Method Article
Использование Дрозофилы S2 клетки для живой визуализации клеточного отдела
В этой статье
Резюме
Клеточные деления могут быть визуализированы в режиме реального времени с помощью флуоресцентно помеченных белков и замедленной микроскопии. Используя представленный здесь протокол, пользователи могут анализировать динамику синхронизации деления клеток, митотические сборки шпинделей, а также хромосомные конгрессионы и сегрегацию. Дефекты в этих событиях после РНК-интерференции (РНК) опосредованного гена нокдаун может быть оценена и количественно.
Аннотация
Дрозофила Клетки S2 являются важным инструментом в изучении митоза в культуре тканей, обеспечивая молекулярное понимание этого фундаментального клеточного процесса быстрым и высокой пропускной способом. S2 клетки оказались поддаются как фиксированной и живой клетки изображений приложений. Примечательно, что живая клеточная визуализация может дать ценную информацию о том, как потеря или нокдаун гена может повлиять на кинетику и динамику ключевых событий во время деления клеток, включая митотический шпиндельный сбор, хромосомный конгрессион и сегрегацию, а также общее время цикла ячейки. Здесь мы используем S2 клетки stably трансфицируется флуоресцентно помечены mCherry: я-тубулин, чтобы отметить митотический шпиндель и GFP: CENP-A (называется "CID" ген в Drosophila) для обозначения центром для анализа последствий ключевых митотических генов на время деления клеток, от профазы (в частности, при разбивке ядерного конверта; НЭБД) к началу анафазы. Этот протокол изображения также позволяет визуализировать динамику микротрубока шпинделя и хромосомы на протяжении всего митоза. В этом и заключается цель обеспечить простой, но всеобъемлющий протокол, который позволит читателям легко адаптировать клетки S2 для живых экспериментов по визуализации. Результаты, полученные в результате таких экспериментов, должны расширить наше понимание генов, участвующих в делении клеток, определяя их роль в нескольких одновременных и динамических событиях. Наблюдения, сделанные в этой системе культуры клеток, могут быть проверены и дополнительно исследованы in vivo с использованием впечатляющего инструментария генетических подходов у мух.
Введение
Cell Division является процессом, критически важным для всех многоклеточных организмов, как в их развитии и гомеостаза1. Дрозофила уже давно используется в качестве модели для изучения деления клеток, с экспериментами в различных типах тканей и генетических условий, обеспечивая ключевое понимание процесса. Хотя многие из этих идей приходят из фиксированных клеточных условий, деление клеток является динамической процедурой со многими движущимися частями, что делает визуализацию живых клеток неотъемлемой частью оценки РНК или генетических эффектов нокаута на многочисленных частях деления клеток, в том числе образование шпинделя, хромосома конгресса и сегрегации, и цитокинез.
Многие протоколы были разработаны и использованы на протяжении многих лет, чтобы визуализировать дризофилы деления клеток in vivo. Различные группы культивировали методы разделения изображений как в личиальных ими, так и в личинок мозгах2,3,4,5,6,7,8 . Эти методы, хотя и полезны для визуализации в конкретных тканях, ограничены в пропускной состоянии и часто требуют генерации и поддержания генетических запасов для генерации флуоресцентных компонентов и изменения экспрессии генов, представляющих интерес. Культивированные клетки S2 из Дрозофилы обеспечивают более высокую пропускную возможность для быстрого тестирования влияния различных генов в делении клеток. Кроме того, с возможностью трансфектировать различные флуоресцентные белки, S2 клетки могут быть быстро изменены, чтобы определить влияние РНК нокдаун на многочисленные компоненты деления клеток. Флуоресцентно помеченные гены, представляющие интерес, также можно наблюдатьв делении клеток, что позволяет динамическую характеристику их функции 9.
Здесь мы предоставляем подробный протокол для живой визуализации митотических клеток S2 с использованием методов, которые мы недавно описали10. Наш метод использует стабилизированные клетки с флуоресцентными маркерами для микротруби и центрометров, выражение которых находятся под контролем медно-индуцируемого промотора (металлотионеин; pMT). Эта методология может быть использована для изображения спинели и хромосомы динамики на всех этапах митоза с использованием относительно простого флуоресцентного микроскопа с основным программным обеспечением изображений. Она может быть дополнительно настроена в соответствии с индивидуальными потребностями исследования, с переходной преображая и РНК предлагает расширенные возможности для определения роли генов-кандидатов в митозе. Из-за относительной простоты протокола, он может быть использован для небольших экранов потери функции для определения генов, для которых дальнейшее изучение in vivo было бы полезным, что позволило бы более целенаправленные усилия и эффективность с последующим генетическим манипуляции мухами.
протокол
1. Подготовка S2 клеток для лечения РНК
-
Из фондовой культуры сопливых pMT:GFP: CID и pMT:mCherry: з-тубулин стабилизированные S2 клетки (80% жизнеспособных; выращенные при 24-28 кв. с), семенные клетки в 6 хорошо стерильной культуры пластины при плотности 1 х 106 клеток / мл в свежем, подогревается, 10% плодовитое сыворотки (FBS) дополнены Шнайдер насекомых средств массовой информации (SIM).
ПРИМЕЧАНИЕ: Stably трансинфицированных S2 клеток могут быть получены после протокола из Дрозофила РНК Скрининг центр (DRSC) (https://dgrc.bio.indiana.edu/Protocols?tab=cells). Хотя специфическая стабильная линия S2, используемая здесь, использует GFP и mCherry, многочисленные альтернативы существуют как в этих флуоресцентных спектрах, так и в других. Подробное обсуждение этих флуоресцентных белков выходит за рамки этого протокола, но их свойства и потенциальные преимущества / недостатки были умело рассмотрены в другом месте 11.- Используя счетчик клеток или гемоцитометр, определить плотность сливая клеточного запаса.
- Определить объем суспензии клеток, необходимых для достижения 1 х 106 клеток / мл в общем объеме 4 мл (например, 4 х 106 общих ячеек). Добавьте этот объем клеточного запаса к объему свежей SIM-карты, чтобы сделать 4 мл/хорошо общий объем. Определите плотность клеточного запаса (клеток/мл), разбавив 100 л клеток непосредственно из косой колбы с 100 злификом носителей и подсчитывая это разбавление 1:2 либо вручную с помощью гемоцитометра, либо с автоматическим счетчиком клеток, если таковая имеется.
ПРИМЕЧАНИЕ: При использовании S2-клеток, не стабилизированных, переходный переходный анализ с флуоресцентно помечены (GFP, mCherry и т.д.) - или -тубулин, и маркер ДНК (CENP-A, Histone H2B и т.д.) может быть выполнена. Кроме того, в Ресурсном центре Drosophila Genetics, который может лучше соответствовать точным экспериментальным потребностям пользователя (https://dgrc.bio.indiana.edu/cells/Catalog, можно получить заведомые клеточные линии с различными митотические шпиндельии и ДНК-маркерами. - Поместите свежесеядные клетки в инкубатор 24-28 градусов по Цельсию на 36-48 ч.
2. Лечение семенных клеток с dsRNA против гена (ы) интереса
ПРИМЕЧАНИЕ: Следующий пример и результаты раздел будет использовать Shortstop (Shot), актин-микротрубуль кроссулят агента в качестве гена интереса. Используйте макет лечения без dsRNA или с dsRNA, направленных против нерелевантного гена (например, бета-галактозидазы, лака)в качестве отрицательного контроля.
- Теплый Шнайдер насекомых средств массовой информации не дополняется FBS (Сыворотка свободных средств массовой информации; SFM) в инкубаторе 24-28 градусов по Цельсию.
-
Переносите клетки из ранее посеянных скважин (от шага 1.1.2) в стерильную трубку 15 мл, отмечая общий объем переданных клеток.
- Сохранить небольшой объем клеток (100 евро) для подсчета в клеточном счетчике или гемоситометре.
- Аккуратно гранулы клетки центрифугировании на 1000 х г в течение 3 мин при комнатной температуре.
- В то время как центрифугии клеток, определить концентрацию клеток на мл с помощью счетчика клеток или гемоцитометра. Умножьте это число на общий объем центрифуги (отмеченный в 1.2.2.), чтобы получить общее количество ячеек.
- Аспирируйте супернатант из пеллетных клеток.
-
Повторно приостановить клетки в разогретых SFM для получения концентрации 3 х 106 клеток / мл.
- Перенесите 1 мл перекрываемых ячеек в новый колодец в 6 скважине.
- Добавьте 10-50 мг dsRNA (разбавленного в 100 л без РНАза воды) против выстрела (или целевого гена интереса) непосредственно к клеткам и закружить пластину для смешивания. Инкубировать пластину в течение 1 ч в инкубаторе 24-28 градусов по Цельсию, чтобы обеспечить прямое поглощение dsRNA в клетки.
- Во время инкубации 1 ч, тепло 10% FBS дополнил SIM в инкубаторе 24-28 градусов по Цельсию.
- После инкубации клеток с dsRNA на 1 ч, добавить 2 мл 10% FBS дополнил SIM непосредственно к колодцу, не удаляя 1 мл носителей.
- Поместите клетки в инкубатор 24-28 градусов по Цельсию в течение 3-7 дней.
ПРИМЕЧАНИЕ: Как и в случае с концентрацией dsRNA, общее время лечения может быть оптимизировано для желаемого гена-мишени. Для оценки эффективности РНК, выполнять западные поклот на всей клетки lysates с помощью антитела против гена-мишени. Если первоначальная последовательность целей dsRNA окажется неэффективной, разработка альтернативных dsRNAs, нацеленных на уникальные последовательности в рамках целевого гена.
3. Индукция флуоресцентного белка выражение и подготовка клеток для визуализации
- Если стабилизированные клетки были созданы с помощью плазмида pMT (содержащего индуцируемый промотор металлотионеин), как описано здесь, вызвать экспрессию флуоресцентных белков путем лечения клеток с сульфатом меди (CuSO4) в конечной концентрации 500 мкм на 24-36 ч до визуализации и через 4 дня после лечения РНК. Использование составных плазмидов выражения, таких как pAct не требует индукции меди.
-
Подготовка клеток для визуализации
- Разогрейте 10% FBS, дополненную SIM-карту в инкубаторе 24-28 градусов по Цельсию на 1 ч.
- Переносите клетки в стерильную трубку объемом 15 мл, отмечая общий объем переданных клеток.
- Сохранить небольшой объем клеток (100 евро) для подсчета в клеточном счетчике или гемоситометре.
- Аккуратно гранулы клетки центрифугировании на 1000 х г в течение 3 мин при комнатной температуре.
- В то время как центрифугии клеток, определить концентрацию (клетки / мл) с помощью счетчика клеток или гемоцитометра. Умножьте это число на общий объем центрифуги (отмечено в 2.2.2.), чтобы получить общее количество ячеек.
- Аспирируйте супернатант из пеллетных клеток.
- Resuspend клетки в свежих, нагревается 10% FBS дополнил SIM для получения концентрации 2 х 106 клеток / мл. Добавьте соответствующее количество CuSO4 для поддержания концентрации 500 км.
- Перенесите 200-500 л повторно взвешенных клеток в один колодец многослойной живой клеточной камеры и поместите на перевернутый флуоресцентный микроскоп. Разрешить клеткам поселиться в камере в течение 15-30 минут до визуализации экспериментов.
ПРИМЕЧАНИЕ: Скважины камерной камеры живых клеток могут быть предварительно покрыты поли-Л-лизином для дополнительного присоединения.
4. Настройка программы визуализации ячеек в реальном времени
ПРИМЕЧАНИЕ: В этом эксперименте Live Cell Imaging был выполнен с использованием перевернутой системы визуализации и связанного с ней программного обеспечения (например, Olympus IX83 с программным пакетом cellSens Dimensions). Детали будут отличаться по производителю микроскопа и пакету программного обеспечения; таким образом, ниже перечислены общие руководящие принципы и операции.
-
Используя программное обеспечение, которое работает перевернутый флуоресцентный микроскоп, подготовить программу для визуализации клетки (или клетки) с течением времени.
- Откройте программное обеспечение, дважды нажав на значок рабочего стола программного обеспечения.
- Создайте новый экспериментальный файл, нажав файл, затем новый экспериментальный файл.
- Во-первых, вставьте цикл промежуток времени, над которым будут сделаны изображения. Сделайте это, нажав значок Стоп-часа (значокпетли Time-lapse) из значка бара. Установите интервал в s на вкладке «Менеджер эксперимента», а затем установите количество циклов в одной и той же вкладке, разделив желаемую общую длину эксперимента (в с) на интервал. Разрешить цикл повторить в течение желаемого общего периода времени.
ПРИМЕЧАНИЕ: Как правило, изображения принимаются каждые 30-60 с в течение 3-4 ч. - В течение промежуток времени слой цикла, вставьте инфракрасную проверку фокусировки для поддержания фокуса цели (если так возможности). Для этого добавьте шаг компенсации в течение времени, нажав на значок «Две стрелки» («Движение XY») и выбрав компенсацию в отношении «З-дрифт» из меню выпадающих данных.
ПРИМЕЧАНИЕ: Термин инфракрасный фокус-проверка относится к системе, с помощью которой инфракрасный импульс используется для поддержания постоянного расстояния между целью и слайд / визуализации камеры. Многие микроскопы имеют такую систему, каждый со своей собственной номенклатурой. Читатели должны проконсультироваться со своим руководством по эксплуатации или представителем для конкретных деталей именования. - После инфракрасной проверки фокусировки добавьте шаг в программу, чтобы сделать многоканальный снимок (например, FITC для GFP и TRITC для mCherry) по 3-5 z-стеков, сначала вставив шаг стекz z, затем указав канал. Сделайте это, добавив слой Multichannel Group в слой петли Time-lapse, нажав на значок Color Wheel (многоканальная группа значок). Затем добавьте слой петли в многоканальный слой группы, нажав на 3-слойную иконку (значокпетли стека). Установите желаемый размер шага и количество фрагментов во вкладке Менеджер эксперимента и установите экспозицию каждого канала как можно ниже, чтобы свести к минимуму фотоотбеление.
ПРИМЕЧАНИЕ: Интервал z-стек, время экспозиции и процентная передача светодиодов могут варьироваться. Типично в этих экспериментах, используйте три z-стеки, принятые в диапазоне 3 мкм, давая интервал в 1 мкм между каждым стеком. Определение центра ячейки, а не верхней и нижней, как правило, приводят к лучшим результатам. Изображения затем собираются для каждого канала (ы) при экспозиции 50 мс и процентной передачи 50% без нейтральной плотности (ND) фильтр включен.
5. Разделение изображений клеток с помощью программы визуализации живых клеток
ПРИМЕЧАНИЕ: S2-клетки не требуют CO2 и растут оптимально при 23-27 градусах Цельсия. Все изображения выполнялись при температуре окружающей среды в хорошо контролируемой комнате.
- Используя окуляры, найдите верхний (или нижний) угол скважины и сфокусите объективное (40-60-кратное погружение масла) на клетки с помощью канала mCherry (я-тубулин).
- Сканирование вдоль верхней (или нижней) скважины, чтобы отойти от вертикального разделителя колодца.
ПРИМЕЧАНИЕ: Изображение слишком близко к разделителям скважины может помешать инфракрасной проверке фокусировки. - Найти ячейку (или ячейки), которые находятся в конце G2 или ранней M-фазы (профазы).
ПРИМЕЧАНИЕ: Эти клетки лучше всего можно определить по наличию ровно двух "звездоподобных" микротрубочных структур (центросомы) и нетронутого ядра (очем круговой области преломленного света внутри клетки), оба легко различимы с помощью к-тубулина (красный) возбуждающего фильтра. Выбор клеток в более раннем межфазе, примечательный одним или нулевым легко видимым центросомами, может привести к расточному времени из-за запаздывания в прогрессии G2/M. И наоборот, отбор клеток после НЭБД предотвратит точный расчет митотической синхронизации и визуализацию ранней сборки шпинделя и динамики хромосомы. Кроме того, S2 клетки часто содержат йgt;2 центросомы. Хотя эти, как правило, кластеров в биполярном шпиндель12, он сообщил, что пользователи избегая этих клеток, если иное желаемого для экспериментального дизайна. Изображения и обсуждение подходящих ячеек для выбора можно найти в разделе Результаты представитель. - Нажмите кнопку Live View, чтобы начать просмотр ячеек на экране программного обеспечения. Используя тонкую ручку фокусировки микроскопа, фокусклетка (или клетки) интерес. Установите инфракрасную проверку фокусировки, нажав кнопку Set Focus.
- Инициировать программу визуализации замедленного времени, нажав кнопку «Пуск». Отрегулируйте гистограммы по мере необходимости, выбрав нужный канал и регулируя средние интенсивности пикселей, чтобы ясно видеть интересную клетку (или клетки).
- Разрешить программу для запуска, проверка ячейки после 15-20 минут для обеспечения ядерного разбивки конверта (NEBD) произошло, что определяется исчезновением круглой темноте, преломленное пятно вблизи клетки центра.
- Продолжайте позволять программе работать, периодически проверяя (каждые 15-20 минут), чтобы определить, как только началась анафаза. Остановите программу на этом этапе, если в течение всего периода времени остается больше времени, и сохраните файл. Кроме того, если события во время телофазы и цитокинеза представляют интерес, позвольте программе запустить достаточновремени времени для того, чтобы также изобразить эти процессы.
-
Выполните анализ NEBD в анафазное время начала, отмечая время NEBD (tNEBD) в мин и время первоначального разделения хромосомы (танафазноеначало) в мин и вычитание тNEBD отt анафазного начала для получения NEBD для анафазного времени начала для данной ячейки.
- Сделайте это, нажав кнопку кадра вверх и отметив кадры, где NEBD и анафаза начала происходят, вычитая их, чтобы определить общее количество проема кадров, и умножение этого на временной интервал между кадрами изображений.
- Продолжить сканирование для предварительного деления ячеек, чтобы получить несколько n для данного состояния. Клетки могут быть изображены в камере живых клеток хорошо до 12 ч после их первоначального урегулирования.
Результаты
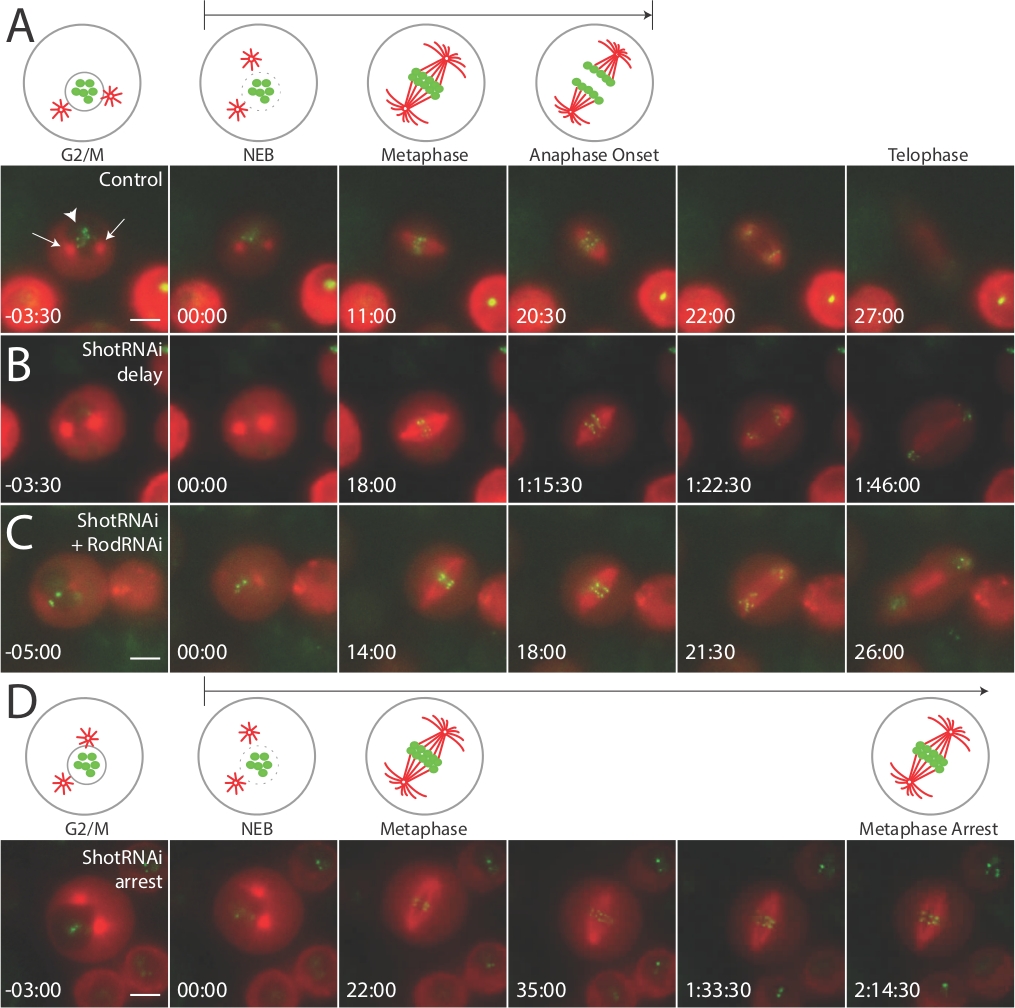
Методы, которые мы описываем выше, приведет к идентификации и визуализации клеток Drosophila S2, проходящих деление клеток. Клетки, которые собираются разделить (т.е. непосредственно перед входом M-фазы) могут быть направлены на наличие двух центросом и нетронутым ядром, указанным преломленным светом и темным пятном внутри клетки при просмотре в канале З-тубулин (Рисунок 1, левые панели, красный канал; наконечник стрелы на рисунке 1A). По нашим оценкам, примерно 2-5% клеток попадают в эту категорию, и между экспериментами по визуализации мы обычно тратим максимум 2-3 минуты на сканирование другой квалифицированной клетки. NEBD может быть визуализирована через исчезновение этого темного пятна, в результате чего равномерное окраску цитоплазмы(рисунок 1, панели с пометкой "00:00", красный). После NEBD, время каждая клетка принимает для формирования шпинделя, конгресс хромосомы, и сегрегатные хромосомы могут быть измерены просто отметив временные точки этих событий по отношению к NEBD.
Мы используем эти подразделения для оценки митотического времени, в частности, NEBD для начала анафазы, отметив точку, в которой хромосомы начинают отделяться. Большинство (90%) медно-индуцированных клеток, выраженных как mCherry: и тубулин и GFP:CID. Небольшой процент ячеек (10%) выразил только один маркер или ни один. Эти клетки удалось избежать при отборе клеток. Как правило, S2 ячейки отображают NEBD в анафазный время начала между 20-30 минут(рисунок 1A, контроль). Затем мы обработали клетки dsRNA, направленные против Shortstop (Shot), актин-микротруббулии перекрестного белка мы подозревали, может повлиять на динамику клеточного цикла. Действительно, после Shot нокдаун клетки выставлены значительные митотические задержки(Рисунок 1B, ShotRNAi задержки), со многими клетками ареста на метафазе и никогда не переход яснее в течение всего 2-3-часового эксперимента изображений (Рисунок 1D, Арест ШотРНАи). Мы рассуждали, что этот фенотип задержки/ареста, вероятно, может быть связан с активацией контрольно-пропускного пункта сборки шпинделя (т.е. контрольно-пропускного пункта M-фазы). Чтобы непосредственно проверить эту гипотезу, мы совместно обработанные клетки с dsRNA против shot и Rough сделки (Rod), важный компонент этой контрольной точки13. Это привело к подавлению фенотипа ареста, в результате чего NEBD в анафазные времена похожи ежефазы, похожие на контроль(рисунок 1C, ShotRNAi-RodRNAi). Таким образом, этот живой протокол визуализации позволил нам сделать вывод, что Shot необходим для своевременной прогрессии M-фазы и что его потеря приводит к активации контрольно-пропускного пункта, которая задерживает или арестовывает митотические клетки.

Рисунок 1: Live изображений показывает дефекты клеточного цикла из-за активации митотической контрольно-пропускной пункт в клетках S2.
Во всех условиях, живо-клеточная визуализация была проведена на S2 клетки, стабилизируемого индуцированным GFP:CID и mCherry: "Тубулин, как описано в этом. (A) Мультфильмы иллюстрируют важные ориентиры во время митоза, и соответствующие изображения отображаются из репрезентативных фильмов указанных генотипов с указанными точками времени по отношению к NEBD (t'00:00) указано. Контрольные клетки обычно прогрессируют до анафазы в течение 20-30 мин. ShotRNAi-обработанные клетки отображают NEBD-анафазную задержку (B) и часто проходят метафазный арест, никогда не ввод явки в рамках эксперимента изображений (D). Совместное лечение клеток с ShotRNAi и RodRNAi, компонент шпинделя контрольно-пропускной пункт сборки, подавляет выстрел фенотипа, ведущих к анафазе начала кинетики похож на контрольные клетки (C). Стрелки в (A) указывают на "звездоподобные" центросомные структуры, а большая стрелка указывает на ядро. Каждая панель масштаба представляет 5 микрон. Эта цифра адаптируется и переиздается с разрешения10 (https://www.molbiolcell.org/info-for-authors). Пожалуйста, нажмите здесь, чтобы просмотреть большую версию этой цифры.
{kind=link}
Видео 1: Покадровая съемка фильма о делении ячеек S2'. Видео показывает репрезентативный фильм о делении ячеек S2 в генотипе «Контроль». Этот фильм соответствует рисунку 1A. Это видео адаптировано и переиздано с разрешения10 (https://www.molbiolcell.org/info-for-authors). Пожалуйста, нажмите здесь, чтобы скачать это видео.
Видео 2: Time-lapse фильм задержки 'ShotRNAi' S2 ячейки разделения. Видео показывает репрезентативный фильм о делении клеток S2 в генотипе 'ShotRNAi', что приводит к фенотипической митотической задержке. Этот фильм соответствует рисунку 1B. Пожалуйста, нажмите здесь, чтобы скачать это видео.
Видео 3: Покадровая съемка фильма о делении S2-клеток S2'ShotRNAi-RodRNAi. Видео показывает репрезентативный фильм о делении клеток S2 в генотипе 'ShotRNAi-RodRNAi'. Этот фильм соответствует рисунку 1C. Это видео адаптировано и переиздано с разрешения10 (https://www.molbiolcell.org/info-for-authors). Пожалуйста, нажмите здесь, чтобы скачать это видео.
Видео 4: Time-lapse фильм арестован 'ShotRNAi' S2 ячейки разделения. Видео показывает репрезентативный фильм о делении клеток S2 в генотипе 'ShotRNAi', что приводит к фенотипическому митотическому аресту. Этот фильм соответствует рисунку 1D. Пожалуйста, нажмите здесь, чтобы скачать это видео.
Обсуждение
Определение соответствующей ячейки
Ключом к визуализации, разделяющей клетки S2, является первое обнаружение правильной клетки. Время может быть потрачено впустую, чтобы элементарные клетки, которые ошибочно считаются готовыми к разделению, но не делают этого в разумные сроки. Клетки должны быть определены, которые имеют две различные и видимые центросомы и неповрежденное ядро. Центросомы должны иметь микротрубочные волокна, иснижающие их, придавая им "звездоподобный" внешний вид. Неповрежденное ядро преломляет свет при регулировке фокуса, а также делает приблизительный центр клетки темнее по внешнему виду. Ядро будет также содержать GFP: CID "точки", еще одна отличительная черта, чтобы помочь в его идентификации. Клетки с юgt;2 центросомы, тубулин пункту без трубулин волокон, исходящих из них, или клетки только с одной центросомой следует избегать. Кроме того, если две центросомы видны, но ядро не является, NEBD уже произошло, и ячейка слишком далеко продвинулись в своем делении, чтобы быть изображены, если полный анализ M-фазы желательно. Как только соответствующая ячейка найдена, скорость в запуске изображения имеет решающее значение, как NEBD происходит быстро (обычно в течение 3-5 минут) и задержки начала изображения может привести к упущенным возможностям. Сосредоточьте ячейку в программном обеспечении для визуализации быстро, так что обе центросомы видны, или если центросомы находятся вне плоскости друг с другом, установите координационный центр между ними, так что каждый из них может быть захвачен, когда стеки собраны выше и ниже определенного центра Точки. После сосредоточения, немедленно установить смещение между этапом и поверхности камеры живой клетки с помощью инфракрасного фокуса проверки (если таковой) и начать программу визуализации. Хотя представленный здесь протокол специально разработан для экспериментов по оценке динамики NEBD-к-анафазе, простые модификации могут подойти читателям, изучающим альтернативные митотические события. Для визуализации NEBD-to-metaphase и анафазы к телофазе мы предлагаем интервалы захвата изображения в 10 с, поскольку эти процессы очень динамичны и быстро происходят в клетках S2 (5-10 мин). Метафазный переход к анафазе (наш типичный экспериментальный фокус) представляет собой более длительный процесс (20-30 мин) в клетках S2, и мы собираем изображения с интервалом в 30 с. Наконец, телофаза через цитокинез происходит довольно медленно в клетках S2, и мы предлагаем 60 с интервалами обеспечения достаточного разрешения для этого приблизительного часового процесса.
Поддержание фокусировки на протяжении всей программы визуализации
Если инфракрасное устройство проверки фокусировки недоступно, не забудьте избежать натыкаясь на микроскоп или стол, на который он сидит, так как это может привести к тому, что клетка выйдет из фокуса во время программы визуализации, что приведет к неоднозначным, неинтерпретируемым результатам. Предварительное покрытие живых клеточных камерных колодцев поли-L-лизин может помочь в присоединении клеток, которые могут помочь избежать движения клеток и особенно полезно для установки без инфракрасных проверок фокусировки. Кроме того, установки, включая ударопоглощающие платформы или воздушные столы, могут дополнительно помочь избежать того, чтобы ячейки не смогли выйти из фокуса. Наконец, некоторые программы имеют функции паузы, позволяющие пользователю переориентироваться и возобновить программу.
Изображение нескольких ячеек
Полезно для накопления данных, что часто несколько клеток, готовых к разделению могут быть помещены в одном поле зрения для визуализации. Это может быть особенно полезно для экспериментов, в которых клетки имеют длительные длительности M-фазы или длительный арест (например, условия, которые вызывают активацию SAC). Для этих клеток, мы обычно изображения до клетки photobleached (примерно 2-3 ч), но полное время арестованных клеток должно быть на усмотрение исследователя. Иногда несколько предразделяющих ячеек могут находиться в разных фокусных плоскостях, и в этом случае может быть полезно добавить z-стеки для размещения всех ячеек. Добавление большего количества стеков z также может помочь улучшить разрешение. Следует проявлять осторожность при этом, однако, как это будет подвергать клетки больше излучения и привести к более быстрой фотоотбелении, а также привести к большим размерам файлов на жестких устройствах хранения дисков.
Как избежать фотоотбелевания и фототоксичности
Наш метод описывает конкретные настройки для времени экспозиции, интервалы изображения, z стеки, и процент передачи для нашего источника светодиодных источников света, которые обычно работают для нашей экспериментальной установки и желаемых результатов. Поскольку микроскопы и экспериментальные цели могут варьироваться, то, что может хорошо работать для нашей системы, может привести к преждевременному отбеливанию фотоилиаза или фототоксичности в других. Фотоповреждение может представлять отсутствие движения шпинделя, фрагментацию микротрубок, неполноценную динамику микротрубок и длительную активацию контрольно-пропускного пункта. Одним из потенциальных методов, чтобы избежать таких ловушек, является ограничение времени воздействия. Большинство современных камер в настоящее время обладают широкими динамическими диапазонами, и гистограммы могут быть скорректированы для визуализации структур даже при очень низких экспозициях. Другой метод заключается в увеличении интервала изображения (например, до нескольких минут между изображениями), так что количество раз, когда клетка подвергается воздействию света в течение всего интервала сбора уменьшается. Это выгодно, особенно в визуализации в течение более длительных периодов времени (много часов), и может дополнительно помочь в уменьшении размера файла таких экспериментов. Ограничение количества z стеков, принятых также может помочь уменьшить воздействие света, используя 2 или даже только 1 вместо 3. Кроме того, регулировка процентной передачи света (для светодиодных источников света) или использование фильтров нейтральной плотности (ND) (как для галогенной лампы, так и для светодиодных источников света) снизит интенсивность света и в сочетании с более длительным временем экспозиции может сохранить видимость . Выбирая клетки с уже разделенными центросомы, общее воздействие может быть ограничено в том, что эти клетки обычно попадают в митоз быстро (в течение минуты или двух) после начала визуализации. Можно также ограничить время, что клетки могут быть изображены до NEBD. Наша лаборатория обычно устанавливает это время на 10 минут, но более консервативный предел может быть легко введен пользователем. Кроме того, более "инвазивной" мерой, которая мы рекомендуем, является лечение dsRNA, нацеленное против компонента SAC (например, Rod или Mad2). Если фенотип ареста вызван неспецифическим повреждением клеток (например, дефектной динамикой микротрубок), такое лечение с меньшей вероятностью подавляет арест по сравнению с добросовестным эффектом гена, интересуемого в первоначальном эксперименте.
Будущие направления
Описанный здесь метод может быть использован на относительно простом эпифлуоресцентном микроскопе для быстрого изображения живых клеточных делений и может быть легко адаптирован в соответствии с конкретным экспериментальным дизайном и целями исследователя. Несколько отличных методов для культивирования, RNAi нокдаун подходы, переходные прерывистой, и флуоресценции микроскопии были опубликованы для S2 и других клеток Drosophila 9,14,15, 16 Год , 17. Наш протокол предлагает несколько преимуществ. (1) Использование двойной стабильной клеточной линии (GFP:CID, mCherry:-Tubulin) одновременно отмечает два ключевых митотических структур, позволяет избежать хлопот и потенциальных осложнений переходного преображенского, и гарантирует, что почти все клетки будут выражать флуоресцентные маркеры как потенциальных кандидатов на визуализацию. (2) Адаптация к митозу добавляет к опубликованным протоколам изучения динамики цитоскелета в подвижной, не разделяющей клетки и распространяется на изучение митотических событий в режиме реального времени. Хотя мы считаем, что наша является мощным методом, который добавляет к репертуару других, всегда могут быть улучшения. Одной из особенностей деления клеток, которую мы обнаружили, трудно представить, является деление центросомного. Это происходит до NEBD и трудно определить, когда одна центросома готова разделить на две части. Использование флуоресцентных маркеров цикла клеток (Циклин А и Циклин B) может помочь решить эту проблему, но происходит за счет каналов, которые могут быть использованы для визуализации компонентов деления клеток. Наша лучшая стратегия для попытки визуализировать разделение центросомного заключается в добавлении многоточечного приобретения в программу визуализации (определение различных точек в плоскости XY к изображению), но это может привести к большим файлам, а также может потребовать (в зависимости от количества точек) увеличение интервала между сбором изображений, уменьшение времени разрешения результирующего фильма. Другим решением может быть синхронизация клеток на желаемом этапе клеточного цикла с использованием препаратов, которые нацелены на регуляторы клеточного цикла, а затем последующее вымывание до визуализации, хотя эти методы не могут быть надежными в клетках S2 в частности.
Представленный здесь протокол также закладывает основу для будущих применений в живой визуализации клеток. Благодаря усовершенствованию технологий визуализации и программного обеспечения, возможно по-настоящему высокой пропускной способности этого протокола с использованием более высокой емкости, многокамерные пластины. Такие нововведения сделали бы крупномасштабные ЭКРАНы RNAi, такие как те, которые уже достигнуты в фиксированной подготовке12,18, более уступчивым в формате живой ячейки. Малые молекулы наркотиков экраны также могут быть предусмотрены в качестве средства выявления новых соединений ориентации клеточного процесса деления. Улучшение флюорофоров и оптических фильтров может также привести к визуализации нескольких митотических компонентов (а не только два мы описываем), что позволяет изображение конкретных митотических регуляторов и их взаимодействия с ДНК и / или шпинделя. Например, генерация клеток с флуоресцентными репортерами, которые выражают после повреждения ДНК или во время апоптоза, будет полезным инструментом для выявления новых генов, участвующих в этих процессах. Аналогичные подходы могут быть использованы для изучения динамики клеточного цикла с использованием выражения флуоресцентно помеченных циклинов19.
Раскрытие информации
Авторам нечего декларировать.
Благодарности
Эта работа финансировалась Национальными институтами здравоохранения (R01 GM108756). Мы благодарны Гэри Карпен (Калифорнийский университет, Беркли) за щедрое предоставление нам GFP: CID S2 ячейки линии запасов, из которых наш GFP: CID/mCherry: "Тубулин линия была создана10.
Материалы
| Name | Company | Catalog Number | Comments |
| Bright-Line Hemacytometer | Sigma-Aldrich | Z359629-1EA | for cell counting |
| cellSens imaging software | Olympus | ||
| CELLSTAR Cell Culture Flask, 50 mL, 25 CM2, PS, Red Filter Screw Cap, Clear, Sterile, 10 PCS/BAG | Greiner Bio-One | 690175 | |
| CELLSTAR Cell Culture Multiwell Plate, 6 well, PS, Clear, TC, Lid with condensation rings, sterile, single packed | Greiner Bio-One | 657160 | |
| Centrifuge 5804 R | eppendorf | Cat. 022623508 | |
| Copper(II) sulfate pentahydrate, minimum 98% | Sigma-Aldrich | C3036-250G | |
| Corning Fetal Bovine Serum | Fisher Scientific | MT35015CV | |
| Effectene Transfection Reagent 1 mL | Qiagen | 301425 | for transient transfection |
| IX-83 Inverted Epifluorescent Microscope | Olympus | ||
| LabTek Chambered Slide Insert | Applied Scientific Instruments | I-3016 | |
| MEGAscript T7 Transcription Kit | Thermo Fisher Scientific | AM1334 | For dsRNA production |
| MOXI Z Mini Automated Cell Counter Kit | ORFLO | MXZ001 | for cell counting |
| MS-2000 XY Flat-Top Automated Stage and Controller | Applied Scientific Instruments | ||
| Nunc Lab-Tek II Chambered Coverglass (no 1.5 borosilicate glass) 8-well | Thermo Fisher Scientific | 155409 | |
| Orca-Flash 4.0 LT Camera | Hammamatsu Photonics K.K. | C11440-42U | |
| pMT/V5-His A Drosophila Expression Vector | Thermo Fisher Scientific | V412020 | |
| Poly-L-Lysine | Cultrex | 3438-100-01 | |
| Purifier Logic+ Class II, Type A2 Biosafety Cabinets | Labconco | 302310000 | |
| Schneider's insect medium | Sigma-Aldrich | S0146-100ML | |
| Spectra Tub Centrifuge Tubes | VWR | 470224-998 | |
| Uner Counter BOD Incubator | Sheldon manufacturing (VWR) | 89409-346 | |
| X-CITE 120 LED | ExcelitasTechnologies | Led light source | |
| Z -Drift Compensator (ZDC) | Olympus | infrared focus check |
Ссылки
- Ragkousi, K., Gibson, M. C. Cell division and the maintenance of epithelial order. Journal of Cell Biology. 207 (2), 181-188 (2014).
- Aldaz, S., Escudero, L. M., Freeman, M. Live imaging of Drosophila imaginal disc development. Proceedings of the National Academy of Science U. S. A. 107 (32), 14217-14222 (2010).
- Cabernard, C., Doe, C. Q. Live imaging of neuroblast lineages within intact larval brains in Drosophila. Cold Spring Harbor Protocols. 2013 (10), 970-977 (2013).
- Lerit, D. A., Plevock, K. M., Rusan, N. M. Live imaging of Drosophila larval neuroblasts. Journal of Visualized Experiments. (89), (2014).
- Morris, L. X., Spradling, A. C. Long-term live imaging provides new insight into stem cell regulation and germline-soma coordination in the Drosophila ovary. Development. 138 (11), 2207-2215 (2011).
- Prasad, M., Jang, A. C., Starz-Gaiano, M., Melani, M., Montell, D. J. A protocol for culturing Drosophila melanogaster stage 9 egg chambers for live imaging. Nature Protocols. 2 (10), 2467-2473 (2007).
- Restrepo, S., Zartman, J. J., Basler, K. Cultivation and Live Imaging of Drosophila Imaginal Discs. Methods in Molecular Biology. 1478, 203-213 (2016).
- Tsao, C. K., Ku, H. Y., Lee, Y. M., Huang, Y. F., Sun, Y. H. Long Term Ex Vivo Culture and Live Imaging of Drosophila Larval Imaginal Discs. PLoS One. 11 (9), e0163744 (2016).
- Rogers, S. L., Rogers, G. C. Culture of Drosophila S2 cells and their use for RNAi-mediated loss-of-function studies and immunofluorescence microscopy. Nature Protocols. 3 (4), 606-611 (2008).
- Dewey, E. B., Johnston, C. A. Diverse mitotic functions of the cytoskeletal cross-linking protein Shortstop suggest a role in Dynein/Dynactin activity. Molecular Biology of the Cell. 28 (19), 2555-2568 (2017).
- Rodriguez, E. A., et al. The Growing and Glowing Toolbox of Fluorescent and Photoactive Proteins. Trends in Biochemical Sciences. 42 (2), 111-129 (2017).
- Kwon, M., et al. Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes. Genes and Development. 22 (16), 2189-2203 (2008).
- Basto, R., Gomes, R., Karess, R. E. Rough deal and Zw10 are required for the metaphase checkpoint in Drosophila. Nature Cell Biology. 2 (12), 939-943 (2000).
- Currie, J. D., Rogers, S. L. Using the Drosophila melanogaster D17-c3 cell culture system to study cell motility. Nature Protocols. 6 (10), 1632-1641 (2011).
- Lu, W., Del Castillo, U., Gelfand, I. V. Organelle transport in cultured Drosophila cells: S2 cell line and primary neurons. Journal of Visualized Experiments. (81), e50838 (2013).
- Yang, J., Reth, M. Drosophila S2 Schneider cells: a useful tool for rebuilding and redesigning approaches in synthetic biology. Methods in Molecular Biology. 813, 331-341 (2012).
- Zhou, R., Mohr, S., Hannon, G. J., Perrimon, N. Inducing RNAi in Drosophila cells by soaking with dsRNA. Cold Spring Harbor Protocols. 2014 (5), (2014).
- Goshima, G., et al. Genes required for mitotic spindle assembly in Drosophila S2 cells. Science. 316 (5823), 417-421 (2007).
- Sakaue-Sawano, A., et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 132 (3), 487-498 (2008).
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеThis article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены