
Для просмотра этого контента требуется подписка на Jove Войдите в систему или начните бесплатную пробную версию.
Method Article
Реактивация нервных стволовых клеток в культивируемых эксплантах мозга дрозофилы
В этой статье
Резюме
Установлен метод реактивации покоящихся нервных стволовых клеток в культивируемых эксплантах мозга дрозофилы . Используя этот метод, роль системных сигналов может быть отделена от тканевых внутренних сигналов в регуляции покоя, входа и выхода нервных стволовых клеток.
Аннотация
Нервные стволовые клетки (НСК) обладают способностью пролиферировать, дифференцироваться, подвергаться апоптозу и даже входить и выходить из покоя. Многие из этих процессов контролируются сложным взаимодействием между внутренними генетическими программами НСК с внешними факторами НБК, локальными и системными. В генетическом модельном организме , Drosophila melanogaster, NSC, известные как нейробласты (NB), переключаются с покоя на пролиферацию во время эмбрионального перехода к личиночному. За это время личинки выходят из своей яичной скорлупы и начинают ползать, ища диетические питательные вещества. В ответ на кормление животных жировое тело, эндокринный орган с емкостью для хранения липидов, вырабатывает сигнал, который системно высвобождается в циркулирующую гемолимфу. В ответ на сигнал, полученный из жирового тела (FBDS), инсулиноподобные пептиды Drosophila (Dilps) производятся и высвобождаются из нейросекреторных нейронов мозга и глии, что приводит к последующей активации сигналов роста PI3-киназы в NB и их глиальной и трахеальной нише. Хотя это текущая модель того, как НБ переключаются с покоя на распространение, природа внешнего сигнала FBDS остается неуловимой. Чтобы лучше понять, как внешние системные сигналы NB регулируют выход из покоя, был разработан метод культивирования раннего личиночного мозга in vitro перед кормлением животных. С помощью этого метода экзогенные факторы могут подаваться в культуральную среду и выходить NB из анализа покоя. Мы обнаружили, что экзогенного инсулина достаточно для реактивации NB из покоя в эксплантах всего мозга. Поскольку этот метод хорошо подходит для крупномасштабных экранов, мы стремимся выявить дополнительные внешние сигналы, которые регулируют NB по сравнению с решениями о распространении. Поскольку гены и пути, которые регулируют решения о пролиферации НСК, эволюционно сохраняются, результаты этого анализа могут дать представление об улучшении регенеративной терапии в клинике.
Введение
Стволовые клетки представляют большой интерес из-за их потенциала для использования в регенеративной медицине 1,2. Многие животные, особенно долгоживущие, поддерживают стволовые клетки во взрослых тканях. Эти резидентные стволовые клетки функционируют для поддержания тканевого гомеостаза и используются для восстановления после физической травмы или заболевания 3,4. Большинство стволовых клеток у взрослых животных находятся в состоянии покоя, относительно спящем состоянии, характеризующемся остановкой клеточного цикла и инактивацией сигналов роста5. В ответ на внешние сигналы стволовые клетки выходят из покоя, входят в клеточный цикл и начинают генерировать дочернее потомство, специфичное для их типа ткани. Например, чтобы установить эффективный иммунный ответ, антигенпрезентирующие клетки индуцируют покоящиеся наивные Т-клетки входить в клеточный цикл и клонально расширяться6. В ответ на повреждение скелетных мышц мышечные сателлитные стволовые клетки входят в клеточный цикл и генерируют дочерние миобласты для замены поврежденных миофибрилл 5,7. Хотя ясно, что покоящиеся стволовые клетки реагируют на внешние сигналы, во многих случаях природа внешнего сигнала остается неясной, а также механизм активации стволовых клеток, индуцированных сигналом. Получение лучшего понимания того, как покоящиеся стволовые клетки реагируют на внешние сигналы и входят в клеточный цикл, поможет в разработке лучших методов лечения стволовыми клетками в клинике и увеличит научные знания.
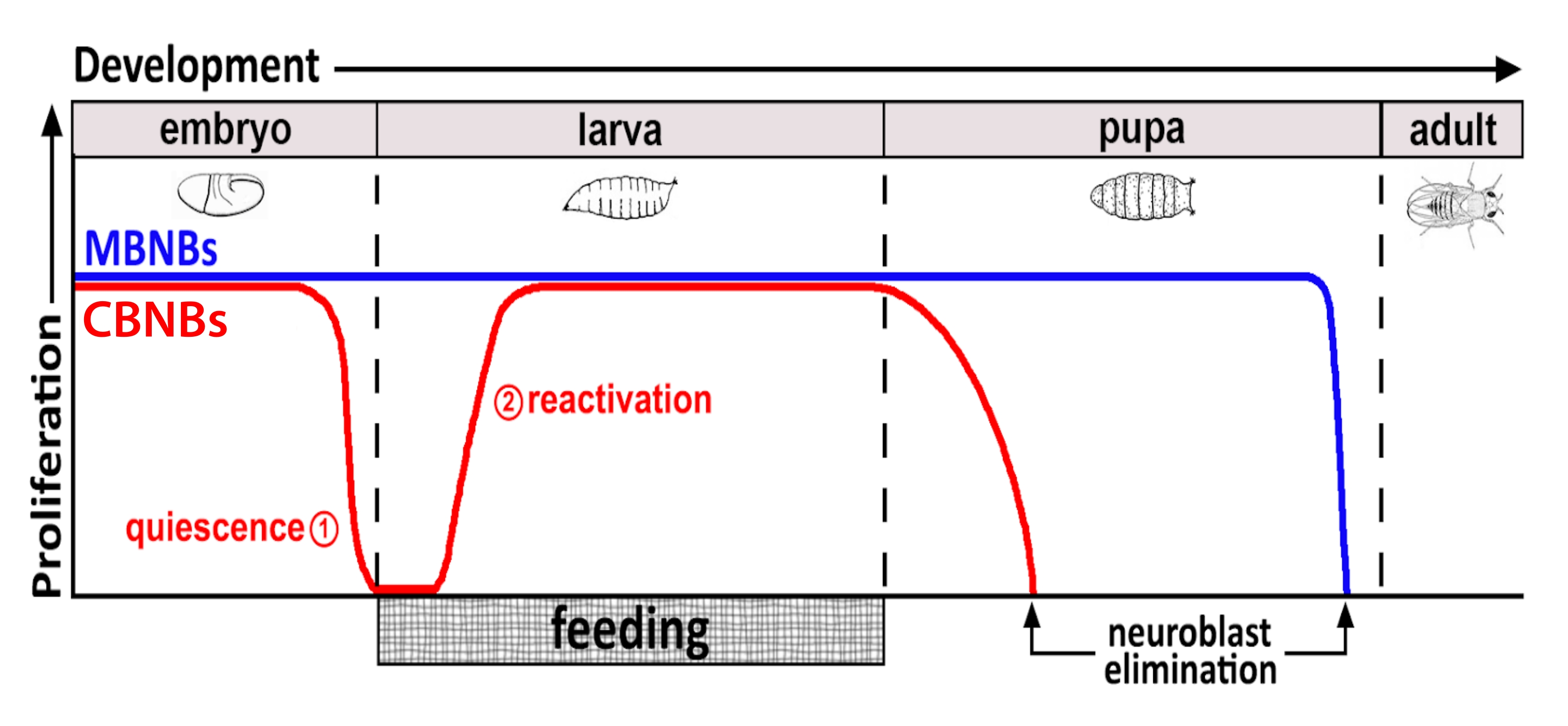
На протяжении десятилетий модельные организмы использовались для выявления генов и клеточных сигнальных путей, которые регулируют пролиферацию стволовых клеток во время развития и во взрослой жизни. У дрозофилы нервные стволовые клетки (НСК), известные как нейробласты (НБ), делятся на протяжении всего развития, чтобы генерировать все нейроны и глию, которые в конечном итоге интегрируются, образуя нейронную цепь, необходимую для функции мозга 8,9. Как и другие стволовые клетки, NB делятся асимметрично для самообновления и, в некоторых случаях, симметрично для расширения пула стволовых клеток. NB определяются во время эмбриогенеза, и большинство из них входят в состояние покоя к концу, что совпадает с уменьшением запасов питательных веществ у матери (рисунок 1). После завершения эмбриогенеза личинки вылупляются и начинают питаться. В ответ на кормление животных NB реактивируются из покоя и возобновляют деление клеток 10,11,12,13,14,15,16. Поскольку дрозофила ЦНС относительно проста и поскольку NB входят и выходят из покоя в определенное время, использование дрозофилы для исследования регуляции покоя, входа и выхода оказывается идеальным.

Рисунок 1: Относительная пролиферация CB NB (центральные нейробласты головного мозга, красный) и MB NBs (нейробласты грибного тела, синий) в течение времени развития. В конце эмбриогенеза большинство НБ (красная линия) прекращают пролиферацию и вступают в состояние покоя. Покой продолжается до тех пор, пока свежевылупившиеся личинки не съедят свой первый полноценный прием пищи. Временные точки фокусировки для этой методологии обозначены красными кругами (1, покоя и 2, реактивация). MB NB (синий) представляют собой подмножество центральных NB мозга, которые постоянно делятся на протяжении всего развития (4 на полушарие мозга). Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}
В ответ на кормление животных сигнальные пути роста PI3-киназы и TOR становятся активными в NB и в их глиальной и трахеальной нише 10,11,15,16. Когда диетические питательные вещества выводятся или когда уровни PI3-киназы снижаются, NB не могут реактивироваться, и рост глии и трахеи также снижаетсяна 10,11,15,16. Текущая модель утверждает, что реактивация NB связана с ростом личинок жировым телом, которое высвобождает системный сигнал в ответ на кормление животных 12,17,18. Этот сигнал, который остается неуловимым, вероятно, способствует экспрессии и высвобождению инсулиноподобного пептида Drosophila (Dilps) в мозге, что приводит к последующей активации PI3-киназы в NB и их глиальной и трахеальной нише. Чтобы лучше понять природу системных сигналов, мы разработали метод реактивации покоящихся NB в культивируемых эксплантах мозга. С помощью этого метода реактивация NB может быть проанализирована при отсутствии системных сигналов всего животного. Экзогенные факторы могут поступать в культуральную среду, а реактивация NB анализируется на основе включения аналога тимидина, EdU. Используя этот метод, мы определили, что экзогенного инсулина достаточно для реактивации покоящихся NB в эксплантах мозга. Будущая работа будет направлена на выявление дополнительных факторов, которые при добавлении обратно либо положительно, либо отрицательно регулируют состояние NB в мозговых эксплантах.
протокол
1. Коллекция личинок дрозофилы
ПРИМЕЧАНИЕ: Подготовьте дрожжевую тарелку, виноградную пасту и кондоминиум Fly перед началом:
- Дрожжевая паста: В небольшой емкости смешайте 5 г активных сухих дрожжей с 10 мл воды, чтобы сформировать пасту, которая имеет консистенцию арахисового масла. Накройте дрожжевую пасту полиэтиленовой пленкой и используйте резинку, чтобы прочно прикрепить ее к контейнеру.
ПРИМЕЧАНИЕ: Свежая дрожжевая паста будет расширяться в своем контейнере и будет отрываться от крышки, если ее не прочно закрепить. Дрожжевая паста прослужит несколько дней при комнатной температуре (RT). - Виноградные тарелки: Следуйте рецепту приготовления виноградных тарелок (таблица 1). При использовании пластин, хранящихся при температуре 4°C, обязательно предварительно нагрейте пластины перед использованием, поместив их в RT на 1 ч.
- Смешайте воду (750 мл) и агар (18,75 г) в колбе 4 л, завихрении и автоклаве в течение 20 мин (жидкий цикл).
- Смешайте виноградный сок (250 мл) и сахарозу (25 г) в колбе объемом 1 л с большим перемешиванием на нагретой тарелке (на медленном огне). Когда сахароза растворится, выключите огонь, подождите, пока колбу можно будет потрогать, прежде чем добавлять Тегосепт (10%, 4 мл) и Пропионовую кислоту (5 мл). Держите перемешивание включенным.
- Когда автоклавирование будет завершено, дайте ему остыть, пока колба не будет затронута (~ 60 ° C), затем смешайте в смеси виноградного сока.
- Смешайте все растворы в одной колбе и дайте помешивать на тарелке.
- Пипетка раствором в крышки небольших по размеру чашек Петри (35 мм). Пипетка примерно по 9 мл на крышку или до получения выпуклого купола.
- ДОПОЛНИТЕЛЬНО: Зажгите крышки, чтобы избавиться от пузырьков.
- Когда пластины затвердеют, уложите виноградные тарелки в коробку с герметичной крышкой и поместите коробку при температуре 4 °C. Тарелки могут храниться до 1 месяца.
- Fly condo: Пробить ~ 20 отверстий в бутылке с полипропиленом дрозофилой с квадратным дном весом 6 унций с помощью иглы весом 18 г.
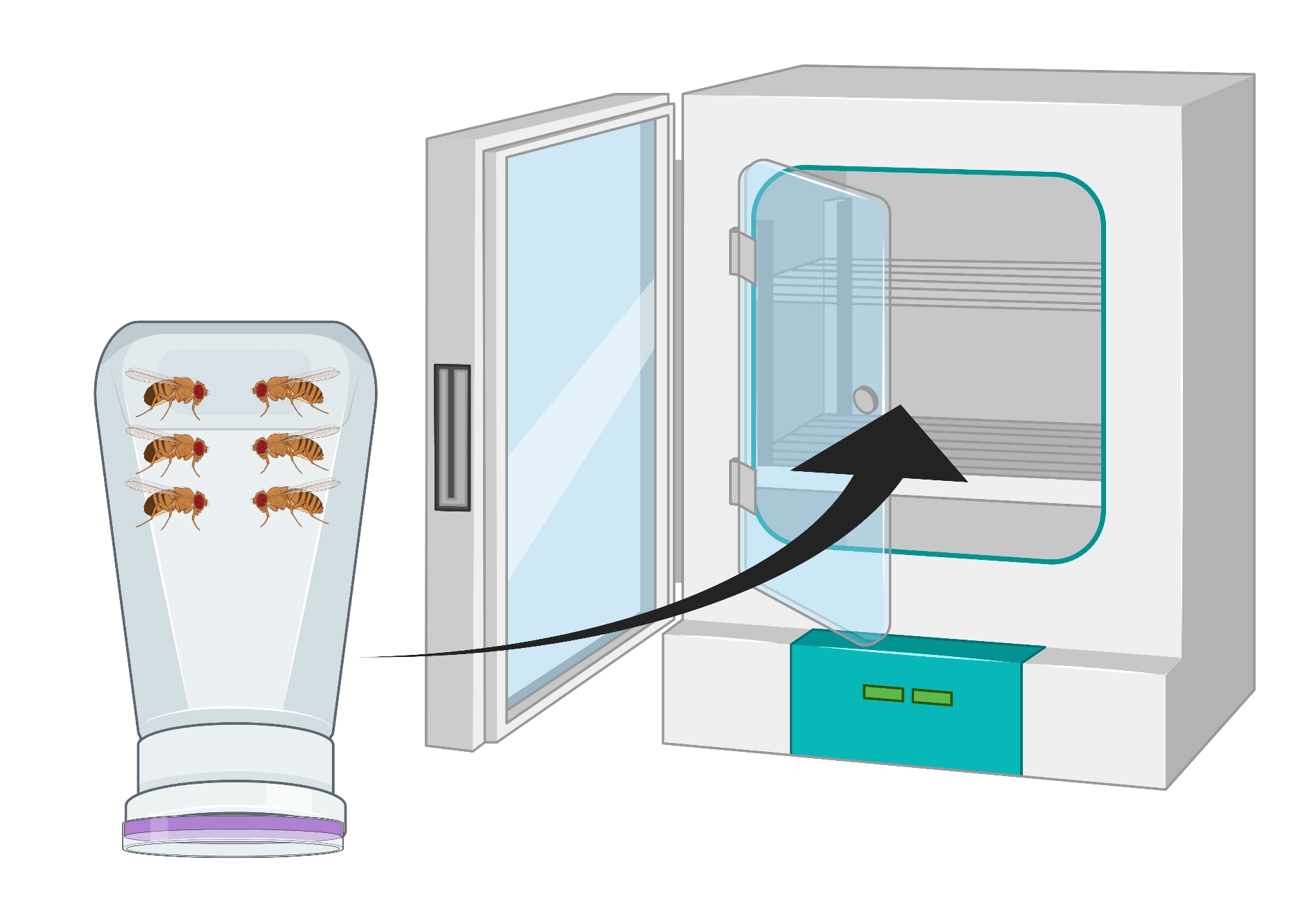
- Перенесите взрослых мух (~ 100 OregonR или любого генотипа) в кондоминиум мух и покройте кондоминиум тарелкой виноградного агара, увенчанной дрожжевой пастой. Поместите мазок к центру пластины и прикрепите пластину к кондоминиуму с помощью лабораторной ленты.
- Переверните контейнер так, чтобы тарелка виноградного агара находилась на дне, и поместите его в инкубатор при температуре 25 °C на 24 ч (рисунок 2).

Рисунок 2: Визуальное представление перевернутой бутылки мухи (кондоминиум) с самцами и самками взрослых дрозофил . Пластиковая бутылка имеет небольшие проколы, генерируемые иглой 18 Г, для кислородного обмена. Горлышко бутылки запечатано крышкой из виноградного сока агара и перевернуто и хранится в инкубаторе при температуре 25 °C. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}
- Через 24 ч поменяйте тарелку виноградного агара и замените ее новой тарелкой, увенчанной дрожжевой пастой. Быстро переключайте две тарелки, непрерывно постукивая по кондоминиуму слегка на скамейке, чтобы взрослые мухи не убежали.
- Осмотрите пластинку на глаз и оцените количество эмбрионов на пластине. Эмбрионы дрозофилы продолговатые и белые с двумя нитевидными придатками.
- Если на тарелке очень мало эмбрионов (менее 100), выбросьте пластину (соскоблите агар в мусорное ведро и сохраните пластиковую крышку для повторного использования). Во многих случаях взрослые самки не будут откладывать много эмбрионов в первую ночь в новой квартире. Если это так, дайте взрослым мухам еще 24 часа, чтобы акклиматизироваться.
- Если на пластине большое количество эмбрионов (не менее 100), сохраните ее, и аккуратно удалите дрожжевую пасту с помощью шпателя с плоским дном.
- После того, как дрожжевая паста удалена, используйте металлическую кирку, чтобы вручную удалить все личинки с виноградной пластины под рассекающим микроскопом. При взгляде на пластинку под рассекающим микроскопом следует наблюдать ползание личинок, а также эмбрионы.
- Удалите все личинки, расчесав металлическую кирку в сторону одной личинки. Личинки липкие и будут прилипать к кирке. Как только одна личинка окажется на кирке, дополнительные личинки можно легко подобрать, используя личинку на инструменте, чтобы прикрепить больше.
ПРИМЕЧАНИЕ: Личинки любят прилипать друг к другу. В этот момент не имеет значения, повреждаются личинки. Эти личинки будут выброшены. - После сбора и удаления всех личинок поместите пластину обратно в инкубатор при температуре 25 °C. Убедитесь, что пластина помещена в контейнер большего размера, который можно запечатать. Поместите влажные бумажные полотенца на дно большего контейнера, чтобы сохранить влагу.
- Через 30-60 мин отнесите пластинку обратно в рассекающий микроскоп и теперь тщательно соберите ~ 20-25 личинок из той же пластины виноградного агара, чтобы убедиться, что собранные личинки только что вылупились в течение 30-60 минут времени.
- Погрузите кончик инструмента с 20-25 свежевылупившимися личинками в чашку Петри (60 мм), заполненную 1-2 мл 1x фосфатного буферизованного физиологического раствора (PBS) в течение 2 минут.
- Через 2 мин опрокидывайте блюдо под углом, чтобы жидкость распылялась на дне. Используя небольшую кисть, вычистите личинок из жидкости вверх по дну чашки Петри.
- Соберите все личинки на кисти и перенесите личинок в новую чашку Петри (60 мм), содержащую 1-2 мл 70% этанола. Повторите шаги 1.15, чтобы собрать личинок кистью и перенести их в новую чашку Петри с 1-2 мл 1x PBS.
2. Питательные среды и инструментальная подготовка
- Опрыскайте скамейку и рабочую зону 70% этанолом и дайте высохнуть.
- Распылите инструменты для рассечения, щипцы и две стеклянные посуды для часов с 70% этанолом и дайте им высохнуть на скамейке.
- Сделайте дополнительный носитель Шнайдера (SSM, таблица 2) и поместите его на лед.
- Пипетка 1 мл SSM в каждую стеклянную посуду для часов.
- Используя микропипетку со стерильным наконечником, перенесите свежевылупившихся личинок с пластины PBS в SSM в первой стеклянной посуде для часов. Используя микропипетку со стерильным наконечником, перенесите свежевылупившихся личинок в SSM во второй стеклянной посуде для часов.
3. Вскрытия и культуры мозга
- Как только личинки окажутся во второй стеклянной посуде с SSM, рассекайте мозг из личинок с помощью щипцов и рассекающего микроскопа. При необходимости отрегулируйте увеличение.
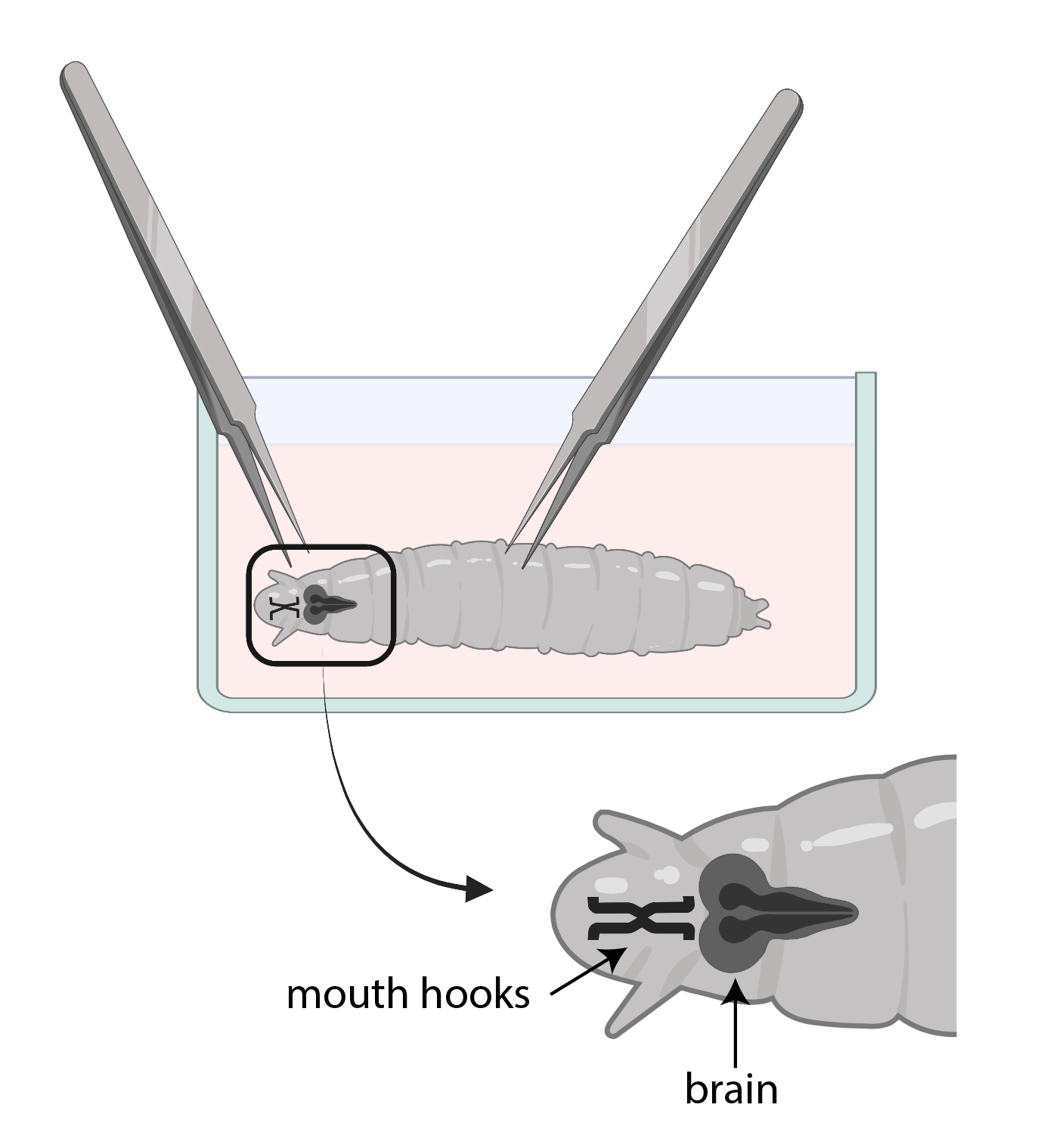
- Используйте один щипц, чтобы схватить рот крючками, а с другим осторожно схватите тело на полпути вниз и потяните в противоположном направлении (рисунок 3), чтобы разделить личинку на две части.
ПРИМЕЧАНИЕ: Мозг будет расположен прямо за крючками рта. Обратите внимание, что могут быть другие ткани, окружающие мозг. Будьте очень осторожны при удалении этих тканей, так как это может привести к повреждению мозга.

Рисунок 3: Личинки дрозофилы в стеклянной посуде для часов с SSM. Щипцы правильно позиционируются для рассечения. Расположение личиночного мозга (темно-серый) находится позади ротовых крючков (черный), и оба показаны внутри личинки. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}
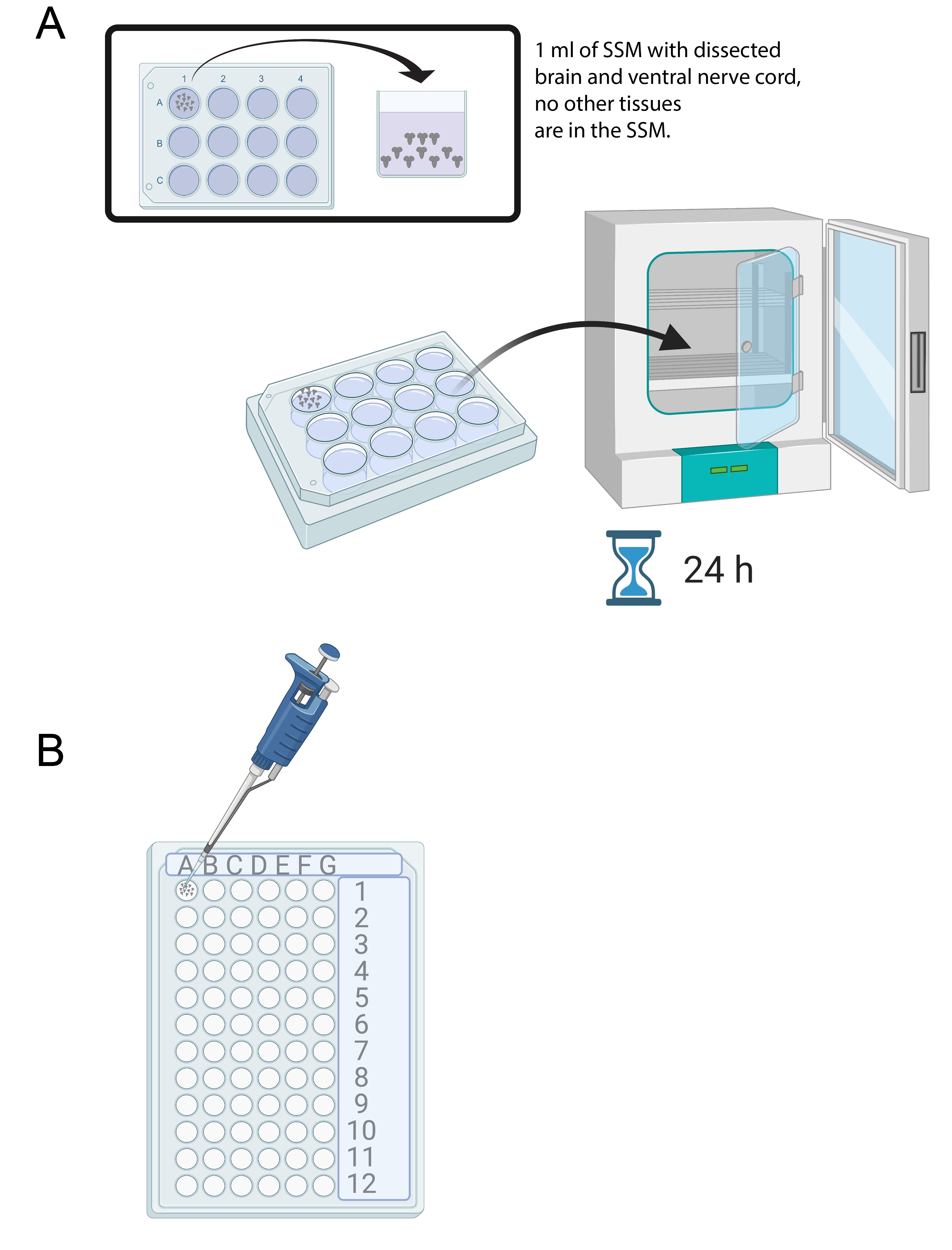
- После вскрытия 15-20 мозгов добавьте 1 мл SSM в один колодец стерильного 12-луночного культурального лотка. Перенесите свежерассеченный мозг в SSM с помощью микропипетки и стерильного наконечника (рисунок 4A).
- Поместите мозг в среду SSM в 12-луночный культуральный лоток в инкубатор при 25 °C в течение 24 ч (рисунок 4A).

Рисунок 4: Культура мозга и иммуноокрашивание. (А) Весь мозг в 12-луночной культуральной чашке, содержащей 1 мл SSM. Затем чашку для культивирования помещают в инкубатор с температурой 25 °C на 24 ч. (B) 72-луночный мини-лоток, в котором хранятся экспланты мозга во время иммуноокрашивания. Мозги промывают, а растворы переносят с помощью микропипетки P20, установленной на 10 мкл. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}
4. Анализ пролиферации, фиксация мозга и окрашивание антител
- На следующий день делают 1 мл раствора EdU SSM. Пипетка 10 мкл запаса 5-этинил-2'-дезоксиуридина (ЭдУ) с 990 мкл SSM (конечная концентрация EdU равна 0,1 мМ) в стерильную микроцентрифужную трубку и смешивают. После завершения 24-часовой инкубации пипетку 1 мл EdU SSM в один колодец стерильного 12-луночного культурального лотка.
- Перенесите мозги с помощью микропипетки со стерильным наконечником из скважины, содержащей SSM, только в новую скважину, содержащую раствор EdU SSM. Инкубировать в течение 1 ч при 25 °C.
- Затем перенесите меченый EdU мозг в другую лунку в том же культуральном лотке, содержащем 1 мл фиксатора (4% параформальдегида, см. Таблицу 3 для рецепта) в течение 20 мин.
ВНИМАНИЕ: Параформальдегид представляет собой биологическую опасность и должен быть утилизирован должным образом. - После фиксации быстро перенесите мозги в колодцы 72-луночного мини-лотка с помощью микропипетки. Каждая скважина может вместить 10 мозгов и 10-15 мкл жидкости (рисунок 4B). После того, как мозг переносится в мини-лоток (не более 10 мозгов на лунку), удалите фиксацию и промывайте мозг 3 раза в 10 мкл 1x PBT (фосфатный буфер, рН 7,4, содержащий 0,1% Triton-X 100).
ПРИМЕЧАНИЕ: Промывание означает пипетирование 10 мкл 1x PBT на мозг, удаление и повторение 3 раза. - Далее промыть мозги 3 раза по 10 мин каждый, снова в 10 мкл 1x PBT. Убедитесь, что мозг всегда покрыт жидкостью.
- После того, как промывания завершены, пипетка 10 мкл блокирующего раствора (1x PBT с 10% нормальной козьей сывороткой) на мозг. Накройте лоток и запечатайте его полоской парапленки по краю.
- После запечатывания поместите мини-лоток в герметичный ящик с влажными полотенцами, чтобы обеспечить влажную среду для предотвращения испарения. Поместите коробку с лотком при температуре 4 °C на ночь.
- На следующий день внесите первичный раствор антител.
ПРИМЕЧАНИЕ: В этом протоколе анти-каракули кролика использовались для маркировки клеточных мембран и крысиного анти-мертвеца для маркировки нейробластов, хотя можно было использовать любое количество других первичных антител.- Чтобы сделать раствор первичного антитела, сначала делают разведения первичных антител в блокирующий раствор. Например, кроличьи анти-каракули используют в конечной концентрации 1:1000. Поэтому сначала разводят антикараковое антитело кролика на 1:100 (1 мкл антитела плюс 99 мкл блокирующего раствора). Крысиный мертвец используется в конечной концентрации 1:100. Поэтому сначала разводят антитело крысиного мертвеца в 1:10 (1 мкл антитела плюс 9 мкл блокирующего раствора).
ПРИМЕЧАНИЕ: Эти разведения могут храниться в течение длительного времени при 4 °C, если азид натрия (0,05%) также добавляется для ингибирования роста бактерий. - Далее подсчитайте количество лунок, которые содержат мозги. Количество лунок определяет объем первичного раствора антител, который необходимо сделать. Например, если имеется 2 скважины мозга, приготовьте 20 мкл раствора первичного антитела (на 10 лунок, 100 мкл и т.д.). Чтобы получить раствор первичного антитела объемом 20 мкл, добавляют 2 мкл каждого первичного разведения антител и 16 мкл блокирующего раствора.
ПРИМЕЧАНИЕ: Конечная концентрация каждого из первичных антител составляет 1:1000 и 1:100 соответственно. Короче говоря, сделайте первое разведение первичных антител в концентрации, чтобы второе разведение всегда составляло 1:10, чтобы достичь соответствующих конечных концентраций. В этом случае 1:1000 для кролика против каракулей и 1:100 для крысиного анти-мертвого.
- Чтобы сделать раствор первичного антитела, сначала делают разведения первичных антител в блокирующий раствор. Например, кроличьи анти-каракули используют в конечной концентрации 1:1000. Поэтому сначала разводят антикараковое антитело кролика на 1:100 (1 мкл антитела плюс 99 мкл блокирующего раствора). Крысиный мертвец используется в конечной концентрации 1:100. Поэтому сначала разводят антитело крысиного мертвеца в 1:10 (1 мкл антитела плюс 9 мкл блокирующего раствора).
- Удаляют блокирующий раствор с микропипеткой, установленной на 10 мкл, и пипеткой 10 мкл раствора первичных антител в каждую лунку.
- Накройте и запечатайте лоток с помощью парапленки и поместите его обратно в герметичный ящик влажными полотенцами. Инкубировать в течение ночи при 4 °C.
ПРИМЕЧАНИЕ: Встряхивание не требуется и настоятельно не рекомендуется. Антитела проникнут в мозг без тряски или смешивания. - На следующий день удалите раствор первичного антитела с помощью микропипетки и промывайте мозг 3 раза 10 мкл 1x PBT. Затем промыть мозги 4 раза 10 мкл 1x PBT в течение 10 мин каждый. В течение 10 мин промывания готовят раствор вторичных антител.
- Чтобы сделать раствор вторичных антител, выбирайте вторичные антитела, которые распознают первичные антитела. В этом протоколе использовались козья анти-кролик Alexa Fluor 488 и козья анти-крыса Alexa 555.
- Пипетка 1 мкл каждого из вторичных антител в микроцентрифужную трубку с 298 мкл блокирующего раствора для получения конечной концентрации 1:300 для каждого вторичного антитела.
- После последних 10 мин промывки удалите 1x PBT и пипетку 10 мкл раствора вторичных антител в каждую лунку. Запечатайте лоток с помощью парапленки и поместите его обратно в коробку с влажными полотенцами. Инкубировать в течение ночи при 4 °C.
ПРИМЕЧАНИЕ: Не беспокойтесь об удалении каждого последнего мкл в лунках между полосканиями, промывками или при добавлении первичных и вторичных растворов антител. Мозг всегда будет оставаться погруженным в несколько мкл жидкости, что просто прекрасно. - На следующий день удалите раствор вторичного антитела с помощью микропипетки и промывайте мозг 3 раза по 10 мкл каждый из 1x PBT. Затем промыть мозги 4 раза по 10 мкл из 1x PBT в течение 10 минут каждый.
- В течение 10 минут промывки приготовьте реакционную смесь EdU для обнаружения включения EdU. Приготовьте реакционную смесь EdU в соответствии с рекомендациями производителя.
- После окончательной промывки удалите 1x PBT и пипетку 10 мкл реакционной смеси EdU в каждую лунку с мозгами. Запечатайте пластину микролунки парапленкой и накройте алюминиевой фольгой. Оставьте тарелку на скамейке на 30 мин.
- Через 30 мин промыть мозги 3 раза по 10 мкл из 1x PBT и промыть мозги 3 раза по 10 мкл из 1x PBT в течение 5 минут каждый.
- После последней стирки извлеките 1 pBT и пипетку объемом 10 мкл из монтажного материала на основе глицерина. Запечатайте пластину и поместите при температуре 4 °C на ночь.
5. Монтаж и визуализация мозга
- На следующий день подготовьте слайды микроскопа (25 мм x 75 мм x 1 мм): Приклейте (например, суперклей) одно квадратное покрытие 22 мм x 22 мм x 1 мм квадратное покрытие на каждый конец слайда микроскопа, чтобы создать «мост», над которым будет размещена большая крышка размером 22 мм x 50 мм x 1 мм, чтобы создать пространство между слайдом и большим чехлом (рисунок 5A). Это пространство позволит мозгу достаточно двигаться, чтобы быть правильно ориентированным, предотвращая его раздавливание.

Рисунок 5: Схема, показывающая слайд микроскопа, ориентацию и типы клеток в личиночном мозге. (A) Визуальное представление слайда микроскопа, на котором установлен личиночный мозг и готов к получению изображения. (B) Показано также, что руководство используется для ориентации тканей. (C) Слайд микроскопа, готовый к визуализации на конфокальном микроскопе. (D) Карикатура, показывающая некоторые типы клеток в личиночном мозге. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}
- После приклеивания покровных стекол размером 22 мм x 22 мм x 1 мм к предметному стеклу микроскопа, пипетке 9,3 мкл монтажного медиа-раствора на основе глицерина, содержащего один мозг из колодца пластины микроэлемента, и поместите его в центр слайда (рисунок 5A).
ПРИМЕЧАНИЕ: Личиночный мозг может прилипать к кончику пипетки, поэтому будьте осторожны. Чтобы избежать адгезии, начните с аспирации антитемения, а затем аспирации одного мозга к концу объема 9,3 мкл. - Как только мозг окажется на слайде, аккуратно поместите сверху крышку размером 22 мм x 50 мм x 1 мм. Позиционируйте мозг, как показано на рисунке 5B. Осторожно перемещайте покрывало, чтобы сориентировать мозг. После этого образец готов к визуализации.
- Используйте конфокальный микроскоп, оснащенный большим увеличением и объективом с высокой числовой диафрагмой (рисунок 5C), чтобы получить лучшие изображения. Например: 60x или 63x, 1.4 NA масляный погружной объектив.
- Представьте себе мозг с дорсальной поверхностью, ближайшей к покровному листу (и объективу). Приобретите Z-стеки через все полушарие мозга, начиная с вентральной поверхности (наиболее удаленной от цели) с интервалом 1 мкм или размером шага Z.
ПРИМЕЧАНИЕ: Используемые лазеры зависят от вторичных антител. В этом протоколе использовались лазерные линии 488 нм для обнаружения окрашивания Scribble, 555 нм для обнаружения Deadpan и 633 нм для обнаружения EdU.
- Представьте себе мозг с дорсальной поверхностью, ближайшей к покровному листу (и объективу). Приобретите Z-стеки через все полушарие мозга, начиная с вентральной поверхности (наиболее удаленной от цели) с интервалом 1 мкм или размером шага Z.
6. Анализ данных
- Используйте программное обеспечение с открытым исходным кодом Fiji для анализа полушарий мозга и используйте плагин счетчика клеток Fiji для подсчета клеток.
Результаты
Свежевылупившиеся мозги дикого типа OregonR были рассечены и культивированы в течение 24 ч в дополненной среде Шнайдера (SSM) инсулином. Ткани фиксировались и окрашивались согласно протоколу. Использовались первичные антитела, генерируемые против Deadpan (Dpn) для обнаружения NB и Scribble для маркир?...
Обсуждение
Описанный здесь способ культивирования эксплантов мозга может быть выполнен в большинстве лабораторных сред. Необходимые инструменты, а также процедура и сбор данных просты и понятны. С помощью этого метода можно проверить различные гипотезы, в том числе связанные с клеточными сигнал...
Раскрытие информации
У авторов нет конкурирующих интересов.
Благодарности
Мы приветствуем программу LSAMP Bridges to Doctorate для финансирования (CNK), а также NIH / NIGMS (R01-GM120421 и R35-GM141886). Мы благодарны доктору Конору Сайпу за рисунок 1. Мы также благодарим всех членов лаборатории Siegrist за их постоянную поддержку и наставничество. Мы особенно благодарим Чхави Суда и Гэри Титерса за внимательное прочтение рукописи и за предоставленные комментарии.
Материалы
| Name | Company | Catalog Number | Comments |
| 10 µL Pipette tips | Denville Sci | P2102 | |
| 1000 µL Pipette tips | Denville Sci | P2103-N | |
| 1000 µL Pipettor | Gilson | P1000 | |
| 16% paraformaldehyde (10 x 10 mL) | Electron Microscopy Sciences | 2912.60.0000 | Used for Fixation of Larval Brains |
| 20 µL Pipette | Gilson | P20 | |
| 200 µL Pipette tips | Denville Sci | 1158U56 | |
| 24-well multiwell culture plates | Fisher Scientific | 50-197-4477 | |
| 35 mm Petri dishes | Fisher Scientific | 08-757-100A | Grape Plate Ingredients |
| 4 °C refrigerator | Fisher Scientific | Provides an ideal temperature for >24 h incubations in antibody solution | |
| 63x Objective | Lecia | ||
| Active dry yeast | Most supermarkets | ||
| Agarose | Fisher Scientific | 214010 | Grape Plate Ingredients |
| Click-iT EdU Cell Proliferation Kit for Imaging, Alexa Fluor 647 dye | Thermo Fisher Scientific | C10340 | to label proliferating cells |
| Confocal Microscope | Leica | SP8 | |
| Coverslips 22 mm x 22 mm x 1 mm , 10 pack of 4 oz | Fisher Scientific | 12-544-10 | Two Coverslips are super glued to the ends of the microscope slide. This creates a space that allows for the brains to float in antifade while being imaged. |
| Coverslips, 22 mm x 50 mm x 1 mm | Fisher Scientific | 12-545E | The coverslip is placed on two square coverslips on the microscope slide ensuring that the brain in the antifade does not move while imaging. |
| Dissecting microscope | Zeiss | Stemi 2000 | |
| Ethanol 200 proof (100%), Decon Labs, 1 gallon bottle | Fisher Scientific | 2701 | Used to wash off the larvae before the 24 hr hold in culture medium |
| Fetal Bovine Serum (10%) | Sigma | F4135-100ML | Supplement for cell culture media. |
| Fine forceps for dissection | Fine Science Tools | 11295-20 | Forcepts used in disections. They work best when sharpened. |
| Fly Bottles for Crossing | Genessee Scientific | 32-130 | This bottle is used as a container that lets the flies lay eggs on the grape plate. |
| Glass Dissection Dish (3 well) | These are no longer available | ||
| Glutathione | Sigma | G6013 | Provides oxidative protection during cell culture. |
| Goat Serum | Sigma | G9023- 10ML | Blocking Agent |
| Grape Plates | Made in house | Made in house | Grape juice/agarose plates for collecting freshly hatched eggs |
| Image J | Imagej.net/fiji/downloads | Free Download: https://fiji.sc | Imaging platform that is used to count cells and Edu reactivation |
| Incubator | Thermo Fisher Scientific | Ensures that the temperature, humidity, and light exposure is exactly the same throughout experiment. | |
| Insulin | Sigma | I0516 | Independant variable of the experiment |
| Laminar flow hood | For aliquoting culture media | ||
| L-Glutamine | Sigma | G7513 | Provides support during cell culture |
| Nunc 72-well Microwell Mini Trays | Fisher Scientific | 12-565-154 | Immunostaining steps are performed in this tray |
| Parafilm | Fisher Scientific | S37440 | Film used to seal plates in order to prevent evaporation |
| Pen-Strep | Sigma | P4458-100ml | Antibiodics used to prevent bacterial contamination of cells during culture. |
| Phosphate Buffer, pH7.4 | Made in house | Made in house | Solvent used to wash the brains after fixing and staining steps |
| Pick | Fine Science Tools | 10140-01 | Used to pick larvae off of the grape plate |
| Propionic acid | Fisher Scientific | A-258 | Grape Plate Ingredients |
| Rabbit 405 | Abcam | ab175653 | Antibodies used for immunostaining |
| Rat 555 | Abcam | ab150166 | Antibodies used for immunostaining |
| Rb Scribble | A Gift from Chris Doe | Antibodies used for immunostaining | |
| Rt Deadpan | Abcam | ab195173 | Antibodies used for immunostaining |
| Schneiders Culture Medium | Life Tech | 21720024 | Contains nutrients that help the cells grow and proliferate |
| SlowFade Diamond Antifade (5 x 2 mL) | Life Tech | S36963 | Reagent that provides protection against fading fluorophores |
| Sterile Water | Autoclave Milli-Q water made in house | Needed for Solutions | |
| Sucrose | Fisher | S2-12 | Grape Plate Ingredients |
| Superfrost Microscope Slides | Fisher Scientific | 12-544-7 | |
| Superglue | Most supermarkets | ||
| Tegosept | Genesee Scientific | 20-259 | Grape Plate Ingredients |
| Triton-X 100 | Sigma | T9284-100ML | PBT |
| Welch's 100% grape grape juice | Most supermarkets | Grape Plate Ingredients |
Ссылки
- Suman, S., Domingues, A., Ratajczak, J., Ratajczak, M. Z. Potential clinical applications of stem cells in regenerative medicine. Advances in Experimental Medicine and Biology. 1201, 1-22 (2019).
- Tabar, V., Studer, L. Pluripotent stem cells in regenerative medicine: challenges and recent progress. Nature Reviews Genetics. 15, 82-92 (2014).
- Daley, G. Q. Stem cells and the evolving notion of cellular identity. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. 370, 20140376 (2015).
- Rodrigues, M., Kosaric, N., Bonham, C. A., Gurtner, G. C. Wound healing: A cellular perspective. Physiological Reviews. 99, 665-706 (2019).
- van Velthoven, C. T. J., Rando, T. A. Stem cell quiescence: Dynamism, restraint, and cellular idling. Cell Stem Cell. 24, 213-225 (2019).
- Chapman, N. M., Boothby, M. R., Chi, H. Metabolic coordination of T cell quiescence and activation. Nature Reviews Immunology. 20, 55-70 (2020).
- Wosczyna, M. N., Rando, T. A. A muscle stem cell support group: Coordinated cellular responses in muscle regeneration. Developmental Cell. 46, 135-143 (2018).
- Homem, C. C., Knoblich, J. A. Drosophila neuroblasts: a model for stem cell biology. Development. 139, 4297-4310 (2012).
- Kang, K. H., Reichert, H. Control of neural stem cell self-renewal and differentiation in Drosophila. Cell and Tissue Research. 359, 33-45 (2015).
- Chell, J. M., Brand, A. H. Nutrition-responsive glia control exit of neural stem cells from quiescence. Cell. 143, 1161-1173 (2010).
- Sousa-Nunes, R., Yee, L. L., Gould, A. P. Fat cells reactivate quiescent neuroblasts via TOR and glial insulin relays in Drosophila. Nature. 471, 508-512 (2011).
- Britton, J. S., Edgar, B. A. Environmental control of the cell cycle in Drosophila: nutrition activates mitotic and endoreplicative cells by distinct mechanisms. Development. 125, 2149-2158 (1998).
- Lin, S., et al. Extremes of lineage plasticity in the Drosophila brain. Current biology : CB. 23, 1908-1913 (2013).
- Sipe, C. W., Siegrist, S. E. Eyeless uncouples mushroom body neuroblast proliferation from dietary amino acids in Drosophila. Elife. 6, 26343 (2017).
- Speder, P., Brand, A. H. Systemic and local cues drive neural stem cell niche remodelling during neurogenesis in Drosophila. Elife. 7, 30413 (2018).
- Yuan, X., Sipe, C. W., Suzawa, M., Bland, M. L., Siegrist, S. E. Dilp-2-mediated PI3-kinase activation coordinates reactivation of quiescent neuroblasts with growth of their glial stem cell niche. PLoS Biology. 18, 3000721 (2020).
- Colombani, J., et al. A nutrient sensor mechanism controls Drosophila growth. Cell. 114, 739-749 (2003).
- Geminard, C., Rulifson, E. J., Leopold, P. Remote control of insulin secretion by fat cells in Drosophila. Cell Metabolism. 10, 199-207 (2009).
- Siller, K. H., Serr, M., Steward, R., Hays, T. S., Doe, C. Q. Live imaging of Drosophila brain neuroblasts reveals a role for Lis1/dynactin in spindle assembly and mitotic checkpoint control. Molecular Biology of the Cell. 16, 5127-5140 (2005).
- Prithviraj, R., Trunova, S., Giniger, E. Ex vivo culturing of whole, developing Drosophila brains. Journal of Visualized Experiments: JoVE. (65), e4270 (2012).
- Bostock, M. P., et al. An immobilization technique for long-term time-lapse imaging of explanted drosophila tissues. Frontiers in Cell and Developmental Biology. 8, 590094 (2020).
- Datta, S. Activation of neuroblast proliferation in explant culture of the Drosophila larval CNS. Brain Research. 818, 77-83 (1999).
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеСмотреть дополнительные статьи
This article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены