número de amostras. Atribua números de lote a todas as amostras na coluna "lote", de forma que a_1 e b_1 de condições sejam atribuídos 1, e que as a_2 e as b_2 de condição sejam atribuídas 2. Atribua todas as designações de condição a todas as amostras da coluna "construto", de tal forma que as amostras de condição são todas "A" e as amostras de condição b são todas "B".
- Defina a variável de lote também, e uma matriz de modelo nulo específica para ComBat, conforme mostrado na Figura S2F. Execute o ComBat com o comando definido na Figura S2F.
- Faça a curadoria dos dados arredondando para o inteiro mais próximo. Também remova genes com um valor negativo. Use os comandos mostrados na Figura S3A.
NOTA: A saída da normalização do lote terá contagem de leitura não-inteiro e alguns genes com valores negativos. Esta etapa é necessária porque a análise de expressão diferencial a jusante não suporta contagens negativas de leitura. - Defina o perfil de expressão diferencial para cada construção usando o DESeq2.
- Insira o design do experimento para DESeq2 como mostrado na Figura S3B. Construa um DESeqDataSet (dds) usando a função DESeqDataSetFromMatrix, estime os fatores de tamanho e execute o DESEq2, como mostrado na Figura S3B.
NOTA: É imprescindível que os dados da coluna inseridos para "condição" esteja na mesma ordem da coluna na matriz de contagem. - Para avaliar a qualidade da análise, extrair as contagens de rlog normalizadas utilizadas pelo DESeq2, conforme mostrado na Figura S3B.
NOTA: Durante a análise, a transformação DESeq2 conta com um "log regularizado", rlog, transformação para reduzir as diferenças amostrais para amostra para genes com baixa contagem (baixa informação) a fim de preservar diferenças em genes com maior contagem entre amostras (altas informações). - Ao extrair os resultados de cada perfil transcricional a partir dos resultados do DESeq2, realize comparações pareiras em relação à condição de knockdown ou vetor vazio da linha de base, conforme mostrado na Figura S3C. Altere ainda mais esses resultados com os símbolos genéticos HGNC, como mostrado na Figura S3D.
- Como visto na Figura S3E,extraia dados dos resultados do DESeq2. Exporte como um único arquivo com o ID genético Ensembl, símbolo HGNC, expressão média base e dados de expressão diferencial para todos os construtos com log2FoldChange e valores p brutos e ajustados.
NOTA: Utilizar um valor p ajustado < 0,05 é o limite recomendado para a expressão diferencial. - Avalie a normalização do lote bem-sucedida e a similaridade intra-amostra. Verifique o agrupamento de amostras com pca e parcelas de distância amostra-a-amostra usando as contagens normalizadas do rlog usando o código mostrado nas Figuras S4A-B.
- Insira o design do experimento para DESeq2 como mostrado na Figura S3B. Construa um DESeqDataSet (dds) usando a função DESeqDataSetFromMatrix, estime os fatores de tamanho e execute o DESEq2, como mostrado na Figura S3B.
- Use os perfis de expressão diferencial para gerar parcelas vulcânicas usando o código na Figura S4C. Avalie mudanças na expressão genética através de construções.
- Use as contagens normalizadas de rlog e o agrupamento hierárquico para identificar assinaturas genéticas exclusivas dos diferentes construtos. Use o código mostrado na Figura S4D.
- Extrair os 1000 genes mais variáveis em todos os construtos em uma matriz. Use o featmap para realizar agrupamento hierárquico não supervisionado de suas amostras com base nesses genes.
- Extrair os aglomerados de interesse do dendrograma decidindo em que nível dos aglomerados de dendrograma aparecem. Definir "k" igual ao número de clusters nesse nível. Replot o mapa de calor ordenado por cluster para determinar quais clusters são de interesse, como mostrado na Figura S5.
- Exporte a lista de genes associados a cada cluster, conforme demonstrado na Tabela S1. Use essas informações para determinar os genes em aglomerados de interesse.
- Identificar os papéis biológicos para diferentes aglomerados de genes identificados e comparar entre as classes. Isso pode ser realizado usando uma variedade de ferramentas de bioinformática. O ToppGene24 é usado aqui e está disponível gratuitamente online.
NOTA: Existem muitas ferramentas gratuitas que requerem apenas uma lista de genes para copiar e colar em um campo em um site. Escolha as ferramentas analíticas mais adequadas para as questões de pesquisa sob investigação. - Opcionalmente, se houver dados disponíveis sobre a ligação genômica que conduza a saída transcricional para fator de transcrição de interesse, compare a resposta transcricional em genes associados a diferentes elementos vinculantes para avaliar ainda mais a função mutante.
6. Comparação com Fenótipos Relevantes
- Compare os fenótipos correlativos com os dados de perfil transcriômico gerados e interprete conforme apropriado.
Resultados
Dados preliminares de qRT-PCR sugeriram que um mutante EWS/FLI chamado DAF, com mutações específicas de tyrosina para alanina na região repetitiva e desordenada do EWS, manteve a capacidade de ativar genes de alvo EWS/FLI, mas não conseguiu reprimir genes-alvo críticos23. Para compreender melhor a relação entre esses resíduos no domínio EWS e na função EWS/FLI, utilizou-se o protocolo descrito acima e descrito na Figura 1. As células de sarcoma A673 Ewing foram viralmente transduzidas com um shRNA direcionado ao 3'UTR de FLI1, resultando no esgotamento do EWS/FLI endógeno. Após quatro dias de seleção, a função EWS/FLI foi resgatada com transdução viral de diferentes construções mutantes EWS/FLI marcadas por 3XFLAG, com vetor vazio como controle para nenhum resgate. Um mutante não funcional sem o domínio EWS, chamado Δ22, foi usado como controle negativo e o EWS/FLI do tipo selvagem, chamado wtEF, foi usado como um controle positivo(Figura 2A). O DAF foi usado como construção de teste, embora mais de uma construção de teste possa ser usada se desejar. As células foram selecionadas por mais 10 dias para permitir que a expressão de construção se estabilize e depois coletada para RNA (com uma etapa de remoção de gDNA), proteína e ensaios de formação de colônias. Quatro réplicas foram coletadas e as manchas representativas qRT-PCR e ocidentais que mostram knockdown e resgate eficazes são mostradas na Figura 2B-D. Deve-se notar que as células resgatadas pelo DAF não conseguiram formar colônias como mostrado na Figura 2E,sugerindo a transformação oncogênica prejudicada.
Após a conclusão da validação da réplica e ensaios fenotípicos, o RNA foi submetido ao Instituto de Medicina Genômica do Hospital Infantil Nacional para preparação da biblioteca e sequenciamento de próxima geração com ~50 milhões de leituras de fim de ano de 150 bp coletadas. Os dados foram devolvidos como arquivos .gz Fastq. Leituras de baixa qualidade foram aparadas desses arquivos com TrimGalore e STAR foi usada para alinhar leituras ao genoma humano hg19 e contar as leituras por gene. hg19 foi utilizado para fins de compatibilidade com os outros conjuntos de dados curados para EWS/FLI utilizados na análise a jusante. Estas contagens de leitura foram combinadas em uma matriz de contagem única para todas as amostras, as primeiras 6 linhas das quais são mostradas na Figura 3.
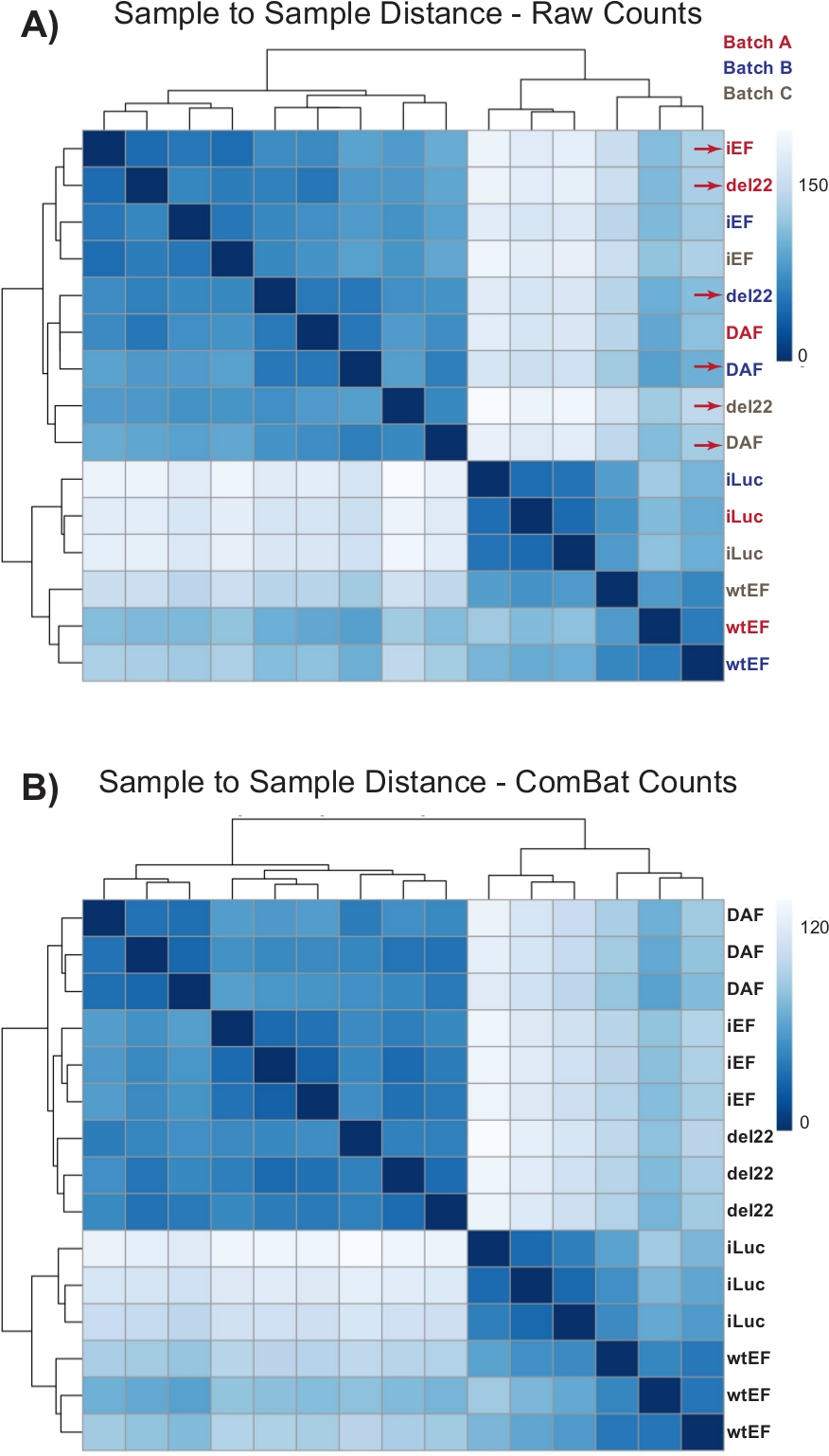
As contagens foram inicialmente executadas através do DESeq2 sem a normalização em lote, no entanto, a inspeção visual da distância amostra-a-amostra mostrou potenciais efeitos de lote de confusão, como mostrado destacado com setas vermelhas na Figura 4A. Isso provavelmente surgiu devido à variabilidade biológica introduzida pela passagem de células na cultura e diferenças no processamento de cada lote. A normalização dos efeitos em lote foi realizada com o ComBat e é geralmente recomendada. As distâncias amostra-a-amostra dos dados normalizados em lote são mostradas na Figura 4B. Após a normalização do lote, o DESeq2 foi usado para gerar perfis transcricionais para os três construtos (wtEF, Δ22 e DAF) relativos à linha de base. Note que enquanto as células A673 "parentais" (knockdown simulado e resgate simulado, chamado "iLuc" aqui) foram incluídas na análise diferencial, a referência para este experimento são as células com EWS/FLI-esgotado, chamados células iEF. O perfil transcricional pode ser gerado para a proteína endógena aqui, comparando a amostra iLuc com iEF, e isso pode interessar em entender como funciona o sistema de resgate, mas esse não é o objetivo desta análise em particular. Os perfis transcricionais gerados para os mutantes incluem controles positivos (wtEF) e negativos (Δ22), com relação ao iEF, de tal forma que estes devem funcionar como referência para outros mutantes. Isso é importante, pois o controle positivo neste exemplo não recapitulava completamente a função do Endógeno EWS/FLI como discutido em outros lugares7,23.
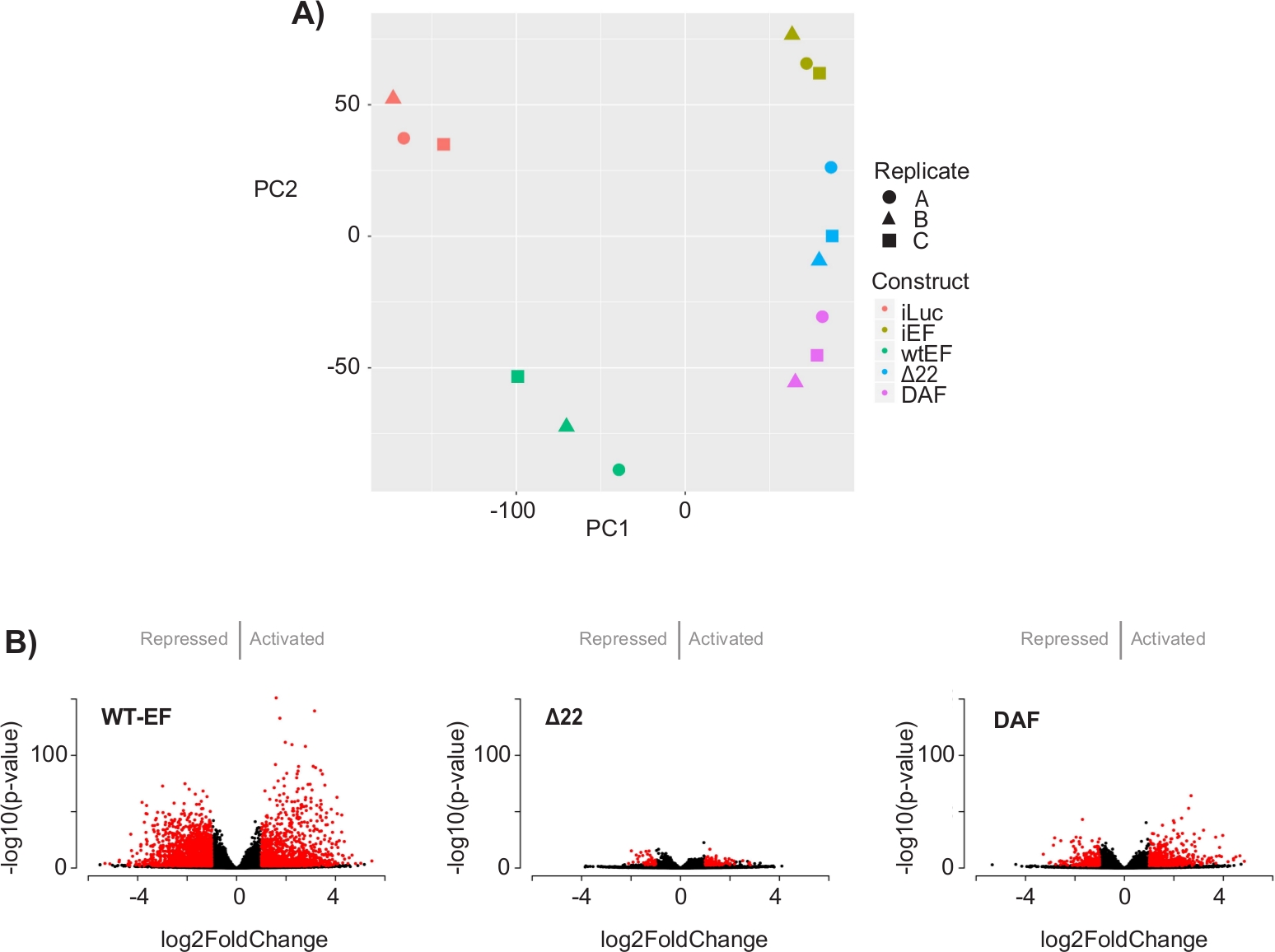
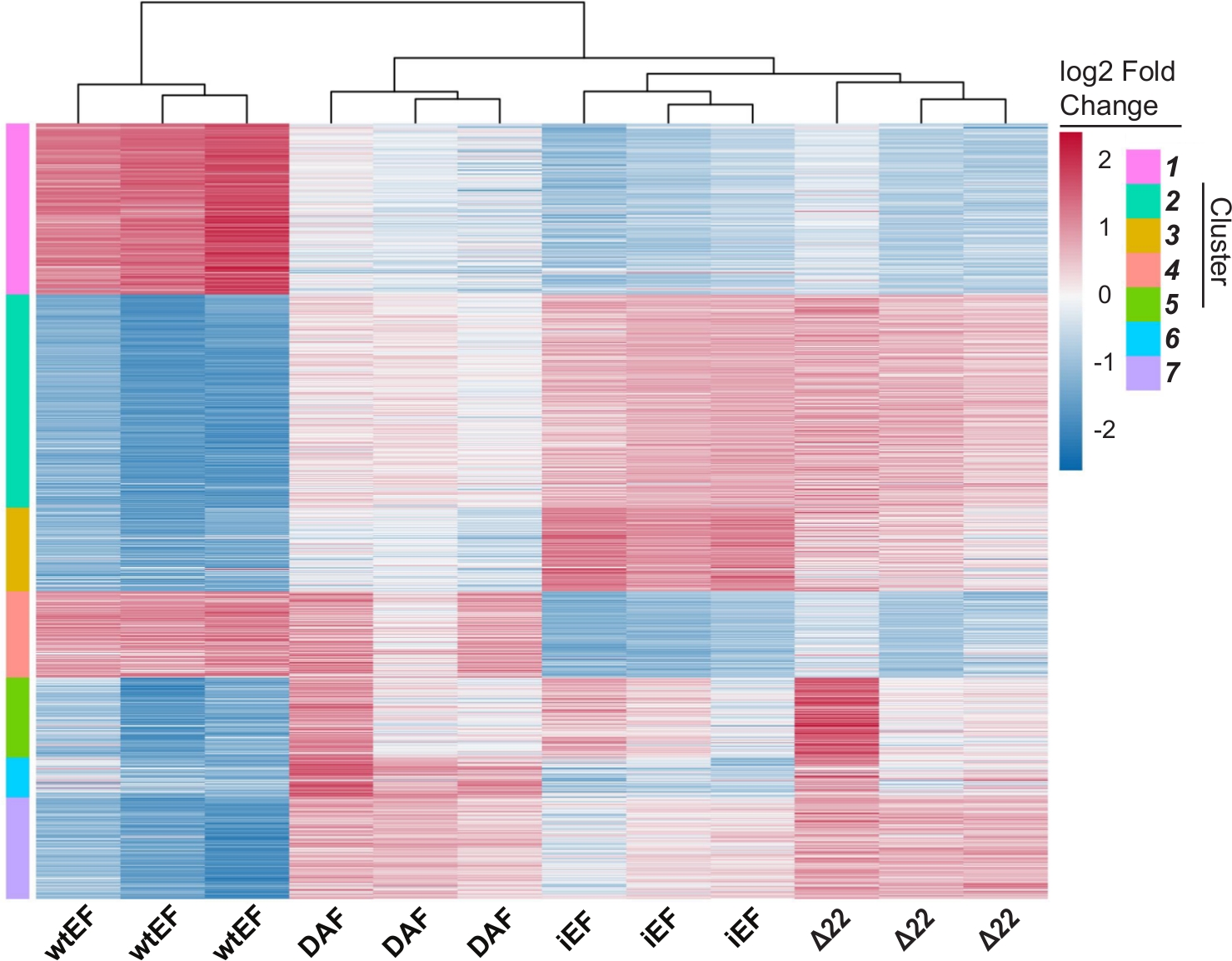
A análise dos componentes principais (PCA) na Figura 5 sugere que o perfil transcricional do DAF é intermediário entre wtEF e Δ22, confirmando a função parcial. Além disso, o agrupamento hierárquico dos 1000 genes mais variáveis entre as amostras mostrou que o DAF falhou em reprimir genes alvo EWS/FLI, e apenas reteve parcialmente a atividade de ativação genética, como mostrado na Figura 6A e Figura S5. A análise do ToppGene sugeriu que as classes de genes que o DAF ativa são funcionalmente distintas daqueles alvos ativados pelo EWS/FLI, onde o DAF não é funcional(Figura 6B). Curiosamente, a função de genes ativados resgatados pelo wtEF, mas não pelo DAF, parecem estar relacionadas ao controle transcricional e à regulação da cromatina. Com base nos resultados dos ensaios de formação da colônia, os genes desta assinatura genética central devem ser analisados por seu papel na oncogênese mediada pelo EWS/FLI. A importância da repressão genética mediada pelo EWS/FLI foi descrita anteriormente17.
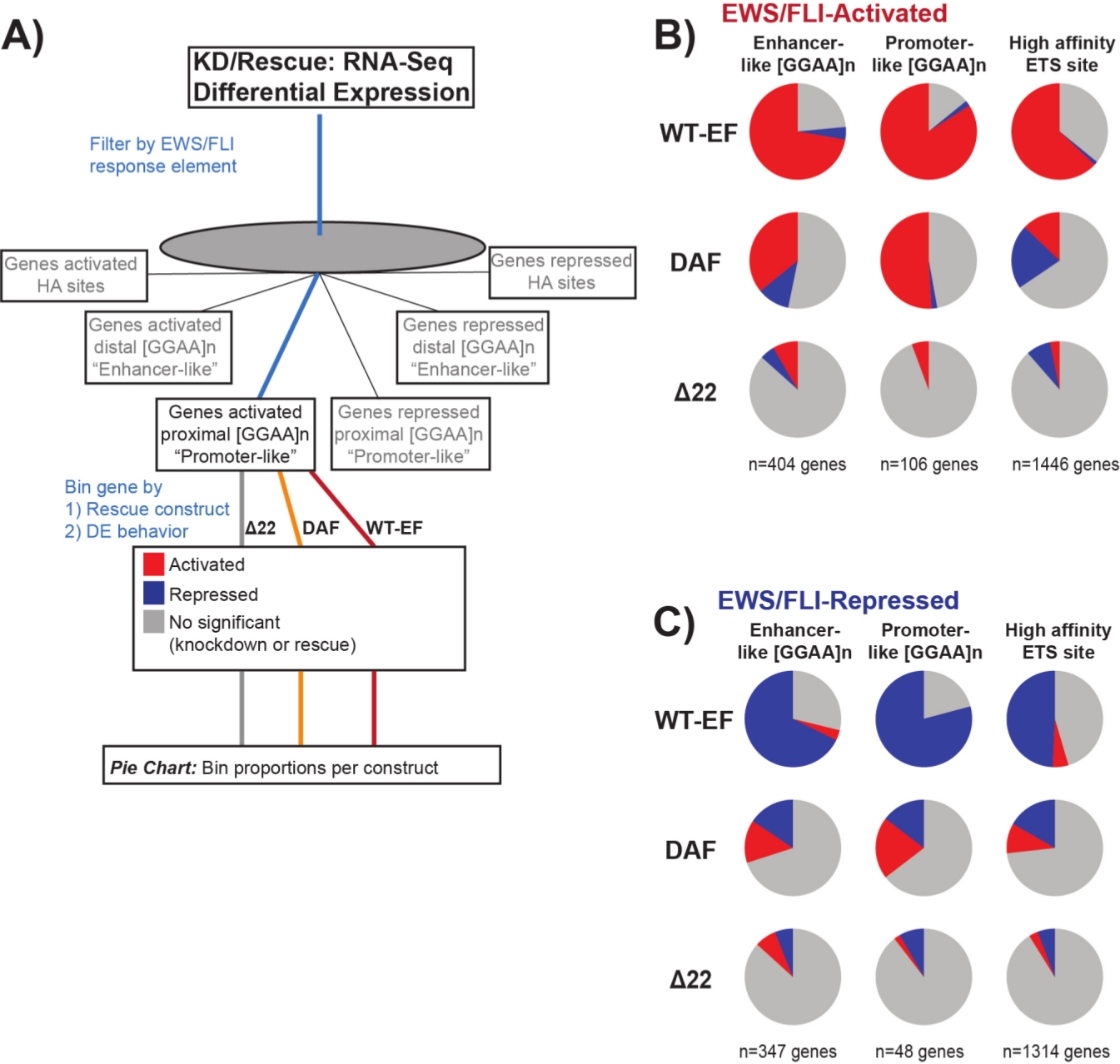
Sabe-se que o EWS/FLI possui uma afinidade vinculante única para os elementos de repetição GGAA-microsatélite19,22, e que a vinculação a esses elementos impulsiona a regulação genética a jusante11,15,18,20,22. Esses microsatélites têm sido caracterizados como associados à ativação ou repressão, e ou proximal a (< 5 kb) TSS ou distal (> 5 kb) TSS25. Além disso, existem genes regulados por EWS/FLI com alto afinidade (HA) ETS, motivos proximais ao TSS23. A fim de analisar melhor as características da função DAF e quais tipos de genes ativados por EWS/FLI, o DAF foi capaz de resgatar, foi analisada a expressão diferencial dos genes associados a essas diferentes classes. Curiosamente, o DAF foi mais capaz de resgatar genes ativados ggaa-microsatélites, mas incapaz de resgatar genes ativados perto de um local ha como visto na Figura 7. Como visto com o agrupamento hierárquico, o DAF falha em resgatar a repressão mediada pelo EWS/FLI em todas as classes de motivos. Esses dados sugerem que o DAF retém características estruturais suficientes do EWS para se ligar e ativar a partir de microsatélites GGAA, tanto proximal quanto distal ao TSS. Isso provavelmente surge do domínio SYGQ intacto considerado importante para a atividade EWS/FLI na GGAA repete11. Esses dados também sugerem que as tyrosinas específicas mutadas no DAF desempenham papéis importantes, mas mal compreendidos, na regulação genética mediada pelo EWS/FLI a partir de sites ha, bem como na repressão genética, destacando uma área importante de investigação mais aprofundada.

Figura 1: Fluxo de trabalho. Representação do procedimento passo-a-passo para realizar o mapeamento de função estrutura por transcriômica. As células foram preparadas para expressar o conjunto de construções necessárias para o mapeamento estrutura-função. Seguindo a expressão, as células foram colhidas para RNA e proteína e avaliadas para fenótipos correlativos. A expressão dos construtos foi validada, e esse processo foi repetido 3-4 vezes para coletar réplicas biológicas independentes. O RNA foi então submetido ao sequenciamento de próxima geração (NGS). Quando os dados foram recebidos, os dados foram aparados para qualidade, alinhados e as contagens por transcrição foram calculadas. Os efeitos em lote foram controlados e as assinaturas transcriômicas e a expressão diferencial foram determinadas por meio do DESeq2. O clusteramento hierárquico e a análise a jusante integrando outros conjuntos de dados -omics e diferentes caminhos ou análises funcionais podem ser incorporados. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 2: Validação da expressão de construção e ensaios correlativos. (A) Esquema representando os construtos testados neste exemplo. (B) Validação do knockdown do EWS/FLI endógeno e expressão de construções marcadas por 3X-FLAG por imunoblot. (C,D) Validação da atividade de construção em um gene alvo ativado EWS/FLI (C), NR0B1, e(D) gene alvo reprimido, TGFBR2, por qRT-PCR. Os dados são apresentados como desvio médio +/- padrão. Os valores P foram calculados com um teste de significância honesto de Tukey. * p < 0,05, ** p < 0,01, *** p < 0,005 (E) Colônia conta a partir de ensaios de ágar macio realizados para avaliar a atividade transformadora de construções. Os valores P foram calculados com um teste de significância honesto de Tukey. * p < 0,05, ** p < 0,01, *** p < 0,005. Esta figura é adaptada de Theisen, et al.23Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 3: Dados finais de contagem de colhidos para análise. Captura de tela das 6 primeiras linhas do arquivo de contagem com contagem de genes para que todas as amostras sejam normalizadas e analisadas em lote. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 4: Mapas de distância de amostra para amostra. (A) Gráfico de distância amostra-a-amostra mostrando o agrupamento amostral dos dados de contagem bruta. As amostras que estão agrupando tanto por lote quanto por amostra são denotadas com setas vermelhas. (B) Parcela de distância amostra-a-amostra após a normalização do lote com o ComBat. Aqui, amostras de todas as réplicas agrupam-se, independente do lote. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 5: Resultados da análise de expressão diferencial. (A) O gráfico de análise de componentes de princípio (PCA) das assinaturas transcriômicas geradas para todas as amostras mostra um forte agrupamento intra-amostra e demonstra que o DAF é intermediado entre os controles positivo (wtEF) e negativo (Δ22). (B) Gráficos vulcânis mostrando o -log(p-value) plotado contra o log2FoldChange para genes em cada construção. Genes com valor p ajustado < 0,05 e |log2(FoldChange)| > 1 são considerados significativos e são mostrados em vermelho. Painel 5B é adaptado de Theisen, et al.23Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 6: Agrupamento hierárquico para identificar classes genéticas. ( A )Oagrupamento hierárquico dos 1000 genes mais variáveis em todas as construções e a linha de base, iEF, mostra que o DAF resgata parcialmente a ativação genética mediada por EWS/FLI. (B) A ontologia genética (função molecular) resulta do ToppGene mostrando o enriquecimento funcional de genes ativados por EWS/FLI que são resgatados ou não pelo DAF. Painel 6B é adaptado de Theisen, et al.23Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 7: Análise detalhada de diferentes elementos de resposta ao fator de transcrição para diferentes construções: (A) Esquema que retrata o processamento de dados usado para gerar painéis (B) e (C) incorporando outros conjuntos de dados disponíveis com os perfis transcriômicos aqui. (B,C) Compilação mostrando o resgate de diferentes classes de alvos diretos EWS/FLI- (B) ativados e(C) alvos reprimidos. Os genes incluídos foram apenas aqueles genes com expressão diferencial detectável por EWS/FLI endógeno. Em cada gráfico de tortas, o cinza retrata a porção de genes que não são resgatados pela construção. O vermelho retrata a porção de genes que são ativados diferencialmente, e o azul retrata a porção de genes que são diferencialmente reprimidos. Esta figura é adaptada de Theisen, et al.23Clique aqui para ver uma versão maior desta figura.
{kind=link}
Figura S1: Carregando os arquivos fastq.gz para o ambiente HPC, aparando e alinhamento. Clique aqui para baixar este número.
{kind=link}
Figura S2: A leitura de collating conta entre amostras e a normalização do lote em execução com o ComBat. Clique aqui para baixar este número.
{kind=link}
Figura S3: Executando o DESeq2 e extraindo resultados de análise de expressão diferencial. Clique aqui para baixar este número.
{kind=link}
Figura S4: Analisando a saída. Clique aqui para baixar este número.
{kind=link}
Figura S5: Agrupamento hierárquico para identificar classes genéticas: Agrupamento hierárquico dos 1000 genes mais variáveis em todos os construtos e na linha de base, iEF, classificados em aglomerados k. Neste caso k=7, mas este parâmetro é definido pelo usuário como mostrado na Figura S4D. Clique aqui para baixar este número.
{kind=link}
Tabela S1: Lista de genes (Ensembl gene ID) com anotação de cluster. Clique aqui para baixar esta tabela.
Discussão
Estudar os mecanismos bioquímicos dos fatores de transcrição oncogênica é extremamente importante para entender as doenças que causam e projetar novas estratégias terapêuticas. Isso é especialmente verdade em malignidades caracterizadas por translocações cromossômicas que resultam em fatores de transcrição de fusão. Os domínios incluídos nessas proteínas quimricas podem não ter interações significativas com domínios regulatórios presentes nas proteínas do tipo selvagem, complicando a capacidade de interpretar informações estruturais-funções no contexto da fusão26,27,28. Além disso, muitas dessas fusões oncogênicas são caracterizadas por domínios intrinsecamente desordenados de baixa complexidade10,13,29,30.
O domínio EWS é um exemplo de um domínio tão intrinsecamente desordenado que está envolvido em uma variedade de fusões oncogênicas10. A natureza intrinsecamente desordenada e repetitiva tem dificultado os esforços para entender os mecanismos moleculares empregados pelo domínio EWS. Esforços anteriores para investigar a estrutura-função têm recorrido em grande parte ao uso de diferentes mutantes no contexto de ensaios genéticos de repórteres ou em fundos celulares que não conseguem recapitular o contexto celular relevante, ou não possuem variações estruturais que produzam uma função parcial significativa11,17,25. O método aqui apresentado aborda essas questões. O mapeamento estrutura-função é realizado em um contexto celular relevante para a doença e o sequenciamento de próxima geração permite que o perfil transcriômico avalie a função do fator de transcrição na configuração da cromatina nativa. No caso específico do mutante DAF da EWS/FLI, daf foi relatado para mostrar pouca atividade em ensaios de repórteres usando elementos de resposta isolados, mas para mostrar atividade no contexto do promotor genético completo, seja em um ensaio repórter ou em cromatina nativa, sugerindo um fenótipointeressante 23. O uso do método aqui descrito resolve mais diretamente a questão de qual tipo de elementos regulatórios em todo o genoma são mais responsivos no cenário da doença. Ao testar todos os genes-alvo do candidato em seu contexto nativo de cromatina simultaneamente, uma abordagem transcriômica é mais provável para identificar construções com função parcial.
A força inerente do uso de um fundo celular relevante para a doença é talvez a maior limitação dessa técnica. Um dos fatores mais importantes é a escolha do sistema celular adequado para esses experimentos. Muitas linhas celulares derivadas de malignidades com fatores de transcrição pathognomônica não toleram prontamente o knockdown desse fator de transcrição, e em muitos casos, particularmente para cânceres pediátricos, a verdadeira célula de origem permanece controversa e a expressão do oncogene em outras origens celulares é proibitivamente tóxica31,32 . Nesses casos, pode ser útil realizar experimentos em um fundo celular diferente, desde que o pesquisador exerça cautela na interpretação dos resultados e valide adequadamente quaisquer achados relevantes em um tipo celular mais relevante para a doença.
É extremamente importante validar cuidadosamente a estabilidade e as consequências fenotípicas da expressão do oncogene e submeter apenas amostras para sequenciamento que atendam a critérios rigorosos. Aqui, isso incluiu a mancha ocidental para confirmar o knockdown e o resgate, e qRT-PCR de um pequeno número de genes alvo conhecidos para validar o controle positivo(Figura 2). Da mesma forma, é crucial diminuir o máximo de variabilidade de lote possível, realizando cuidadosamente as preparações de célula e RNA da forma mais semelhante possível através de cada lote.
O método aqui descrito torna-se especialmente poderoso quando emparelhado com outros tipos de dados genômicos que falam da função genoma do fator de transcrição em estudo. As direções futuras para este tipo de análise estrutura-função se expandiriam para incluir ChIP-seq e ATAC-seq para determinar a vinculação do fator de transcrição e quaisquer alterações induzidas na acessibilidade da cromatina. Como um conjunto, esse tipo de dados pode apontar para onde diferentes componentes estruturais de um fator de transcrição oncogênica contribuem para diferentes aspectos da função (ou seja, vinculação de DNA vs. modificação de cromatina versus recrutamento co-regulador). No geral, o uso de abordagens baseadas em NGS para mapear as relações estrutura-função dos fatores de transcrição de fusão pode revelar novos insights nos determinantes bioquímicos da função oncogênica dessas proteínas. Isso é importante para aprofundar nossa compreensão das doenças que causam e possibilitar o desenvolvimento de novas estratégias terapêuticas.
Divulgações
A SLL declara um conflito de interesses como membro do conselho consultivo e detentor de capital da Salarius Pharmaceuticals. SLL também é um inventor listado em Patentes dos Estados Unidos No. EUA 7.393.253 B2, "Métodos e composições para o diagnóstico e tratamento do Sarcoma de Ewing", e US 8.557.532, "Diagnóstico e tratamento do sarcoma resistente a medicamentos Ewing". Isso não altera nossa adesão às políticas da JoVE no compartilhamento de dados e materiais.
Agradecimentos
Esta pesquisa foi apoiada pelo Centro de Computação de Alto Desempenho do Instituto de Pesquisa Abigail Wexner do Nationalwide Children's Hospital. Este trabalho foi apoiado pelo Instituto Nacional de Saúde Instituto Nacional de Câncer [U54 CA231641 para SLL, R01 CA183776 para SLL]; Alex's Lemonade Stand Foundation [Prêmio Jovem Investigador para ERT]; Pelotonia [Bolsa para ERT]; e o Conselho Nacional de Saúde e Pesquisa Médica CJ Martin Overseas Biomedical Fellowship [APP1111032 to KIP].
Materiais
| Name | Company | Catalog Number | Comments |
| Wet Lab Reagents | |||
| anti-FLI rabbit pAb | Abcam | ab15289 | 1:500 |
| anti-lamin B1 rabbit pAb | Abcam | ab16048 | 1:2000 |
| Cell-based system for introduction of mutant constructs | Determined by cell system used | ||
| Cryotubes | For viral aliquots | ||
| DMEM | Corning Cellgro | 10-013-CV | For viral production |
| Fetal bovine serum | Gibco | 16000-044 | For viral production |
| G418 | ThermoFisher | 10131027 | For viral production |
| HEK293-EBNAs | ATCC | CRL-10852 | For viral production |
| HEPES | Gibco | 15630106 | |
| Hygromycin B | ThermoFisher | 10687010 | |
| M2 anti-FLAG mouse mAb | Sigma | F3165 | 1:2000 |
| Near IR-secondary antibodies | Li-Cor | ||
| Optimem | Gibco | 31985062 | For viral production |
| Penicillin/Streptomycin/Glutamine | Gibco | 10378-016 | For viral production |
| Polybrene | Sigma | TR-1003-G | For viral transduction |
| Puromycin | Sigma | P8833 | Stored at 2 mg/mL stock |
| RNeasy Plus kit | Qiagen | 74136 | Has gDNA removal columns |
| Selection reagents | As dictated by cell system used | ||
| Sodium Pyruvate | Gibco | 11360-070 | For viral production |
| Tissue culture media | Determined by cell system used | ||
| TransIT-LT1 | Mirus | MIR 2304 | For viral production |
| Software | |||
| Access to HPC environment | |||
| AnnotationDbi | 1.38.2 | ||
| Cairo | 1.5-10 | ||
| DESeq2 | 1.16.1 | ||
| genefilter | 1.58.1 | ||
| ggbiplot | 0.55 | ||
| ggplot2 | 3.1.1 | ||
| org.Hs.eg.db | 3.4.1 | ||
| pheatmap | 1.0.12 | ||
| PuTTY | |||
| R | 3.4.0 | ||
| RColorBrewer | 1.1-2 | ||
| reshape2 | 1.4.3 | ||
| rgl | 0.100.19 | ||
| R-studio | |||
| STAR | Version 2.6 or later | ||
| sva | 3.24.4 | ||
| TrimGalore! | |||
| WinSCP |
Referências
- Miettinen, M., et al. New fusion sarcomas: histopathology and clinical significance of selected entities. Human Pathology. 86, 57-65 (2019).
- Knott, M. M. L., et al. Targeting the undruggable: exploiting neomorphic features of fusion oncoproteins in childhood sarcomas for innovative therapies. Cancer and Metastasis Reviews. 38, 625-642 (2019).
- Yoshihara, K., et al. The landscape and therapeutic relevance of cancer-associated transcript fusions. Oncogene. 34, 4845-4854 (2015).
- Duesberg, P. H. Cancer genes generated by rare chromosomal rearrangements rather than activation of oncogenes. Medical Oncology and Tumor Pharmacotherapy. 4, 163-175 (1987).
- Dupain, C., Harttrampf, A. C., Urbinati, G., Geoerger, B., Massaad-Massade, L. Relevance of Fusion Genes in Pediatric Cancers: Toward Precision Medicine. Molecular Therapy - Nucleic Acids. 6, 315-326 (2017).
- Mitelman, F., Johansson, B., Mertens, F. The impact of translocations and gene fusions on cancer causation. Nature Reviews Cancer. 7, 233-245 (2007).
- Smith, R., et al. Expression profiling of EWS/FLI identifies NKX2.2 as a critical target gene in Ewing's sarcoma. Cancer Cell. 9, 405-416 (2006).
- Davicioni, E., et al. Identification of a PAX-FKHR gene expression signature that defines molecular classes and determines the prognosis of alveolar rhabdomyosarcomas. Cancer Research. 66, 6936-6946 (2006).
- Gröbner, S. N., et al. The landscape of genomic alterations across childhood cancers. Nature. 555, 321-327 (2018).
- Kim, J., Pelletier, J. Molecular genetics of chromosome translocations involving EWS and related family members. Physiological Genomics. 1, 127-138 (1999).
- Boulay, G., et al. Cancer-Specific Retargeting of BAF Complexes by a Prion-like Domain. Cell. 171, 163-178 (2017).
- Lessnick, S. L., Braun, B. S., Denny, C. T., May, W. A. Multiple domains mediate transformation by the Ewing’s sarcoma EWS/FLI-1 fusion gene. Oncogene. 10, 423-431 (1995).
- Leach, B. I., et al. Leukemia fusion target AF9 is an intrinsically disordered transcriptional regulator that recruits multiple partners via coupled folding and binding. Structure. 21, 176-183 (2013).
- Ng, K. P., et al. Multiple aromatic side chains within a disordered structure are critical for transcription and transforming activity of EWS family oncoproteins. Proceedings of the National Academy of Sciences U.S.A. 104, 479-484 (2007).
- Riggi, N., et al. EWS-FLI1 Divergent Chromatin Remodeling Mechanisms to Directly Activate or Repress Enhancer Elements in Ewing Sarcoma. Cancer Cell. 26, 668-681 (2014).
- Tomazou, E. M., et al. Epigenome Mapping Reveals Distinct Modes of Gene Regulation and Widespread Enhancer Reprogramming by the Oncogenic Fusion Protein EWS-FLI1. Cell Reports. 10, 1082-1095 (2015).
- Sankar, S., et al. Mechanism and relevance of EWS/FLI-mediated transcriptional repression in Ewing sarcoma. Oncogene. 32, 5089-5100 (2013).
- Gangwal, K., et al. Microsatellites as EWS/FLI response elements in Ewing's sarcoma. Proceedings of the National Academy of Sciences U.S.A. 105, 10149-10154 (2008).
- Gangwal, K., Close, D., Enriquez, C. A., Hill, C. P., Lessnick, S. L. Emergent Properties of EWS/FLI Regulation via GGAA Microsatellites in Ewing's Sarcoma. Genes & Cancer. 1, 177-187 (2010).
- Guillon, N., et al. The Oncogenic EWS-FLI1 Protein Binds In Vivo GGAA Microsatellite Sequences with Potential Transcriptional Activation Function. PLoS One. 4, 4932 (2009).
- Chong, S., et al. Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science. 361, (2018).
- Johnson, K. M., et al. Role for the EWS domain of EWS/FLI in binding GGAA-microsatellites required for Ewing sarcoma anchorage independent growth. Proceedings of the National Academy of Sciences U.S.A. 114, 9870-9875 (2017).
- Theisen, E. R., et al. Transcriptomic analysis functionally maps the intrinsically disordered domain of EWS/FLI and reveals novel transcriptional dependencies for oncogenesis. Genes & Cancer. 10, 21-38 (2019).
- Chen, J., Bardes, E. E., Aronow, B. J., Jegga, A. G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Research. 37, 305-311 (2009).
- Johnson, K. M., Taslim, C., Saund, R. S., Lessnick, S. L. Identification of two types of GGAA-microsatellites and their roles in EWS/FLI binding and gene regulation in Ewing sarcoma. PLOS One. 12, 0186275 (2017).
- Kim, P., Ballester, L. Y., Zhao, Z. Domain retention in transcription factor fusion genes and its biological and clinical implications: a pan-cancer study. Oncotarget. 8, 110103-110117 (2017).
- de Mendíbil, I. O., Vizmanos, J. L., Novo, F. J. Signatures of Selection in Fusion Transcripts Resulting from Chromosomal Translocations in Human Cancer. PLOS One. 4, 4805 (2009).
- Frenkel-Morgenstern, M., Valencia, A. Novel domain combinations in proteins encoded by chimeric transcripts. Bioinformatics. 28, 67-74 (2012).
- Hegyi, H., Buday, L., Tompa, P. Intrinsic Structural Disorder Confers Cellular Viability on Oncogenic Fusion Proteins. PLoS Computational Biology. 5, 1000552 (2009).
- Latysheva, N. S., Babu, M. M. Discovering and understanding oncogenic gene fusions through data intensive computational approaches. Nucleic Acids Research. 44, 4487-4503 (2016).
- Deneen, B., Denny, C. T. Loss of p16 pathways stabilizes EWS/FLI1 expression and complements EWS/FLI1 mediated transformation. Oncogene. 20, 6731-6741 (2001).
- Kendall, G. C., et al. PAX3-FOXO1 transgenic zebrafish models identify HES3 as a mediator of rhabdomyosarcoma tumorigenesis. eLife. 7, 33800 (2018).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados