
Method Article
Imagem 4-Dimensional da Morfogense da Copa Óptica de Zebrafish
Neste Artigo
Resumo
Este protocolo descreve uma abordagem para rotulagem e imagem multidimensional de zebrafish desenvolvimento precoce dos olhos. Descrevemos rotulagem, incorporação e imagens quadridimensionais (4D) usando microscopia confocal de varredura a laser, e considerações para otimizar a aquisição de conjuntos de dados para dissecção de mecanismos de morfogênese de copo óptico.
Resumo
A função do sistema visual requer o estabelecimento de estruturas precisas de tecidos e órgãos. No olho vertebrado, defeitos estruturais são uma causa comum de deficiência visual, mas os mecanismos de morfogênese ocular ainda são mal compreendidos. A organização básica do olho embrionário é conservada em todos os vertebrados, assim, a imagem viva dos embriões de zebrafish tornou-se uma abordagem poderosa para observar diretamente o desenvolvimento dos olhos em tempo real sob condições normais e patológicas. Processos celulares dinâmicos, incluindo movimentos, morfologias, interações, divisão e morte podem ser visualizados no embrião. Desenvolvemos métodos para rotulagem uniforme de estruturas subcelulares e microscopia confocal timelapse do desenvolvimento precoce dos olhos em zebrafish. Este protocolo descreve o método de geração de mRNA tampado para injeção no embrião de zebrafish de 1 célula, montagem de embriões em estágio de vesícula óptica (~12 horas após fertilização pós-fertilização, hpf), e realização de imagens multidimensionais timelapse de morfognese de copo óptico em um microscópio confocal de varredura a laser, de modo que vários conjuntos de dados são adquiridos sequencialmente na mesma sessão de imagem. Tal abordagem produz dados que podem ser usados para uma variedade de propósitos, incluindo rastreamento celular, medições de volume, renderização tridimensional (3D) e visualização. Nossas abordagens nos permitem identificar os mecanismos celulares e moleculares que impulsionam o desenvolvimento de copos ópticos, tanto em condições selvagens quanto genéticas mutantes. Esses métodos podem ser empregados diretamente por outros grupos ou adaptados para visualizar muitos aspectos adicionais do desenvolvimento dos olhos dos zebrafish.
Introdução
O desenvolvimento do olho vertebrado começa com o surgimento, ou evaginação, das vesículas ópticas do potencial neuroepithelium cerebral. As vesículas ópticas passam então por uma série de alterações na forma tecidual, alongando e, em seguida, invaginando para gerar o copo óptico. No copo óptico, a retina neural e o pigmento da retina, ambos derivados do neuroeplílio, envolvam a lente nascente, que é derivada do ectoderme superficial. Todo o processo requer uma série complexa de movimentos celulares e tecidos e sinalização molecular, coordenada entre as populações de neuroefidelo, ectoderme e células mesenquimais. Esses eventos iniciais estabelecem a estrutura básica do olho, e passos posteriores de desenvolvimento ocular, incluindo a formação de íris e córneas, são elaborações sobre organização precoce. Interrupções no desenvolvimento precoce dos olhos e morfogênese estão por trás de inúmeras condições de deficiência visual em humanos, incluindo anophthalmia, microftalmia e coloboma. Desbloquear os mecanismos celulares e moleculares que regem a morfogênese do copo óptico é crucial para entender melhor o desenvolvimento do sistema visual e as condições patológicas que resultam quando esses processos dão errado.
Nossa compreensão do desenvolvimento dos olhos vertebrados e da morfogênese emergiu de uma enorme quantidade de trabalho que abrange estudos histológicos clássicos para abordagens embriológicas e genéticas em uma variedade de organismos modelo, incluindo camundongos, filhotes, sapos e peixes1,2,3,4,5. Enquanto este corpo de trabalho estabeleceu mecanismos moleculares que regulam o desenvolvimento precoce dos olhos, historicamente existe uma má compreensão da morfogênese do olho: o surgimento de sua estrutura 3D. A maior parte desses achados veio de embriões seccionados por imagem em pontos de tempo discretos. Embora isso seja suficiente para fornecer uma visão da morfologia tecidual em 2 dimensões, a morfogênese é um processo dinâmico e 3D. Para determinar como a forma do tecido muda em 3 dimensões ao longo do tempo, como as células individuais se comportam e como esses comportamentos contribuem para mudanças na forma do tecido 3D, diferentes abordagens são necessárias.
Uma solução para resolver essa lacuna significativa no conhecimento é a imagem viva, que permite a observação dinâmica de células e tecidos em tempo real à medida que o órgão toma forma. Infelizmente, isso não é facilmente viável em muitos organismos modelo devido às restrições do desenvolvimento embrionário. Por exemplo, embriões de camundongos e filhotes (desenvolvendo-se no útero ou dentro de uma casca de ovo) não são facilmente acessados, e muitos embriões vivos não são opticamente transparentes, causando dispersão significativa de luz e limitando a profundidade a que as imagens podem ser adquiridas. O zebrafish, com desenvolvimento externo e embriões transparentes, proporcionam uma oportunidade única para realizar imagens vivas de morfogênese ocular6,7,8,9,10,11,12,13,14,15,16,17,18,19. Amplas linhas transgênicas e mutantes também estão disponíveis, bem como ferramentas para gerar novos transgênicos e mutantes20,21,22,23,24. Além disso, a morfogênese do copo óptico ocorre rapidamente em zebrafish, durante um período de 12h (12-24 horas após fertilização, hpf), viabilizando a imagem de todo o processo.
Os esforços de imagem ao vivo têm sido acelerados pela expansão e otimização da família de proteínas fluorescentes, que permitem rotulagem vital geneticamente codificada de estruturas subcelulares, bem como melhorias e inovações nos métodos de microscopia. O protocolo descrito aqui usa microscopia confocal de escaneamento a laser, em vez de outras abordagens atuais para a imagem de embriogênese de zebrafish, incluindo microscopia confocal de disco giratório, microscopia de iluminação de plano seletivo (SPIM e suas variantes), e outros métodos mais especializados de microscopia. Para o olho de zebrafish em desenvolvimento, descobrimos que a microscopia confocal de disco giratório não era suficiente para imagens mais profundas no tecido. Embora o SPIM tenha um tempo de imagem extremamente rápido e esteja se tornando mais amplamente utilizado, lidar com os grandes conjuntos de dados para visualização e análise continua sendo um desafio. Em contraste, a microscopia confocal de varredura a laser é facilmente acessível, especialmente para indivíduos que não têm experiência na montagem de hardware óptico. Esperamos que a ampla disponibilidade de microscopia confocal de escaneamento a laser torne nosso protocolo útil para muitos laboratórios.
Aqui, descrevemos nosso método para capturar conjuntos de dados 4D de morfogênese de copo óptico, usando a rotulagem do embrião para membranas e cromatina, e um microscópio confocal de varredura a laser para aquisição de imagens (esquematizado na Figura 1). As proteínas fluorescentes utilizadas aqui (EGFP-CAAX e H2A. F/Z-mCherry) foi escolhido para fornecer rotulagem de tecido com resolução única celular. Usamos conjuntos de dados gerados com este protocolo para uma variedade de funções de análise e visualização de imagens. Este protocolo pode ser facilmente adaptado se outras estruturas subcelulares forem desejadas. A rotulagem da membrana plasmática foi selecionada para visualização da forma celular: utilizamos EGFP-CAAX, no qual os últimos 21 aminoácidos de H-ras, servindo como uma sequência de sinal de prenylation, são fundidos ao termo C do EGFP13. Outras proteínas fluorescentes direcionadas à membrana plasmática (por exemplo, fusão transmembrana ou myristoylated) provavelmente funcionarão tão bem. Para marcar núcleos, selecionamos H2A. F/Z-mCherry, no qual mCherry é fundido a uma proteína histona13. Isso garante que a divisão celular, incluindo a orientação do fuso mitotístico, seja facilmente visualizada.
Com qualquer abordagem de imagem ao vivo, deve-se considerar trocas entre aumentar a velocidade de imagem e maximizar a relação sinal-ruído, resolução axial e preservação da amostra. Otimizamos nossos métodos para maximizar a qualidade da imagem e o número de embriões que podem ser imagens em uma única execução. Muitas vezes, o objetivo é imaginar a morfogênese do copo óptico em um embrião mutante homozigoso, que pode ser fenotipicamente indistinguível do tipo selvagem no início da morfogênese de copo óptico e a prole de um incross heterozigoso (25% dos embriões são o genótipo desejado). Otimizando e, em seguida, multiplexando a aquisição de imagens, há uma maior probabilidade de capturar um conjunto de dados de um embrião mutante homozigoso.
A resolução temporal, ou com que frequência os dados de volume (Z-stacks) são adquiridos, é um aspecto fundamental da imagem timelapse. Dependendo do propósito para tais conjuntos de dados, existem diferentes requisitos para velocidade. Inicialmente este protocolo foi desenvolvido para rastreamento manual de células 4D. O rastreamento de células individuais dentro de um tecido uniformemente rotulado requer resolução temporal alta o suficiente para fornecer confiança de que qualquer célula está sendo continuamente rastreada ao longo do tempo. Descobrimos que as pilhas de z-stacks de morfogênese de copo óptico de zebrafish devem ser adquiridas pelo menos a cada 3,1 min ao longo de 12 h; aqui, otimizamos nossa aquisição em um microscópio confocal de varredura a laser de modo que podemos adquirir pilhas Z de 4-5 embriões a cada 2,5 min.
Estabelecer o tamanho da etapa Z foi um passo crucial na otimização do protocolo: para renderização e visualização 3D, os dados isotrópicos são ideais, nos quais o tamanho do passo Z é igual à dimensão de pixel XY. Na realidade, é extremamente difícil adquirir tais dados timelapse com amostras ao vivo, dadas restrições com tempo de imagem e fotobleaching. Portanto, determinar o tamanho adequado do passo Z é importante para as necessidades de renderização e visualização do experimento e, especificamente, qual a relação voxel X:Y:Z é necessária para maximizar as informações axiais, mantendo a velocidade e prevenindo o fotobleaching. Para este protocolo, a relação voxel estabelecida foi de 1:1:3.5 (0,6 μm x 0,6 μm x 2,1 μm tamanho passo Z usando um objetivo de imersão de água de longa distância de trabalho de 40x). Ao adquirir uma profundidade Z de 130-140 μm, isso produz dados de volume com resolução temporal adequada e pouco fotobleaching.
Como discutido acima, este protocolo é específico para imagens 4D de morfogênese de copo óptico de zebrafish, usando embriões em toto rotulados para membrana plasmática e cromatina, e um microscópio confocal de varredura a laser. O protocolo abaixo pode ser facilmente adaptado para uma variedade de experimentos e necessidades. Primeiro, em relação às estruturas subcelulares, qualquer estrutura para a qual exista um marcador de célula viva pode ser imagen. Em seguida, embora o foco aqui seja exclusivamente na morfogênese do copo óptico, a imagem timelapse pode ser adaptada para outros estágios de desenvolvimento ocular, por exemplo, neurogênese25,26,27,28,29,30,31,32,33. Para o desenvolvimento posterior da imagem, pode-se precisar considerar a imobilização de embriões (já que a atividade muscular espontânea começa em torno de 24 hpf), pigmentação (que começa a emergir em torno de 24 hpf), tamanho do tecido (o olho cresce significativamente em volume durante a neurogênese) e velocidade de imagem (que deve ser ajustada dependendo da velocidade do processo de interesse). Todas essas considerações podem ser prontamente gerenciadas. O protocolo é bastante flexível; além dos detalhes do protocolo específico aqui, existem princípios gerais que ajudarão os interessados em imagens vivas de outros aspectos do desenvolvimento dos olhos.
Protocolo
Todos os métodos descritos aqui usando zebrafish estão cobertos pelo protocolo animal da Dra. Kristen Kwan, "Mecanismos Celulares e Moleculares de Desenvolvimento de Sistemas Visuais em Zebrafish", e aprovados pelo Comitê Institucional de Cuidados e Uso de Animais (IACUC) da Universidade de Utah.
1. Síntese de RNA tampada
- Gere o modelo de DNA para transcrição in vitro.
- Linearize o modelo de DNA digerindo 10 μg de DNA em 100 μL de volume da mistura de reação. Uma reação típica é montada da seguinte forma: digerir 10 μg de DNA usando 3 μL de enzima e 10 μL de tampão, trazer o volume de reação para 100 μL com água.
NOTA: Um modelo típico para digestão é um vetor pCS2, por exemplo, pCS2-EGFP-CAAX ou pCS2FA-H2A.F/Z-mCherry para a rotulagem de membrana e cromatina descrita aqui. Neste caso, os DNAs plasmídeos são digeridos usando a enzima NotI. Os usuários devem ser cautelosos com a atividade estelar; neste caso, uma enzima de alta fidelidade é recomendada e comercialmente disponível. - Incubar a reação a 37 °C durante a noite para garantir que o DNA seja digerido até a conclusão.
- Limpe o digestor de restrição usando um kit de purificação pcr seguindo o protocolo do fabricante. Elute o DNA com 30 μL ddH2O. Armazene o DNA linearizado a -20 °C e use conforme necessário.
NOTA: A quantidade de DNA digerido e o volume de eluição produzem aproximadamente 0,3 μg/μL; isso é suficiente para ~5 rodadas de transcrição in vitro, cada rodada usando ~2 μg de DNA como modelo.
- Linearize o modelo de DNA digerindo 10 μg de DNA em 100 μL de volume da mistura de reação. Uma reação típica é montada da seguinte forma: digerir 10 μg de DNA usando 3 μL de enzima e 10 μL de tampão, trazer o volume de reação para 100 μL com água.
- RNA transcrito in vitro usando um kit de transcrição in vitro. Para modelos pCS2 (como descrito aqui), use um kit SP6.
- Monte a reação de transcrição in vitro, da seguinte forma: Digerir 2 μg de modelo de DNA ou até 6 μL com 2 μL de tampão de reação de 10x, 10 μL de 2x Mistura de Ribonucleotídeos e 2 μL de 10x Enzima Mix.
- Incubar a 37 °C por 2-4 h ou mais (tempos mais longos levarão a maior rendimento). Se desejar, 1 μL de Enzima Mix pode ser suplementado na metade do período de incubação.
- Digerir o modelo de DNA adicionando 1 μL de DNase livre de RNase e incubando por 15 min a 37 °C.
- Purifique o RNA tampado usando um kit de purificação de RNA seguindo o protocolo do fabricante (ver Tabela de Materiais). Elute com 100 μL de H2O sem RNase.
NOTA: A adição de β-mercaptoetanol não é necessária para esta aplicação. Embora o método descrito neste protocolo seja simples, reagentes alternativos também podem ser usados para purificar o RNA. - Precipitar o RNA.
- Adicione 10 μL de NaOAc 3 M RNase sem RNase e 2,5 volumes (~275 μL) de RNase-free 100% EtOH sem gelo ao RNA elucido.
- Incubar a reação a -20 °C por 15-30 min. Gire por 15 min em alta velocidade a 4 °C para pelotar o RNA.
NOTA: A pelota deve ser visível contra a parede do tubo. É útil notar como o tubo está alinhado na centrífuga nesta etapa, a fim de prever onde a pelota deve estar no final do giro. - Remova o EtOH cuidadosamente com uma seringa, tomando cuidado para evitar o aquecimento, a secagem excessiva ou a desalojação da pelota. Resuspend a pelota em 20 μL de RNase-free H2O.
- Verifique o RNA para garantir que a síntese seja bem sucedida. Use 1 μL para avaliar a concentração em um espectotômetro e execute 0,5 μL em um gel de 1% de agarose para verificar se há uma ou duas bandas discretas, em vez de uma mancha de baixo peso molecular. O rendimento da reação de transcrição in vitro deve ser de ~1 μg/μL.
- Alíquota e armazenamento RNA a -20 °C ou -80 °C. O RNA não é diluído até imediatamente antes das injeções.
2. Microinjeção de embriões de zebrafish de 1 célula
NOTA: Injete 200-300 pg por RNA para obter expressão onipresente de cromatina e membranas celulares ao longo da morfogênese de copo óptico. Prepare 5-10 μL da diluição do RNA e carregue 2,5-5 μL/agulha; plano para extra no caso de a agulha quebrar.
- Preparação antecipada para injeções.
- Prepare um prato de molde de injeção para facilitar o alinhamento e orientação de embriões de 1 célula para microinjeção. Derreta 2% de agarose em E3 (meio de embrião padrão) e despeje em uma placa de Petri. Flutue cuidadosamente o molde de injeção (ver Tabela de Materiais) na parte superior da agarose quente para gerar a impressão dos cochos na agarose à medida que se solidifica. Uma vez que a agarose solidifice, remova o molde de injeção.
NOTA: Os pratos de molde de injeção podem ser usados por vários meses; cobrir em E3 e armazenar a 4 °C quando não estiver em uso. - Puxe agulhas de microinjeção. Puxe capilares de vidro para um taper longo para fazer agulhas de microinjeção usando uma máquina de puxar agulhas. Programe a máquina específica para o tipo de capilares utilizados. Para 1,0 x 0,78 mm borossilicate capilares use o seguinte programa: calor = 546 °C, puxar = 130, velocidade = 70, tempo = 90 (Figura 2F). Guarde as agulhas em uma placa de Petri e fixe-as com argila de modelagem.
NOTA: A receita de puxar vai variar, dependendo da máquina e do filamento, e novos filamentos devem ser sempre calibrados. Cada rodada de puxar produz duas agulhas(Figura 2G); fazer isso várias vezes para produzir agulhas suficientes em caso de quebras acidentais e experimentos futuros. - Diluir o RNA tampado para injeção. Diluir tanto a concentração de EGFP-CAAX quanto h2A-mCherry RNAs para 200-300 ng/μL de concentração usando H2O sem RNase e 1 μL de vermelho fenol em um volume total de 5 μL (concentração final de vermelho fenol é de 0,1%). Gire a mistura e gire brevemente para baixo para coletar o volume total. Mantenha o RNA diluído no gelo antes de injetar.
- Prepare um prato de molde de injeção para facilitar o alinhamento e orientação de embriões de 1 célula para microinjeção. Derreta 2% de agarose em E3 (meio de embrião padrão) e despeje em uma placa de Petri. Flutue cuidadosamente o molde de injeção (ver Tabela de Materiais) na parte superior da agarose quente para gerar a impressão dos cochos na agarose à medida que se solidifica. Uma vez que a agarose solidifice, remova o molde de injeção.
- Injeção de embriões de 1 células
- Uma vez que os peixes comecem a procriar, permita ~15-20 min para garantir que os ovos se tornem fertilizados. Durante este tempo, recarregue a agulha com 2,5-5 μL da diluição do RNA usando uma ponta microcarregadora P10 e P10.
- Utilizando um picoinjector, calibrar a agulha usando um tique-trópria (um slide de calibração de micrômetro também é suficiente) para medir o volume da gotícula (volume de uma esfera = (4/3) * pi * raio^3). Ajuste o tempo de injeção e a pressão de modo que o volume de injeção seja de 1 nL(Figura 2I).
- Com uma pipeta de rolo/transferência, carregue cuidadosamente os ovos no molde de injeção(Figura 2J). Se útil, use fórceps para enrolar embriões de tal forma que a célula única seja visível, antes ou durante a injeção. É a preferência do usuário usar um micromanipulador.
- Injete embriões no estágio de 1 célula, visando a célula e não a gema(Figura 2M). Isso garantirá a rotulagem uniforme do embrião em desenvolvimento.
- Elevar os embriões para o estágio desejado (antes de 12 cvf, de acordo com o estágio padrão34). Embriões injetados terão um desenvolvimento atrasado, por isso elevá-los a uma temperatura ligeiramente mais alta (29.0-29.5 °C) para compensar o tempo. Durante a tarde, verifique os embriões e remova os que estão mortos para preservar a saúde da embreagem.
3. Montagem de embriões de zebrafish estágio vesícula óptica para imagens timelapse
- Antes de montar, prepare 1,6% de baixa derretimento na E3. Se planejar vários experimentos de imagem, prepare ~20 mL de agarose de baixo derretimento e armazene à temperatura ambiente. No dia da montagem do embrião, derreta-o para gerar uma alíquota fresca (1-5 mL) em um tubo que pode ser colocado em um bloco de calor de 42 °C antes da montagem.
- Embriões de tela para injeção bem sucedida e brilho geral de fluorescência usando um microscópio de fluorescência antes da montagem. Uma amostra ideal terá fluorescência forte de EGFP e mCherry e estará no estágio correto de desenvolvimento(Figura 3B - B'').
- Embriões de tela usando um microscópio de fluorescência. Selecione embriões que expressem fortemente fluorescência de EGFP e mCherry para montagem.
- Selecione embriões de 11 hpf. Conte somites para encenar adequadamente os embriões34; a 11 cv deve haver 3 somites, e por 12 hpf há 6 somites.
NOTA: Montagem de embriões antes de 12 cvf (a 11 hpf), as amostras serão adequadamente encenadas a 12 cvf quando o timelapse começar.
- Embriões descorrionatos antes da montagem. Faça isso manualmente com fórceps finos ou usando quimicamente pronase (2 mg/mL). Com embriões tão jovens, é essencial que a descorção seja realizada dentro de um prato revestido de ágar e que os embriões não entrem em contato com a interface ar-água.
- Usando uma pipeta de rolo de vidro, suge um embrião e ejete o máximo de E3 possível, de tal forma que o embrião fique na ponta da pipeta pasteur de vidro.
- Jogue o embrião no tubo de agarose do bloco de calor. Deixe-o afundar na agarose por alguns segundos, depois sugou algumas agaroses primeiro e depois o embrião, certificando-se de que o embrião permaneça na ponta da pipeta(Figura 3C - C').
- Coloque um prato de fundo de vidro para imagens sob um escopo de dissecação. Ejete o embrião e se agarose em uma gota de agarose no prato de fundo de vidro. Use fórceps para muito rapidamente orientar, de tal forma que o embrião seja dorsal-down (topo da cabeça no fundo do vidro). Permita que a gota de agarose endureça(Figura 3D - D'). Este passo deve ser feito rapidamente, mas com cuidado, pois os embriões nesta fase são frágeis. Embriões danificados não sobreviverão ao processo de imagem timelapse.
NOTA: É importante orientar todos os embriões de forma consistente para este experimento. Ao realizar o timelapse, os embriões devem caber dentro do campo de visão atribuído, mantendo espaço adicional para que a vesícula óptica cresça. Uma orientação ideal é alinhar o eixo anterior-posterior de todos os embriões "verticalmente" ou ao longo das 12 e 6 horas em uma face do relógio(Figura 3G). - Monte entre 10-12 embriões, que é mais do que o dobro que será imageado, para que as melhores amostras possam ser selecionadas uma vez avaliadas no confocal (Figura 3E - E'). As amostras serão avaliadas para idade, uniformidade da rotulagem fluorescente e precisão da orientação de montagem.
- Após a montagem dos embriões, pipeta mais agarose para cobrir completamente o fundo do prato, envolvendo assim todas as gotículas de agarose separadas em um único grande disco de agarose(Figura 3F - F'). Uma quantidade suficiente de agarose garantirá que as gotículas individuais de embriões ou todo o disco não levantem da parte inferior do prato e flutuem para fora da vista.
- Uma vez endurecido, sobreponha a agarose com E3 para manter as amostras hidratadas durante a duração do experimento de imagem timelapse (uma preocupação dependendo da umidade do edifício e do ambiente).
NOTA: O tricaine não é necessário para esses experimentos específicos de imagem timelapse, pois esses embriões são suficientemente jovens; no entanto, se desejar, e se trabalhar com estágios mais antigos de embriões, o tricaine pode ser adicionado na própria agarose e sobreposto na mídia. - No papel, desenhe um mapa dos embriões para facilitar a referência durante a configuração do timelapse. Isso é crucial para associar mais tarde a posição com o genótipo de cada amostra.
4. Timelapse confocal de posição múltipla com um microscópio confocal de varredura a laser
NOTA: Este protocolo de imagem timelapse foi projetado para ser usado com um microscópio confocal de varredura a laser equipado com o software do fabricante (ver Tabela de Materiais). Este sistema é equipado com um dispositivo de estágio Z piezo que permite a aquisição rápida de pilhas Z. As linhas laser usadas neste protocolo são um laser de íon argon de 488 nm e um laser DPSS 561 nm. O laser de 561 nm é adequado para a imagem do fluorophore mCherry: ele está perto o suficiente do pico de excitação mCherry (587 nm) e bem alimentado.
- Configure o confocal.
- Ligue o confocal e faça login no computador de mesa.
- Instale a inserção de estágio Z piezo (Figura 4B),e inicie o software de aquisição.
- No software dentro do modo Aquisição, ligue os lasers 488 e 561 usando o menu suspenso(Figura 4F). Os lasers levam vários minutos para aquecer, por isso este passo é realizado cedo para economizar tempo.
NOTA: Os parâmetros de aquisição são os seguintes: Verifique as caixas Z-stack, Time Seriese Position. Definir tamanho do quadro para 512 (X) x 384 (Y), Velocidade de varredura para 9, Modo de Varredura para bidirecional, Zoom para 0,7, Pinhole para 60,2 (1,63 Unidades Arejadas ~= seção de 1,6 μm) e intervalo Z para 2,1 μm. Isso renderá um tamanho de imagem de 303,6 μm x 227,7 μm, e tamanho de pixel de 0,59 μm. Os lasers 488 e 561 devem ser verificados e atribuídos à mesma faixa; a faixa de detecção do EGFP é de 494-545 nm e a faixa de detecção mCherry é 598-679(Figura 4F). - Reveste liberalmente a parte inferior do prato de vidro com meio de imersão que corresponda ao índice de refração da água, tomando cuidado para evitar bolhas de ar(Figura 4C). Selecione o objetivo de água 40x e aplique uma pequena gota de meio de imersão ao objetivo(Figura 4D). Fixar o prato com fundo de vidro na inserção do palco, aplique E3 para manter os embriões úmidos durante a noite e, em seguida, use argila modeladora para selar a tampa plástica sobre o prato(Figura 4E). Levante o objetivo de fazer contato com o prato.
- Tela amostras e prepare-se para timelapse.
- Mude da guia Aquisição para a guia Localizar e clique no símbolo da lâmpada para acender a luz transmitida. Com a luz transmitida, localize a primeira amostra com o joystick: por olho, primeiro mova a amostra para o feixe de luz, depois use as oculares para o centro e foque em uma vesícula óptica.
- Retorne à guia Aquisição. Certifique-se de que as caixas Z-stack, Série tempoe posições sejam verificadas, e os lasers sejam aquecidos para uso.
- Defina o tamanho do quadro para 512 (X) x 384 (Y), escaneie velocidade para 9, modo de varredura para bidirecional e defina Zoom para 0,7.
- Sob a posição Canais, comece atribuindo os lasers 488 e 561 à Power 2.0 e ao Gain 550 (isso pode ser ajustado posteriormente). Coloque o orifício em 60,2 (1,63 Unidades Arejas ~= seção de 1,6 μm).
- Comece a digitalizar usando o botão Contínuo. Com o botão de foco fino, localize a amostra no eixo Z e posicione o eixo X-Y na estrutura. Pare de digitalizar.
- Sob o título Posições, clique em Adicionar para salvar as informações do XYZ na primeira amostra (Figura 4F). Faça isso apenas para que as amostras sejam cronometradas. Selecione amostras para sua fluorescência forte e montagem ideal.
- Mude para a guia Localizar, mova-se para a próxima amostra e repita as etapas 4.2.5-4.2.6, sendo seletiva sobre a escolha de amostras.
- Finalize as posições da amostra, atribua o intervalo z e, em seguida, inicie o timelapse. Uma vez selecionadas todas as amostras (é possível visualizar 4-5 amostras dentro do intervalo de tempo total de 2,5 minutos) e as posições são salvas, passar por cada amostra e ajustar a posição XYZ dentro do quadro de visualização.
- Destaque a primeira posição e clique em Mover para. Durante a varredura contínua, alinhe o vesículo óptico dentro do quadro. Deixe amplo espaço nas regiões anterior e distal em relação à vesícula óptica e ao cérebro.
- Em seguida, atribua a primeira e última fatias Z selecionando Set First e Set Last enquanto se move através da direção Z com o botão de foco fino. Mantenha um número total de fatias de ~63, pois isso acomodará o crescimento do copo óptico. Forneça espaço extra no lado ventral do vesículo óptico para permitir espaço para crescimento.
- Uma vez definidas as primeiras e últimas fatias Z, clique no botão C para mover-se para o centro da pilha Z. Ajuste a potência do laser e ganho para ambos os lasers; se possível, mantenha a potência laser abaixo de 5 e use um ganho maior, se necessário. Pare de digitalizar. Atualize as informações de posição clicando em Atualizar.
- Mova-se para a próxima posição clicando na Posição 2. Repetir as etapas 4.3.1-4.3.3 para cada posição atribuída. A potência e o ganho do laser devem ser definidos de tal forma que todas as amostras estejam suficientemente iluminadas.
- Uma vez finalizadas as informações de posição e as configurações do laser, atribua as configurações da Série Time. Sob a posição Série time, atribua intervalo de tempo a 2,5 min e ciclos a 300(Figura 4F). O número de ciclos é calculado de tal forma que o timelapse prossiga após o estágio de 24 hpf, quando o desenvolvimento do copo óptico estiver completo.
- Para iniciar o timelapse, clique em Iniciar. Monitore o primeiro ciclo completo: use um temporizador para garantir que um ciclo completo (imagem de todas as posições) seja inferior a 2,5 min e verifique se cada posição parece correta. O microscópio funciona de forma independente durante a noite e não precisa ser monitorado.
NOTA: Embora a sala confocal esteja controlada pela temperatura, a temperatura ambiente é de 20-25 °C. Com o equipamento funcionando e os lasers escaneando, a temperatura no palco parece estar mais próxima de 28,5 °C, uma vez que o desenvolvimento embrionário prossegue nesta taxa esperada, como avaliado visualmente com marcos morfológicos. - Com essas configurações, o timelapse durará 12,5 h. Salve o arquivo assim que o timelapse estiver completo. Ele será grande (~40 GB) e pode ser separado em posições individuais depois de ser salvo usando o software de aquisição.
Resultados
Injeção de RNAs fluorescentes permite rotulagem subcelular
As RNAs que rotulam a membrana plasmática da célula e as histonas são injetadas a fim de capturar as morfologias e movimentos individuais das células que estão por trás da morfogênese do copo óptico. A Figura 2 demonstra cada etapa do processo de microinjeção de embriões de zebrafish no estágio de 1 célula. Resumidamente, os zebrafish são acasalados(Figura 2E),e os embriões são coletados e carregados no molde de injeção(Figura 2J). Uma agulha de microinjeção é reabastecida(Figura 2H), e os embriões são injetados diretamente na célula única (Figura 2K-M), que é necessária para obter rotulagem uniforme (Figura 2M, Figura 4I-I''', Filme1). Além da rotulagem uniforme, observa-se fluorescência robusta e o desenvolvimento segue sem perturbação (Figura 3B-B'').
Montagem eficaz é essencial para a imagem timelapse OCM de 12a24 hpf
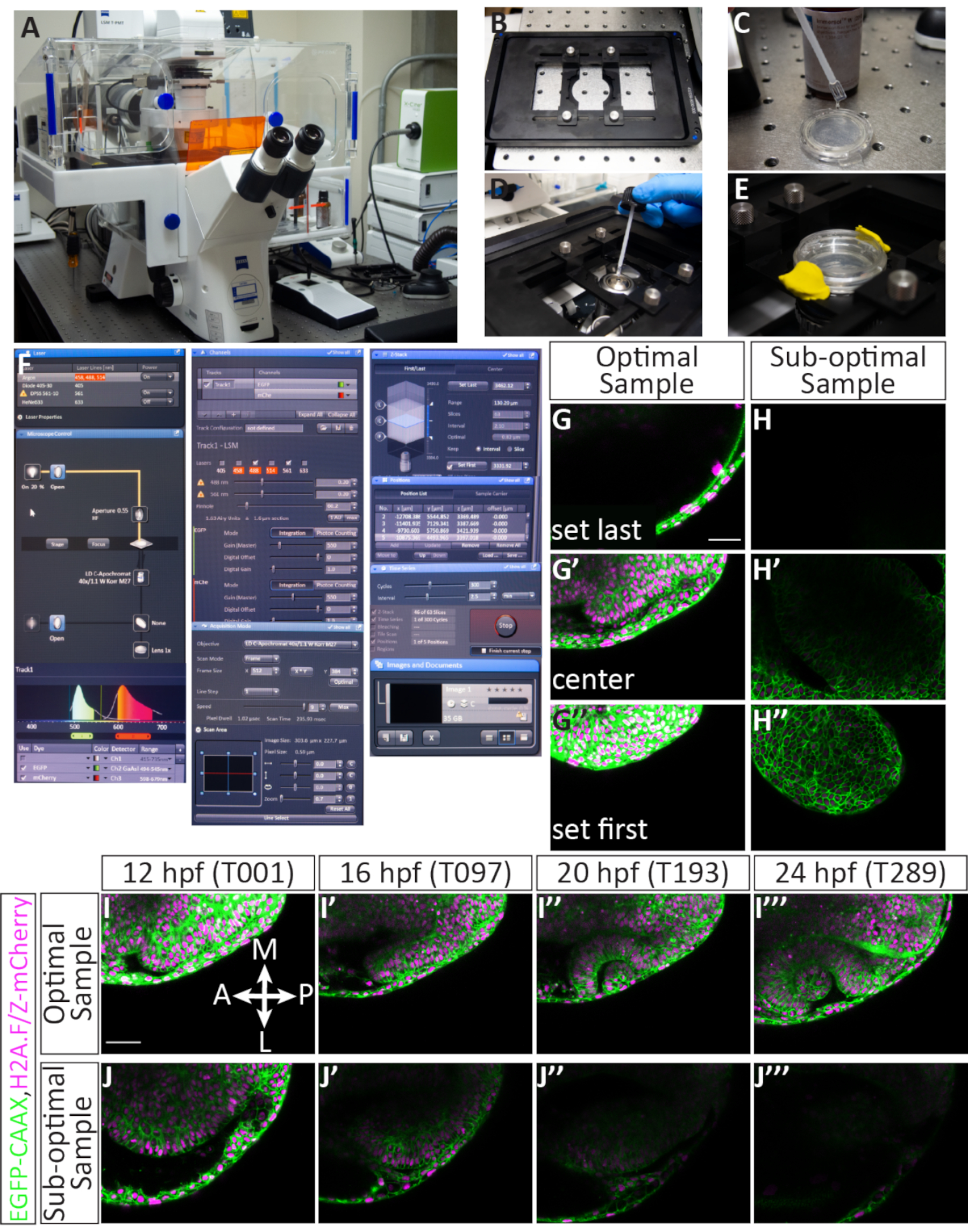
Para a morfogênese da xícara óptica, que ocorre durante um longo período de tempo, é fundamental selecionar embriões ideais e posicionar adequadamente cada amostra durante a incorporação. A Figura 3 mostra uma conta detalhada para a montagem bem sucedida de embriões de 11 hpf. Os embriões injetados são primeiro examinados para fluorescência suficiente (Figura 3B',B'',F'',F''''). Embriões individuais são imersos em agarose(Figura 3C,C)e montados dorsalmente em uma gota individual de agarose(Figura 3D,D). Todas as amostras são montadas em um disco coletivo de agarose(Figura 3E-F). Quando os embriões forem encenados corretamente, suficientemente fluorescentes e adequadamente montados(Figura 3G),as amostras permanecerão no quadro de imagem durante o timelapse permitindo que todo o órgão seja imageado(Figura 4G-G'',I-I''', Filme1). A não realização de qualquer uma dessas etapas resultará em um embrião montado que não é uma amostra ideal para imagens timelapse. A menor variação no grau de rotação terá um efeito dramático na direção do crescimento do embrião durante o timelapse. Rotações sub-ideais são mostradas na Figura 3, incluindo um embrião que é sobre-girado(Figura 3I), que resultará em um timelapse mais posterior, e um embrião que é sub-girado(Figura 3J) no qual apenas o tecido mais anterior pode ser observado. Além da rotação adequada durante a montagem, amostras que são montadas com relação à orientação do quadro são mais propensas a produzir um timelapse bem sucedido (Figura 4G-G'', I-I''', Filme 1). As amostras ideais são aquelas que são orientadas verticalmente no prato, com o eixo anterior-posterior em linha com 12 e 6 horas em uma face de relógio(Figura 3G). Amostras sub-ideais são aquelas que não são orientadas neste eixo, como aquelas que são horizontais ou diagonais(Figura 3H). Essas amostras crescerão fora do quadro de imagem à medida que o timelapse prossegue e não podem ser usadas para análise suplementar(Figura 4H-H'',J-J''').
Aquisição de um conjunto de dados timelapse adequado para análise futura
Embriões injetados, criados e montados com precisão farão amostras de imagem confocal bem sucedidas. A Figura 4 mostra uma amostra ideal para timelapse: há até fluorescência, e a amostra é de 12 cvf. A pilha z para imagem é atribuída de tal forma que a primeira fatia é apenas dorsal para o vesículo óptico, enquanto a última fatia deixa várias fatias além do limite ventral do vesículo óptico de 12 hpf(Figura 4G-G''). Esse viés em direção ao lado ventral proporciona espaço excessivo para o crescimento do tecido na direção ventral. Esses fatores, quando atendidos, resultam em um timelapse ideal (Figura 4I-I''', Filme 1). Uma amostra sub-ideal tem mosaico ou fluorescência fraca, já desenvolveu além de 12 hpf (quando a morfogênese de copo óptico está em andamento), ou está mal posicionada na pilha Z ou no quadro XY(Figura 4H-H''). Quando esses critérios não forem atendidos, o timelapse não capturará mais todo o volume 3D do tecido à medida que se desenvolve e não pode ser usado para análise suplementar(Figura 4J-J''').

Figura 1: Fluxo de trabalho de imagens de timelapse 4D de morfogênese de copo óptico de zebrafish. (A) Para rotular fluorescentemente estruturas subcelulares de interesse, sintetize os mRNAs tampados apropriados. (B) Microinjetar mRNA(s) em embriões de zebrafish no estágio de 1 célula. (C) Os embriões são montados dorscamente, ou cabeça para baixo para um microscópio confocal invertido, no estágio vesícula óptico, ~11 h pós fertilização ou 3 estágios de somite. (D) Embriões múltiplos podem ser imagens sequencialmente durante uma única sessão de imagem timelapse. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 2: Um guia visual para microinjeção de embriões de zebrafish de 1 célula. (A) Itens a serem adicionados em um kit de injeção (sentido horário): caixa de tamanho médio que pode segurar todo o kit com luvas de nitrilo descartáveis; um prato contendo agulhas de microinjeção; fórceps; uma alíquota de óleo mineral e corte quadrados de parafilm; dicas de microcarregamento; RNA diluído no gelo; molde de injeção de agarose; marcador; pipeta rolo/transferência; e pipeta P10. (B) Configuração de microinjeção: um microscópio dissecando ao lado de um pico-injetor conectado a uma fonte externa de gás comprimido. Um pedal de pé e um micromanipulador são ligados ao pico-injetor. O micromanipulador é equipado com um suporte de agulha e posicionado no lugar acima do estágio do microscópio. (C) Um molde de injeção de ágarose contendo seis linhas de ranhuras com um ângulo de 90 graus de um lado e um ângulo de 45 graus do outro lado. (D) O painel frontal do pico-injetor. O visor de pressão lê 7,8 PSI; isso pode ser ajustado com o botão rotulado Pinject. Abaixo, há botões para disparar manualmente a pressão para injetar, para alterar o tempo de injeção, ou para limpar, encher ou segurar o líquido na agulha. O tempo de injeção é definido para 08 (equivalente a 8 x 10 msec). Uma mangueira de saída é conectada a Pout e é anexada à agulha de injeção. O interruptor de origem do medidor de pressão é ajustado parap injetar enquanto define a pressão de injeção, e pode ser comutado parap equilíbrio para ajustar a pressão basal aplicada à agulha quando as injeções não estão sendo feitas. O regulador deequilíbrio P está ao lado do reguladorde injeção P. O botão de alimentação está aceso para indicar que a máquina está ligada. (E) Os zebrafish adultos são acasalados em pequenos tanques de reprodução que contêm um tanque aninhado com um fundo de malha que separa peixes adultos de ovos fertilizados e permite uma fácil coleta. O nível de água é reduzido para imitar condições de águas rasas e os divisores de tanques são removidos para iniciar o comportamento de acasalamento. (F) O painel frontal do puxador de micropipette (agulha): contém um display que mostra as condições específicas usadas para puxar agulhas. A configuração de pressão é P = 500, seguida pela última data e hora em que o programa foi editado, o número do programa, HEAT = 546, PULL = 130, VEL = 70 e TIME = 90. (G) Um capilar de vidro é inserido no puxador de micropipette (agulha) e fixado no lugar. Cada tira programada resulta em duas agulhas longas e afiladas (pontas de flecha vermelha). (H) A diluição do RNA é carregada em uma agulha; o corante vermelho fenol permite uma visualização fácil da solução. (I) Depois de quebrar a ponta da agulha, o bolus de RNA é medido usando uma retílação da ocular no escopo de dissecação. Para este estereótipo, a ampliação total de 30x 6 hash marks é igual a 1 nL de volume. (J) Os ovos fertilizados são carregados no molde de injeção de agarose e distribuídos ao longo dos cochos do molde usando uma pipeta de rolo. (K) O molde de injeção contendo embriões de estágio unicelulares é posicionado sob o microscópio e os embriões são injetados sequencialmente. (L) A agulha de injeção é inserida em cada embrião visando a única célula. (M) Uma vez na célula, o pedal do pé é usado para desencadear uma quantidade regulada de pressão e a liberação de 1 nL de RNA na célula do embrião (como visualizado pelo vermelho fenol). Barras de escala: I = 0,1 mm; L, M = 0,5 mm. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 3: Um guia visual para montagem de embriões para imagem. (A) Configuração de montagem (da esquerda para a direita): um bloco de calor programado a 42 °C, tubos de aquecimento de baixa derretimento agarose; uma pipeta de rolo; um par de fórceps; um prato revestido de ágarose contendo embriões descorrioados; e um estereómico dissetante conectado a uma lâmpada fluorescente de halide metálico. (B,B',B'') Um embrião de 11 hpf injetado com MRNA EGFP-CAAX e mCherry-H2A mRNA, mostrado sob brightfield, fluorescência GFP e fluorescência mCherry, respectivamente. (C,C') Uma pipeta de pasteur de vidro contendo agarose e um embrião sentado na ponta; C' é uma visão ampliada. (D,D') Um embrião montado em uma gota de baixo derretimento surgiu em um prato de fundo de vidro, visto tanto do estágio do microscópio quanto através das oculares do microscópio. (E,E') 12 embriões montados em gotículas individuais de baixo derretimento surgiram em um prato com fundo de vidro, visto tanto do estágio do microscópio quanto através das oculares do microscópio. (F,F',F'',F''') Todos os embriões montados são sobrepostos com uma camada completa de agarose para encher a parte inferior do prato, visto tanto do estágio do microscópio quanto das oculares do microscópio mostrados em campo brilhante; F'' Fluorescência GFP; e f''' mCherry fluorescence. (G) Uma amostra ótima de 12 hpf é montada dormente e orientada verticalmente em linha com 12 e 6 horas com ambas as vesículas ópticas no mesmo plano. (H) Uma amostra sub-ideal: embora as vesículas dorsais e ópticas estejam em plano entre si, esta amostra é orientada em um eixo diagonal e crescerá fora do tamanho do quadro à medida que o desenvolvimento prossegue. (I) Uma amostra sub-ideal: esta amostra é sobre-girada e não um suporte dorsal, como resultado, a porção anterior da vesícula óptica não será capturada no timelapse. (J) Uma amostra sub-ideal: esta amostra é sub-girada e não um suporte dorsal, como resultado a porção posterior da vesícula óptica não será capturada no timelapse. Linhas pontilhadas indicam cada vesícula óptica. Barras de escala: B = 0,1 mm; D' = 1,5 mm; E', F' = 2,5 mm; G = 0,1 mm. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 4: Configuração de um timelapse de várias posições e resultados potenciais. (Um) Microscópio confocal de varredura a laser. (B) Inserção de estágio Z piezo. (C) A parte inferior de um prato de fundo de vidro contendo embriões embutidos em agarose é revestida em meio de imersão que corresponde ao índice de refração da água. (D) Uma gota de meio de imersão é colocada no objetivo de 40x W (longa distância de trabalho). (E) O prato é mantido no lugar com a inserção do palco, o E3 é adicionado em cima da camada agarose, e a tampa é fixada com argila de modelagem. (F) Configurações de software de aquisição: As linhas laser desejadas são ligadas a partir do menu suspenso. Para embriões injetados com RNA EGFP-CAAX e H2A-mCherry RNA, os lasers Argon e DPSS 561-10 são ligados. O destaque vermelho indica que o laser de Argon está aquecendo. Para localizar a amostra sobre o objetivo, a luz transmitida é ligada através do painel de controle do microscópio. Tanto o EGFP quanto o mCherry são designados para a Faixa 1 (para imagens simultâneas) e o alcance de cada detector é definido. Aqui, a faixa EGFP está definida para 494-545 nm, e a gama mCherry está definida para 598-679 nm. Sob o Canais menu, energia laser pode ser definida. Depois que os lasers se aquecerem, a potência pode ser aumentada até 5.0, e o ganho (mestre) pode ser ajustado. O orifício é fixado em 60,2, o que equivale a 1,63 Unidades Aéreas ou seção de 1,6 μm. Sob o Aquisição menu, o tamanho do quadro é definido para 512 x 384, a velocidade de varredura é 9.0, a média é bidirecional, e o zoom é definido para 0,7. Sob o Z-Stack menu, a primeira e última posição no eixo Z são definidas enquanto o confocal está escaneando. O intervalo (tamanho da etapa) é definido para 2,1 μm. Ao escolher a primeira e a última posição Z, um número padrão de 63 fatias é geralmente mantido; isso acomoda o crescimento geral do olho. A opção "use piezo" é selecionada. Sob o Posições menu, cada posição selecionada é listada, incluindo um número atribuído e posição X, Y e Z. Existem botões adicionais para controlar o número de embriões (posições separadas) à imagem, incluindo adicionar, atualizar ou remover. Sob o Série temporal menu, o ciclo é definido para 300 (o número de pilhas Z que serão adquiridas) e o intervalo (etapa de tempo) é definido para 2,5 min. Uma vez que as configurações são finalizadas, a aquisição do timelapse é iniciada pressionando o início. O software mostrará a fatia da pilha Z, o ciclo e a posição que está sendo digitalizada no momento. Quando o timelapse estiver concluído, o arquivo é salvo clicando no ícone disquete no ícone do disquete no Imagens e Documentos painel. (G-J) Membranas celulares (green) são rotulados com EGFP-CAAX e núcleos celulares (cromatina, magenta) são rotulados com RNAs H2A-mCherry. (G,G', G'') Um exemplo de definir a primeira e última fatia Z para uma amostra bem montada. A primeira fatia Z (no lado dorsal) vem às custas do ectoderm dorsal, enquanto a última fatia Z (no lado ventral) está bem abaixo do vesículo óptico, para acomodar seu crescimento na direção ventral. (H,H', H'') Um exemplo de definir a primeira e última fatia Z de uma amostra abaixo do ideal. A amostra tem fluorescência mCherry fraca e é montada na diagonal, de tal forma que é improvável que o copo óptico em desenvolvimento permaneça no quadro durante a duração do timelapse. (Eu, eu', eu',eu'', eu''') Um exemplo de um timelapse de uma amostra ideal. Imagens de fatia única do meio da pilha Z são retiradas de todo o volume de 63 fatias em 4 pontos de tempo (valor T). Br, cérebro; NR, retina neural; RPE, epitélio pigmentado da retina; Le, lente. (J,J',J'',J''') Um exemplo de um timelapse de uma amostra subótima. Neste caso, a amostra saiu do plano Z e apenas uma parte oblíqua do copo óptico anterior foi capturada. Barras de escala: G, I = 50 μm. Clique aqui para ver uma versão maior desta figura.
{kind=link}
Filme 1: Timelapse de morfogênese de copo óptico em um embrião do tipo selvagem ideal. Uma visão dorsal de um embrião tipo selvagem que é rotulado com EGFP-CAAX (membranas plasmáticas, verdes) e H2A. F/Z-mCherry (cromatina, magenta). O timelapse é de ~12-24 hpf e contém uma única seção confocal de um conjunto de dados 4D inteiro. O intervalo de tempo é de 2,5 min entre as pilhas z e é mostrado a 22,5 quadros por segundo. Clique aqui para baixar este filme.
Discussão
Aqui, descrevemos um protocolo para rotulagem em toto e imagem de timelapse 4D de morfogênese de copo óptico. Passamos pelo processo de geração de proteínas fluorescentes de codificação de RNA tampadas para marcar diferentes compartimentos subcelulares; injetáveis embriões de zebrafish 1 células; incorporação de embriões de 11 hpf em agarose para imagens multiplexais; e aquisição de conjuntos de dados 4D de múltiplos embriões durante a duração da morfogênese de copo óptico (12-24 hpf).
Uma miríade de perguntas pode ser respondida com esses conjuntos de dados informativos densos. Os dados 4D podem ser visualizados e analisados quantitativamente de várias maneiras. Embora fora do escopo deste protocolo, incluímos aqui alguns de nossos objetivos e aplicações padrão como um exemplo dos tipos de coisas que podem ser realizadas. É claro que métodos quantitativos de análise de imagens estão sendo constantemente desenvolvidos, e ferramentas comercialmente disponíveis e personalizadas podem ser usadas. Se não se já usou tais métodos antes, deve-se estar preparado para trabalhar através de alguma otimização para garantir que os conjuntos de dados adquiridos sejam adequados para a abordagem de análise quantitativa de escolha.
Visualizar e avaliar quantitativamente os conjuntos de dados 4D pode ser desafiador, devido ao tamanho dos arquivos. O software de aquisição pode ser usado para separar os conjuntos de dados em embriões individuais, e imageJ/Fiji pode ser usado para converter arquivos confocal de formatos comerciais para pilhas de tif mais padrão, nas quais cada ponto de tempo é salvo como um arquivo separado. Isso diminuirá o tamanho dos arquivos e padronizará os formatos de arquivo. Seções ópticas individuais de cada ponto de tempo podem ser montadas como um timelapse 2D (XY) usando ImageJ/Fiji, permitindo visualização rápida 2D e avaliação dos dados. O filme 1 é um exemplo precisamente disso: uma única seção óptica ao longo do tempo montada como um timelapse da amostra ideal mostrada na Figura 4I–I'''. A partir daí, para visualização 3D e 4D, geralmente usamos FluoRender35,36, que está livremente disponível, mas tem requisitos específicos de placas gráficas. Usando o FluoRender, pode-se girar dados renderizados em 3D em qualquer eixo, visualizar o conjunto de dados em 4D ao longo do tempo, gerar recortes em qualquer plano e salvar as rotações e visualizações como um filme ou série de imagens tif.
Em termos de análise quantitativa, há inúmeras perguntas a serem respondidas. Desenvolvemos software internamente para ajudar nossos objetivos específicos de entender os comportamentos celulares subjacentes à morfogênese do copo óptico. Nosso programa, LongTracker, usa o sinal nuclear como proxy para posição para rastrear células13. Com esses dados, podemos determinar quando, onde e como as células se movem; quão rápido e quão longe as células se movem; e com que frequência as células se dividem. Além do nosso próprio software, existem várias opções comercialmente disponíveis e personalizadas para rastreamento de células 4D. Também desenvolvemos o programa LongAxis para realizar a segmentação celular e quantificar a forma e organização celular dentro da retina neural37. Os conjuntos de dados inseridos no LongAxis, no entanto, são pilhas Z únicas, tomadas em alta resolução. Um desafio persistente tem gerado conjuntos de dados timelapse com resolução suficiente para que as células possam ser segmentadas com confiança e sua morfologia extrapolada. Uma alternativa é a rotulagem de mosaico (esparsa) utilizando um fluoróforo fotoconvertível como Kaede para visualização direta da forma celular, como nós e outros realizamos no olho em desenvolvimento8,11,13. Isso simplifica o problema de segmentação celular, e a forma e o tamanho da célula podem ser facilmente quantificados através da renderização 3D em FluoRender.
Cada etapa deste protocolo foi otimizada especificamente para nossos propósitos. A especificidade deste protocolo resulta em várias limitações: o protocolo, como escrito, não é otimizado para a imagem de outros aspectos do desenvolvimento dos olhos de zebrafish (como neurogênese da retina), outras estruturas oculares, outros estágios de desenvolvimento ou outros compartimentos subcelulares. A orientação da incorporação, a velocidade da imagem e os rótulos são todos projetados para nos permitir responder às nossas perguntas biológicas. Para a imagem da neurogênese da retina, por exemplo, a orientação dorsal do embrião como descrito aqui pode não permitir a visualização de características específicas de interesse, e a velocidade da imagem pode não ser apropriada. Muitos aspectos do protocolo podem ser adaptados e modificados para uma variedade de necessidades, dependendo de seus interesses e objetivos específicos. Primeiro, o uso de injeções de RNA torna o processo de rotulagem muito flexível. Construtos de fusão de proteínas fluorescentes podem ser usados para rotular estruturas subcelulares de interesse, e a quantidade de injeção de RNA pode ser variada para otimizar a rotulagem. Com base em nosso trabalho com o fluorohore fotoconvertível Kaede, as injeções de RNA parecem suportar uma explosão de tradução que acabou em 12 hpf11,13. Altos níveis de expressão de proteína fluorescente a partir da injeção de RNA poderiam combater o fotobleaching, mas tal abordagem não suporta a expressão sustentada do rótulo fluorescente de interesse. Se for necessária uma expressão sustentada, como quando se imagem de embriões em estágios posteriores, as linhas transgênicas são uma opção, e construir novas linhas em zebrafish é simples24.
Em seguida, o protocolo pode ser adaptado para estágios posteriores de desenvolvimento. Como o pigmento pode impedir a imagem em estágios posteriores, os embriões podem ser tratados com fenilhiourea (PTU) para inibir a formação de pigmentos, ou mutantes genéticos para síntese de pigmentos podem ser usados. Para evitar a contração de embriões, a tricaine pode ser adicionada às soluções de sobreposição de mídia de agarose e embriões, e a porcentagem de agarose pode ser ajustada conforme necessário. À medida que o olho cresce, pode ser necessário mudar a orientação de montagem; aqui, montamos embriões dorsalmente, mas em estágios posteriores, pode fazer mais sentido montar lateralmente ou anteriormente, dependendo da estrutura de interesse. Como diferentes processos ocorrem em diferentes escalas espaciais e temporais, também pode-se otimizar a etapa Z e a etapa de tempo da aquisição de imagens. Esses recursos só podem ser determinados empiricamente para as necessidades de cada laboratório.
Finalmente, este protocolo foi desenvolvido especificamente para um microscópio confocal de escaneamento a laser, com embriões incorporados em uma porcentagem relativamente alta de agarose em um estágio inicial de desenvolvimento ocular. Se estiver em horários diferentes ou em locais diferentes durante o desenvolvimento ocular, este protocolo precisará ser adaptado para o processo de interesse. Muitas abordagens de imagem diferentes são atualmente possíveis, com mais sendo desenvolvidas por engenheiros ópticos. Cada abordagem traz seu próprio conjunto de desafios, desde diferentes maneiras de incorporar e montar embriões para imagens, até diferentes tamanhos e formatos de arquivos. Delineamos considerações na introdução para orientar o processo de otimização, no qual a resolução espacial e temporal máxima é equilibrada com fotobleaching, fototoxicidade e imensos tamanhos de arquivos. Esperamos que esses princípios gerais, além das informações práticas descritas acima, ajudem outros a estabelecer abordagens de imagem timelapse para estudar as muitas questões abertas no desenvolvimento dos olhos.
Divulgações
Os autores não têm nada a revelar.
Agradecimentos
Somos gratos aos membros passados e atuais do Laboratório Kwan pelo trabalho sobre abordagens e discussões de timelapse deste protocolo. Este trabalho foi apoiado pelo NIH (R01 EY025378 e R01 EY025780 a KMK; F31 EY030758 para SL; e T32 GM007464 para MAC).
Materiais
| Name | Company | Catalog Number | Comments |
| Delta TPG Dish 0.17mm clear | Bioptechs | 0420041500C | coverglass bottom for optical compatibility |
| Disposable Pasteur Pipettes | VWR | 14672-608 | overall length 14.6 cm (53/4") |
| Dumont #5, #55, or #5SF Forceps | Fine Science Tools | 11254-20 | For style #5: straight tip, 0.05 x 0.01 mm tip, inox, 11 cm long |
| Flaming/Brown Micropipette Puller | Sutter | P-97 | |
| Immersol W | Carl Zeiss Microscopy | 4449690000000 | immersion fluid for water-emission objectives, halogen-free, low fluorescence |
| Microinjection Mold | Adaptive Science Tools | TU-1 | Six ramps, one 90 degree and one 45 degree beveled side. |
| Microloader Tips | Eppendorf | 930001007 | Microloader, tip for filling glass microcapillaries, 0.5 – 20 µL |
| mMessage mMachine SP6 Transcription Kit | Invitrogen | AM1340 | contains SP6 Enzyme Mix, 10X Reaction Buffer, 2XNTP⁄CAP Solution, GTP Solution, pTRI-Xef, TURBO DNase, Ammonium Acetate Stop Solution, Lithium Chloride Precipitation Solution, and Gel Loading Buffer and are all stored at –20°C. Nuclease-free Water may be stored at any temperature. |
| Nikon SMZ645 Stereo Microscope | Nikon | SMZ645 | used for injecting embryos (see Fig. 2B) |
| NotI-HF enzyme | NEB | R3189S | comes with Gel Loading Dye, Purple (6X) |
| NuSieve GTG Agarose | Lonza | 50081 | fine resolution, low melt agarose |
| Objective LD "C-Apochromat" 40x/1.1 W Korr M27 | Carl Zeiss Microscopy | 421867-9970-000 | |
| Olympus SZX16 Stereo Microscope | Olympus | SZX2-ZB16 | used for screening, embedding embryos (see Fig. 3A) |
| Phenol red solution | Sigma-Aldrich | P0290 | 0.5% in DPBS, sterile filtered, endotoxin tested, cell culture tested |
| Pico-liter Microinjector | Harvard Apparatus | PLI-100 | Femtoliter to microliter injections; digital readouts for injection count, time, and pressure; contains 5 pressures including inject, balance, clear, fill, and hold |
| Pipette Pump Pipettor | SP Bel-Art | F37898 | fits a disposable pasteur pipette, contains thumbwheel on side for aspirating or dispensing and plunger for quick emptying |
| Premium Thin Wall Borosilicate Glass Capillaries | Warner Instruments | G100T-4 | 1.0 x 0.78 mm |
| QIAquick PCR Purification Kit | Qiagen | 28106 | contains QIAquick Spin Columns, Buffers, Collection Tubes (2 ml) |
| RNase-free H2O | Invitrogen | AM9906 | DNase-Free, Molecular Biology Grade, RNase-Free |
| RNeasy Mini Kit | Qiagen | 74104 | contains RNeasy Mini Spin Columns, Collection Tubes (1.5 ml and 2 ml), RNase-free Reagents and Buffers |
| Stage Attachment Z PIEZO WSB 500 | Carl Zeiss Microscopy | 432339-9000-000 | |
| Stage Insert Z PIEZO WSB Universal | Carl Zeiss Microscopy | 432339-9030-000 | |
| Zeiss LSM 880 with Airyscan | Carl Zeiss Microscopy | N/A | inverted Axio Observer microscope |
| Zen Black 2.3 SP1 software | Carl Zeiss Microscopy | N/A | Zeiss Efficient Navigation (ZEN) controls all light microscope systems from Zeiss |
Referências
- Adler, R., Canto-Soler, M. V. Molecular mechanisms of optic vesicle development: complexities, ambiguities and controversies. Developmental Biology. 305 (1), 1-13 (2007).
- Chow, R. L., Lang, R. A. Early eye development in vertebrates. Annual Review of Cell and Developmental Biology. 17, 255-296 (2001).
- Fuhrmann, S. Eye morphogenesis and patterning of the optic vesicle. Current Topics in Developmental Biology. 93, 61-84 (2010).
- Martinez-Morales, J. R., Wittbrodt, J. Shaping the vertebrate eye. Current Opinion in Genetics and Development. 19 (5), 511-517 (2009).
- Yang, X. J. Roles of cell-extrinsic growth factors in vertebrate eye pattern formation and retinogenesis. Seminars in Cell & Developmental Biology. 15 (1), 91-103 (2004).
- Bogdanovic, O., et al. Numb/Numbl-Opo antagonism controls retinal epithelium morphogenesis by regulating integrin endocytosis. Developmental Cell. 23 (4), 782-795 (2012).
- Bryan, C. D., Casey, M. A., Pfeiffer, R. L., Jones, B. W., Kwan, K. M. Optic cup morphogenesis requires neural crest-mediated basement membrane assembly. Development. 147 (4), (2020).
- Bryan, C. D., Chien, C. B., Kwan, K. M. Loss of laminin alpha 1 results in multiple structural defects and divergent effects on adhesion during vertebrate optic cup morphogenesis. Developmental Biology. 416 (2), 324-337 (2016).
- Cavodeassi, F., Ivanovitch, K., Wilson, S. W. Eph/Ephrin signalling maintains eye field segregation from adjacent neural plate territories during forebrain morphogenesis. Development. 140 (20), 4193-4202 (2013).
- England, S. J., Blanchard, G. B., Mahadevan, L., Adams, R. J. A dynamic fate map of the forebrain shows how vertebrate eyes form and explains two causes of cyclopia. Development. 133 (23), 4613-4617 (2006).
- Gordon, H. B., et al. Hedgehog signaling regulates cell motility and optic fissure and stalk formation during vertebrate eye morphogenesis. Development. 145 (22), (2018).
- Ivanovitch, K., Cavodeassi, F., Wilson, S. W. Precocious acquisition of neuroepithelial character in the eye field underlies the onset of eye morphogenesis. Developmental Cell. 27 (3), 293-305 (2013).
- Kwan, K. M., et al. A complex choreography of cell movements shapes the vertebrate eye. Development. 139 (2), 359-372 (2012).
- Martinez-Morales, J. R., et al. Ojoplano-mediated basal constriction is essential for optic cup morphogenesis. Development. 136 (13), 2165-2175 (2009).
- Miesfeld, J. B., et al. Yap and Taz regulate retinal pigment epithelial cell fate. Development. 142 (17), 3021-3032 (2015).
- Nicolas-Perez, M., et al. Analysis of cellular behavior and cytoskeletal dynamics reveal a constriction mechanism driving optic cup morphogenesis. eLife. 5, (2016).
- Picker, A., et al. Dynamic coupling of pattern formation and morphogenesis in the developing vertebrate retina. PLoS Biology. 7 (10), e1000214 (2009).
- Rembold, M., Loosli, F., Adams, R. J., Wittbrodt, J. Individual cell migration serves as the driving force for optic vesicle evagination. Science. 313 (5790), 1130-1134 (2006).
- Sidhaye, J., Norden, C. Concerted action of neuroepithelial basal shrinkage and active epithelial migration ensures efficient optic cup morphogenesis. eLife. 6, (2017).
- Driever, W., et al. A genetic screen for mutations affecting embryogenesis in zebrafish. Development. 123, 37-46 (1996).
- Haffter, P., et al. The identification of genes with unique and essential functions in the development of the zebrafish, Danio rerio. Development. 123, 1-36 (1996).
- Nusslein-Volhard, C. The zebrafish issue of Development. Development. 139 (22), 4099-4103 (2012).
- Hoshijima, K., et al. Highly efficient CRISPR-Cas9-based methods for generating deletion mutations and F0 embryos that lack gene function in zebrafish. Developmental Cell. 51 (5), 645-657 (2019).
- Kwan, K. M., et al. The Tol2kit: a multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Developmental Dynamics. 236 (11), 3088-3099 (2007).
- Almeida, A. D., et al. Spectrum of Fates: a new approach to the study of the developing zebrafish retina. Development. 141 (9), 1971-1980 (2014).
- Baye, L. M., Link, B. A. Interkinetic nuclear migration and the selection of neurogenic cell divisions during vertebrate retinogenesis. The Journal of Neuroscience. 27 (38), 10143-10152 (2007).
- Clark, B. S., et al. Loss of Llgl1 in retinal neuroepithelia reveals links between apical domain size, Notch activity and neurogenesis. Development. 139 (9), 1599-1610 (2012).
- Das, T., Payer, B., Cayouette, M., Harris, W. A. In vivo time-lapse imaging of cell divisions during neurogenesis in the developing zebrafish retina. Neuron. 37 (4), 597-609 (2003).
- Kay, J. N., et al. Transient requirement for ganglion cells during assembly of retinal synaptic layers. Development. 131 (6), 1331-1342 (2004).
- Norden, C., Young, S., Link, B. A., Harris, W. A. Actomyosin is the main driver of interkinetic nuclear migration in the retina. Cell. 138 (6), 1195-1208 (2009).
- Suzuki, S. C., et al. Cone photoreceptor types in zebrafish are generated by symmetric terminal divisions of dedicated precursors. Proceedings of the National Academy of Sciences of the United States of America. 110 (37), 15109-15114 (2013).
- Wan, Y., et al. The ciliary marginal zone of the zebrafish retina: clonal and time-lapse analysis of a continuously growing tissue. Development. 143 (7), 1099-1107 (2016).
- Yoshimatsu, T., et al. Presynaptic partner selection during retinal circuit reassembly varies with timing of neuronal regeneration in vivo. Nature Communications. 7, 10590 (2016).
- Kimmel, C. B., Ballard, W. W., Kimmel, S. R., Ullmann, B., Schilling, T. F. Stages of embryonic development of the zebrafish. Developmental Dynamics. 203 (3), 253-310 (1995).
- Wan, Y., Otsuna, H., Chien, C. B., Hansen, C. An interactive visualization tool for multi-channel confocal microscopy data in neurobiology research. IEEE Transactions on Visualization and Computer Graphics. 15 (6), 1489-1496 (2009).
- Wan, Y., Otsuna, H., Chien, C. B., Hansen, C. FluoRender: an application of 2D image space methods for 3D and 4D confocal microscopy data visualization in neurobiology research. IEEE Pacific Visualization Symposium. , 201-208 (2012).
- Carney, K. R., Bryan, C. D., Gordon, H. B., Kwan, K. M. LongAxis: a MATLAB-based program for 3D quantitative analysis of epithelial cell shape and orientation. Developmental Biology. 458 (1), 1-11 (2020).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados