
Method Article
Phage-mediated Delivery of Targeted sRNA Constructs to Knock Down Gene Expression in E. coli
In This Article
Summary
We describe a method to knock down gene expression in a growing population of E. coli cells using sequence-targeted sRNA expression cassettes delivered by an M13 phagemid vector.
Abstract
RNA-mediated knockdowns are widely used to control gene expression. This versatile family of techniques makes use of short RNA (sRNA) that can be synthesized with any sequence and designed to complement any gene targeted for silencing. Because sRNA constructs can be introduced to many cell types directly or using a variety of vectors, gene expression can be repressed in living cells without laborious genetic modification. The most common RNA knockdown technology, RNA interference (RNAi), makes use of the endogenous RNA-induced silencing complex (RISC) to mediate sequence recognition and cleavage of the target mRNA. Applications of this technique are therefore limited to RISC-expressing organisms, primarily eukaryotes. Recently, a new generation of RNA biotechnologists have developed alternative mechanisms for controlling gene expression through RNA, and so made possible RNA-mediated gene knockdowns in bacteria. Here we describe a method for silencing gene expression in E. coli that functionally resembles RNAi. In this system a synthetic phagemid is designed to express sRNA, which may designed to target any sequence. The expression construct is delivered to a population of E. coli cells with non-lytic M13 phage, after which it is able to stably replicate as a plasmid. Antisense recognition and silencing of the target mRNA is mediated by the Hfq protein, endogenous to E. coli. This protocol includes methods for designing the antisense sRNA, constructing the phagemid vector, packaging the phagemid into M13 bacteriophage, preparing a live cell population for infection, and performing the infection itself. The fluorescent protein mKate2 and the antibiotic resistance gene chloramphenicol acetyltransferase (CAT) are targeted to generate representative data and to quantify knockdown effectiveness.
Introduction
RNA-mediated gene knockdowns proceed in two stages. First, an RNA molecule is introduced to a cell line or organism of study. Second, endogenous RNA-binding proteins facilitate RNA-target recognition and produce the silencing effect. All RNA knockdown technologies benefit from the customizable nature of synthetic sRNAs, which can be easily produced to match a specific target of interest. However, the molecular details of RNA uptake and silencing vary widely across model system, constraining where and how RNA knockdowns can be applied.
In nematodes, double-stranded RNA (dsRNA) molecules can be introduced directly in the media or by feeding the worms with a population of dsRNA-expressing E. coli cells1,2. In Drosophila, RNAi can be achieved by microinjecting embryos with dsRNA3, or implemented in cell lines by simply adding dsRNA to the culture medium4. In mammalian cell lines, synthetic small interfering RNAs (siRNAs) may be delivered to living cells by electroporation1,2,5, packaged in liposomes3,6, or expressed from DNA plasmid vectors4,7. Once the RNA species reaches the cytosol, the RNAi pathway relies on the RISC complex to process dsRNA, facilitate antisense recognition of the target, and catalyze translational repression, mRNA degradation, or heterochromatin formation, depending on the host.
Because of these requirements, classical RNAi can be performed only in organisms that take up exogenous RNA efficiently and express RISC or a RISC-like activity. Notably, this excludes the model bacterium E. coli, which lacks the RNAi pathway. However, recent advances in synthetic biology provide the tools to solve both the delivery problem and the silencing problem.
In this protocol, sRNA constructs are expressed in E. coli from a DNA vector delivered to living cells using the M13 phagemid/helper system. A phagemid is any plasmid with a phage-derived f1 origin of replication. A helper plasmid, in this case M13KO, carries all the machinery required to produce viral particles, but is not itself competent for replication and packaging. When a phagemid and helper plasmid are co-transformed, the phagemid alone is replicated at the f1 origin, packaged and secreted. The vectorized phagemid is then competent to infect live E. coli via the F pilus.
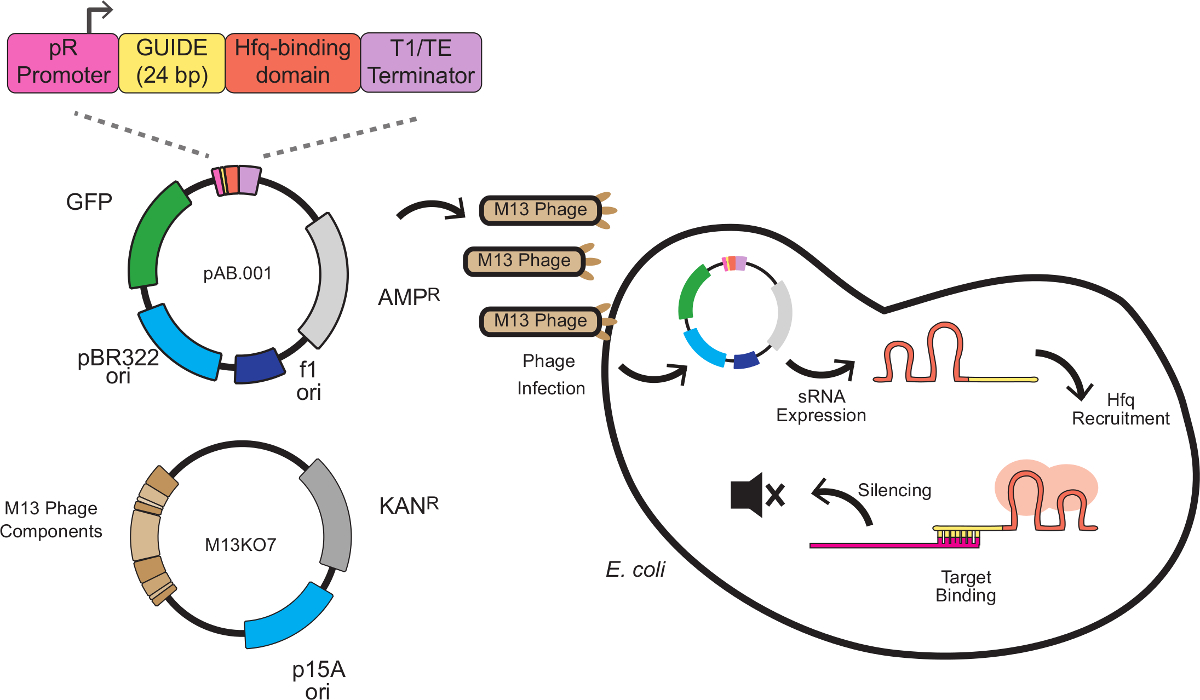
In this system, the silencing effect is produced by custom sRNA cassettes combining a scaffold sequence with a target-binding sequence. The target-binding sequence is 24 base pairs antisense to an mRNA target, typically at the ribosome-binding site (RBS). The scaffold sequence, developed by Na and colleagues8, contains an Hfq-binding motif extracted from MicC, a small regulatory RNA endogenous to E. coli. The Hfq protein stimulates RNA-RNA binding and mRNA degradation, serving a role in this system similar to RISC in RNAi. Figure 1 depicts a complete scheme for phage-mediated sRNA knockdowns, including the sRNA cassette structure, phagemid vectorization, and silencing mechanism.
As a method to modulate gene expression in E. coli, sRNA silencing is simple, rapid and versatile. The targeted E. coli is not burdened beyond propagating the phagemid and expressing the sRNA. This may be relevant in the context of synthetic biology or basic research where the expression of larger heterologous constructs can strain cellular resources9. Phagemids with new targets can be produced with a single PCR and harvested one day after phagemid transformation. Finally, nearly any mRNA can be targeted. The sRNA regulation cassette (on a standard plasmid) has been shown to work on a variety of targets in metabolism with typical repression levels >90%8.
This protocol reproduces and expands on earlier work using phagemid-born sRNA cassettes10. First, a packaged phagemid is introduced to a batch culture of E. coli cells and used to silence expression of the fluorescent protein mKate2. Subsequent fluorescence changes are monitored in real time. Second, knocking down the CAT gene is shown to reduce phenotypic chloramphenicol resistance on agar plates. In both cases, the phagemid itself carries a GFP marker, allowing infection rate to be measured independently of the knockdown efficiency.
Protocol
1. Design and Construction of Phagemid Vectors Bearing sRNA Silencing Cassettes
- De Novo Design of sRNA Silencing Cassettes8
- Identify the complete sequence of the mRNA to be silenced using a DNA sequence database. To generate the TARGET sequence, note the first 24 bp of coding sequence, from position +1 to +24 beginning with the start codon (e.g., ATG).
Note: Silencing is less efficient when other sites or segments of the mRNA are targeted8. - Take the reverse complement of the TARGET sequence to produce the GUIDE sequence for the sRNA cassette. See Table 1 for examples of TARGET and GUIDE sequences for chloramphenicol acetyltransferase (CAT).
- To design the 292 bp complete sRNA expression cassette, arrange the pR promoter, GUIDE sequence, Hfq protein binding domain and T1/TE transcriptional terminator sequences in series (Table 2).
- Add additional cloning sites of choice to facilitate cloning of the sRNA cassette into the target vector.
- Obtain the complete sRNA cassette through commercial gene synthesis or a similar method and clone it into any phagemid vector with a functional f1 replication origin11. See Supporting Information for the complete sequence of the final phagemid vector.
- Identify the complete sequence of the mRNA to be silenced using a DNA sequence database. To generate the TARGET sequence, note the first 24 bp of coding sequence, from position +1 to +24 beginning with the start codon (e.g., ATG).
- Altering the Sequence Targeted by an Existing sRNA Expression Cassette using PCR-based Site Directed Mutagenesis12
- Identify the 24 bp GUIDE sequence in an existing sRNA expression cassette. Note: The annotated pAB.001 plasmid, used in this work, is available as a supplemental sequence file.
- Design forward and reverse primers with short regions of homology to the existing sRNA cassette flanking the new 24 bp GUIDE sequence. Obtain the primers through commercial oligonucleotide synthesis.
Note: Primer design for site-directed mutagenesis is depicted in Figure 2. Exact primer template sequences are provided in Table 3. - Prepare a 5 ml culture of E. coli carrying the template sRNA expression phagemid. Grow the cells overnight at 37 °C with shaking, in LB media with appropriate antibiotics.
- Extract and purify the template sRNA expression phagemid from the 5 ml bacterial culture using a DNA miniprep kit or similar method12.
- Prepare two PCR reactions using the template sRNA expression phagemid and high fidelity polymerase, one with the forward and one with the reverse primer (Table 4). Use PCR conditions as recommended by the polymerase supplier (Table 5). Increase the template concentration to 10-50x higher than a standard reaction to account for the fact that the single primer reaction does not produce exponential amplification.
- Combine the two above PCR reactions in a microcentrifuge tube. Anneal the products by heating to 98 °C in a boiling water bath. Immediately after placing the microcentrifuge tube in the water bath, remove the heat source and allow the bath to slowly return to room temperature over 1-2 hr.
- To eliminate the unmutated template sRNA expression phagemid, add 1 µl. DpnI restriction enzyme to the mixture and incubate at 37 °C for 1 hr, or the time recommended by the manufacturer for complete digestion.
Note: DpnI digests only methylated target sites, which are present on host-replicated phagemids but not PCR products. - Transform purchased or prepared13 chemically competent E. coli with 1-5 µl of the annealed PCR product. Isolate single colonies of the transformed strain by selective plating on LB agar plates containing appropriate antibiotics.
- To verify the incorporation of the correct GUIDE sequence, screen the resulting colonies by colony PCR. Using a 200 µl pipette tip, collect a small quantity of cells from a single transformed colony. Mark and preserve the original colony for downstream use after verification.
- Add the collected cells to 50 µl of nuclease-free water in a microcentrifuge tube. Mix by pipetting up and down.
- Using a bench-top thermocycler or a boiling water bath, lyse the cells by heating to 95 °C for 2 min.
- PCR-amplify the phagemid region using 1 µl of heat-lysed cells as a DNA template. PCR conditions and thermocycler protocols are provided in Tables 6 and 7. See the supplemental pAB.001 sequence file for verification primer sequences.
- Sequence the PCR product to verify incorporation of the correct GUIDE sequence.
- Inoculate a 5 ml culture of the E. coli clone carrying the sequence-verified sRNA expression phagemid. Grow the cells overnight at 37 °C with shaking, in selective LB media.
- Prepare a glycerol stock of the sequence-verified clone. Add 750 µl of the overnight culture to 250 µl of 60% glycerol in a screw-cap cryo tube.
- Store the glycerol stock at -80 °C indefinitely. The remainder of the overnight culture may be used as a source for the sRNA expression phagemid in step 2.
2. Production and Harvest of M13-packaged Phagemid Stocks
- Prepare a 5 ml culture of E. coli carrying the sRNA expression phagemid. Grow the cells overnight at 37 °C with shaking, in LB media with appropriate antibiotics. Note: The sRNA expression phagemid may be obtained through de novo cloning as described in step 1.1.5, or modified from an existing phagemid and harvested in step 1.2.16.
- Similarly prepare a 5 ml culture of E. coli carrying the M13KO7 helper plasmid. Grow the cells overnight at 37 °C with shaking, in selective LB media.
- Extract and purify the sRNA expression phagemid and helper plasmid using a DNA extraction kit or similar method12.
- Cotransform purchased or prepared13 chemically competent E. coli with 1 µl each of sRNA expression phagemid and helper plasmid. Select for cotransformants by plating on LB agar with selective antibiotics for both constructs.
- Prepare a 10 ml culture from a single colony of the cotransformed strain in LB with selective antibiotics. Incubate at 37 °C with shaking for 8-12 hr or overnight.
- Centrifuge the culture at 3,300 x g for 10 min. Collect the supernatant and filter through a 0.2 µm filter. Caution: In case of media spill, clean the area with dilute bleach (0.5%) to destroy infective phage particles.
- Store the packaged phagemid filtrate at 4 °C. Note: Samples can be maintained for days to weeks without loss of activity.
3. Preparation of F+ Target Cells for Silencing
- Determine if the cells to be targeted for silencing express the F pilus14. If the F pilus is already present, proceed to step 4.
Note: Common laboratory strains of E. coli are annotated as F+ or F' to indicate the presence of the F pilus in their genome or on a plasmid. - Obtain an F+ strain of E. coli such as TOP10F'.
Note: Ensure that the target strain carries a unique resistance marker in order to be separated from the F-plasmid donor after conjugation. - To introduce F pilus by conjugation with a F+ strain, prepare 5 ml cultures of both the target strain and F-plasmid donor14. Grow the cells overnight at 37 °C with shaking, in LB media with appropriate antibiotics.
- The following day, dilute both strains 1:100 in 5 ml of selective LB and continue culturing at 37 °C with shaking.
- Determine the growth phase of the cells by measuring the optical density of the culture at 600 nm (OD600) using a benchtop spectrophotometer. Culture the cells for about 2 hr until an OD600 of 0.3 is attained, indicating log-phase growth15.
- Prepare 3 conjugation reactions in microcentrifuge tubes: 0.5 ml F-plasmid donor + 0.5 ml target strain, 0.5 ml F-plasmid donor + 0.5 ml LB media (negative control) and 0.5 ml target strain + 0.5 ml LB media (negative control). Allow the conjugation to proceed for 2 hr at 37 °C with shaking.
- Plate 100 µl of each conjugation reaction on selective LB agar with antibiotics specific to the F-plasmid (typically tetracycline) and target strain. Plate the negative control reactions to confirm that neither donor or recipient strain express both antibiotic resistances.
4. Infection with Packaged Phagemids for Silencing
- Inoculate a single colony of F+ target cells into a 5 ml culture of LB media with appropriate antibiotics. Incubate overnight at 37 °C with shaking.
- The following day, dilute the F+ target cells 1:100 in 5 ml of selective LB media and continue to culture at 37 °C with shaking.
- Determine the growth phase of the cells by measuring the optical density of the culture at 600 nm (OD600) using a benchtop spectrophotometer. Culture the cells for about 2 hr until an OD600 of 0.3 is attained, indicating log-phase growth15. Note: Expression of the F pilus and infection efficiency is highest in log phase.
- Add the M13-packaged phagemids (from step 2.6) to the target cells in a volume ratio of 1:100 to achieve nearly 99% infection of the target population. Allow the infection to proceed at 37 °C with shaking for 30-60 min.
- Assay the sRNA-silencing phenotype according to the method of choice.
Note: For a fluorescent protein target, the silencing effect can be quantified directly by fluorometry10. Alternately, phenotypic assays may be used to observe the phenotypic consequences of gene knockdown8. - Prepare a glycerol stock for the sRNA-expressing phagemid host following steps 1.2.14-1.2.16. Note: the phagemid will propagate in the host strain indefinitely and can be maintained with antibiotic similar to a conventional plasmid.
Results
Silencing of mKate2 Fluorescence in Liquid Media
Figure 1 depicts the scheme for sRNA-mediated knockdowns described in this work, including the sRNA cassette design, phagemid vectorization, and silencing mechanism. Following protocol 1.2, the sRNA silencing cassette of plasmid pAB.001 was altered to target mKate. The sRNA cassette was synthetized and cloned into phagemid Litmus28i_J23115-B0032-GFP, a gift from Monica Ortiz and Drew Endy11. This phagemid carries markers for GFP expression and kanamycin resistance, allowing successful infections to be tracked. Packaged phagemid stocks were prepared following protocol 2.
A derivative of E. coli K12 MG1655 carrying a constitutively expressed, chromosomally integrated mKate2 marker was prepared for phage infection by conjugation with an F plasmid donor strain following protocol 3. The cells were grown to mid log phase and the phagemid introduced following protocol 4. After phagemid infection, 200 μl cultures were transferred to a fluorescence plate reader and fluorescence was monitored continuously for 24 hr.
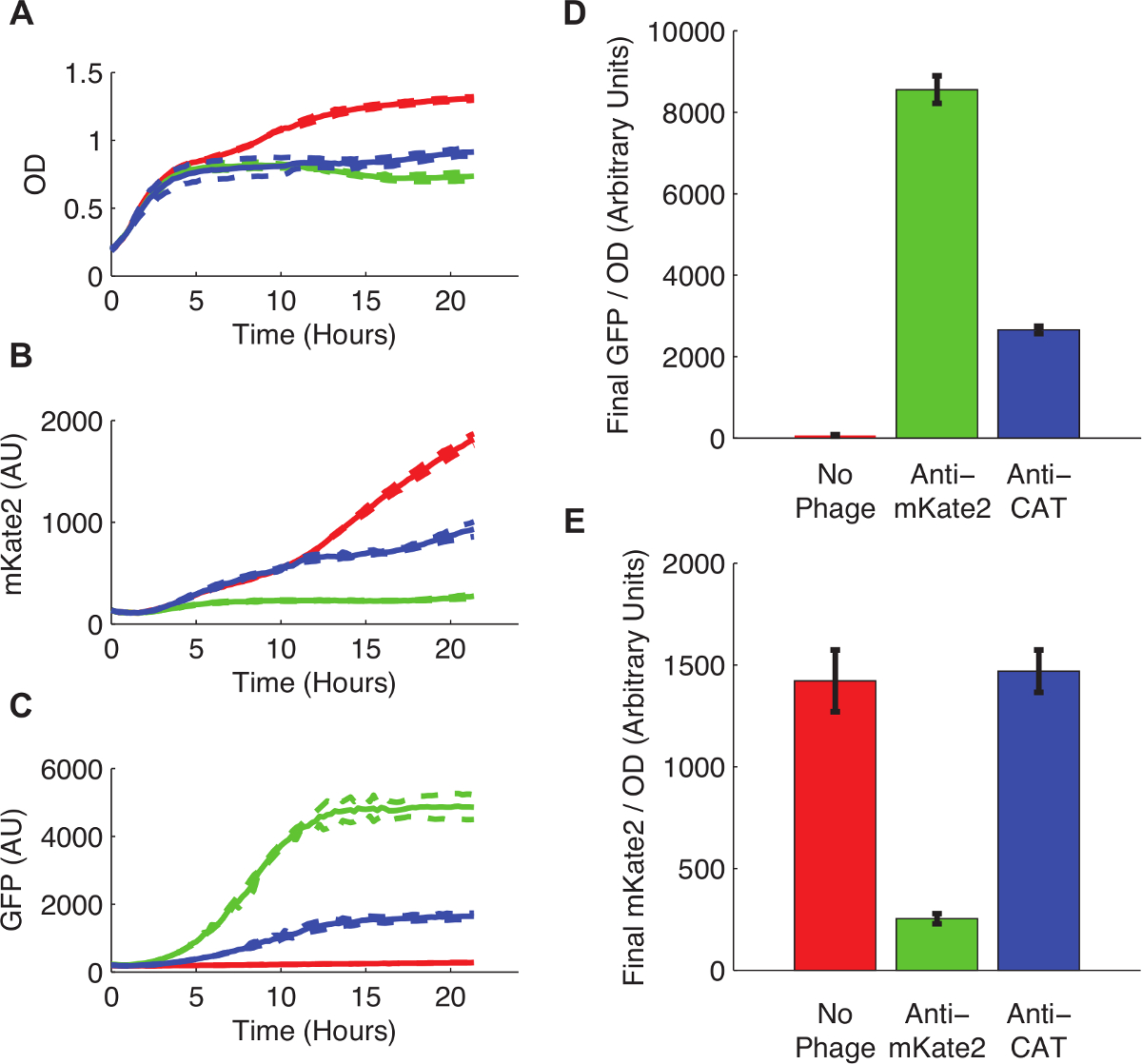
Figure 3 shows the effect of sRNA-mediated silencing on mKate2 expression. The strain infected with an anti-mKate2 phagemid showed no detectable mKate2 fluorescence over background. In contrast, this strain did express the GFP marker, indicating successful uptake of the phagemid. Uninfected control cells produced mKate2 fluorescence but not GFP. An additional control, in which the anti-mKate2 targeting domain was replaced with a sequence targeting CAT, had no effect on mKate2 fluorescence.
Silencing of Chloramphenicol Resistance on Agar Plates
Following protocol 1.1, an sRNA silencing cassette was produced to target CAT. A derivative of E. coli K12 MG1655 carrying a constitutively expressed, chromosomally integrated CAT marker was prepared for phage infection by conjugation with an F-plasmid donor strain following protocol 3. The cells were grown to mid log phase and the phagemid introduced following protocol 4. After 1 hr of incubation at 37 °C, infected cells were serially diluted and plated at a range of chloramphenicol concentrations. Plates were incubated overnight, and the proportion of resistant cells at each chloramphenicol concentration was determined by counting colony forming units (CFUs) the next day.
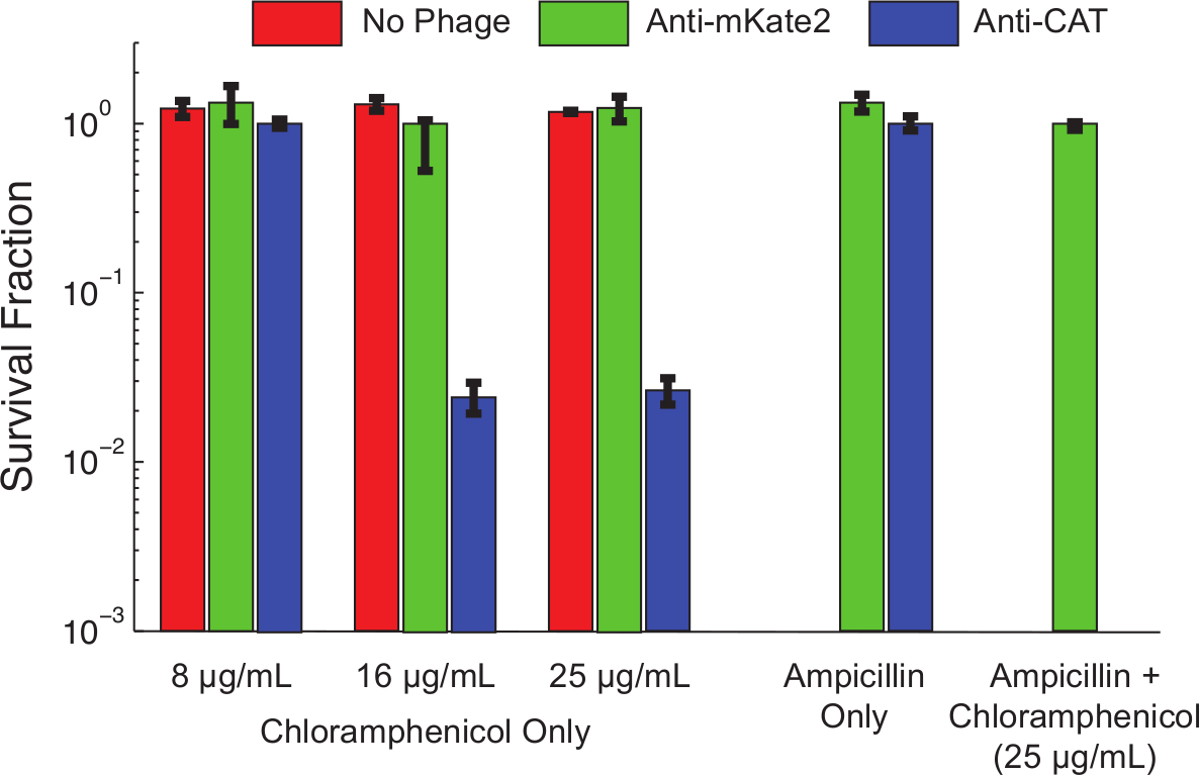
Figure 4 shows the effect of sRNA-mediated silencing on the chloramphenicol resistance phenotype. Uninfected cells, or cells infected with phagemid targeting mKate2, were resistant to chloramphenicol at all concentrations tested. In contrast, cells infected with phagemid targeting CAT showed reduced survival at low chloramphenicol concentrations, and nearly 99% killing at higher concentrations. The addition of ampicillin, to select for only bacteria carrying the phagemid, reduced chloramphenicol survival to undetectable levels. This is consistent with earlier work showing that escape from phage infection is a common route to escape silencing10.

Figure 1: Gene Silencing in E. coli with sRNA Expression Cassettes Delivered by M13 Phage. The sRNA cassette is composed of 4 modules: the pR promoter (a constitutive promoter derived from bacteriophage lambda), a 24 bp targeting domain, an Hfq binding domain extracted from MicC and a transcriptional terminator8. The cassette is incorporated into a phagemid bearing the M13 f1 replication origin and packaged into the M13 capsid using the helper phage M13KO7. Packaged phagemids are able to infect E. coli expressing the F pilus, where sRNA expression begins. The sRNA then recruits the Hfq protein (depicted in red) and binds an antisense mRNA target near the ribosome binding site, resulting in translational repression and mRNA degradation. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Primer Design and Site-Directed Mutagenesis of the sRNA Target Site. Two primers are designed with partial homology to an existing sRNA cassette. The forward primer contains 20 bp homologous to the pR promoter at the 5' end, followed by 24 bp representing the new GUIDE sequence, then 18 bp homologous to the Hfq-binding domain at the 3' end. The reverse primer is the exact reverse complement of the forward primer, with regions of homology to the existing sRNA cassette flanking the reverse complement of the new GUIDE sequence. Exact primer sequences are given in Table 3. Separate single-primer PCR reactions with the forward and the reverse primers produce linear, single-stranded DNA with the desired modified sequence. Annealing the forward and reverse reaction products, following clean-up as described in the protocol, results in double-stranded plasmid DNA bearing the desired modified sRNA cassette. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Knockdown of a Chromosomally Integrated mKate2 Fluorescent Reporter. E. coli MG1655 K12 expressing mKate2 were either left untreated (red lines and bars), infected with an anti-mKate2 phagemid (green lines and bars), or infected with a control phagemid targeting CAT (blue lines and bars). (A) Untreated E. coli grew to higher saturation densities, indicating a metabolic cost to phage infection. Dotted lines indicate standard deviations of 3 replicates. (B) The mKate2 signal was reduced to near background levels in the anti-mKate2 treated strain, but not in control strains. (C) GFP fluorescence, also carried by the phagemid, was detectable only in phagemid-treated controls. (D, E) Final fluorescence readings after 24 hr of growth were normalized to OD. GFP signal, indicating phagemid infection, was absent in untreated controls, but also substantially reduced following anti-CAT phagemid treatment. This may indicate off-target effects of the phagemid. The mKate2 signal was reduced by anti-mKate2 phagemid treatment compared to untreated controls. The CAT-targeted control phagemid showed no effect on mKate2 fluorescence. Error bars represent standard deviation of 3 replicates. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Knockdown of CAT Restores Chloramphenicol Sensitivity to a Genetically Resistant Population. E. coli MG1655 K12 expressing a chromosomally integrated CAT gene were left untreated (red bars), treated with a control phagemid targeting mKate2 (green bars), or treated with a phagemid expressing an anti-CAT sRNA (blue bars). After 1 hr of infection, viability on the indicated antibiotics was assessed by serial dilutions and plating. Strains treated with anti-CAT phagemid were significantly killed (>90%) by chloramphenicol at higher concentrations, while control treatments were unaffected. Adding ampicillin to the culture plates positively selects for phagemid infection and eliminates uninfected cells. Under these conditions, no chloramphenicol resistant colonies were observed after anti-CAT treatment. This indicates that most survivors represent a failure of infection, rather than a failure of silencing. Error bars represent the standard deviation of 3 replicates. Please click here to view a larger version of this figure.
{kind=link}
| CAT TARGET sequence | 5' - ATGGAGAAAAAAATCACTGGATAT - 3' |
| CAT GUIDE sequence | 5' - ATATCCAGTGATTTTTTTCTCCAT - 3' |
Table 1: An example TARGET and GUIDE Sequence for the CAT Gene. Note the reverse complement relationship.
| pR promoter | TAACACCGTGCGTGTTGACTATTTTACCTCTGGCGGTGATAATGGTTGC | ||||
| GUIDE sequence | ATATCCAGTGATTTTTTTCTCCAT | ||||
| Hfq binding domain | TTTCTGTTGGGCCATTGCATTGCCACTGATTTTCCAACATATAAAAAGACAAGCCCGAACAGTCGTCCGGGCTTTTTT TCTCGAG | ||||
| T1/TE terminator | CTCGAGCCAGGCATCAAATAAAACGAAAGGCTCAGTCGAAAGACTGGGCCTTTCGTTTTATCTGTTTTTGTCGGTGAA CGCTCTCTACTAGAGTCACACTGGCTCACCTTCGGGTGGGCCTTTCTGCGTTTATA | ||||
Table 2: Sequence Components of the sRNA Cassette. Each sequence is written 5'-3'. The complete cassette is the concatenation of these 4 elements in order and comprises 292 bp.
| Forward Primer | 5' - CTGGCGGTGATAATGGTTGC[GUIDE]TTTCTGTTGGGCCATTGC - 3' |
| Reverse Primer | 5' - GCAATGGCCCAACAGAAA[TARGET]GCAACCATTATCACCGCCAG - 3' |
Table 3: Primer Design to Alter Existing GUIDE Elements. The forward primer includes the last 20 bp of the pR promoter, the new GUIDE sequence, and the first 18 bp of the Hfq-binding domain. The TARGET sequence is the exact reverse complement of the GUIDE sequence. The reverse primer is the exact reverse complement of the forward primer.
| Component | Volume |
| Template plasmid | 500 ng |
| 10 µM Primer | 2.5 μl |
| 10 mM dNTPs | 0.4 μl |
| High-fidelity Polymerase (2 U/μl) | 0.5 μl |
| 5x PCR Buffer | 10 μl |
| Total volume | 50 μl |
Table 4: Suggested Conditions for the Single-Primer Mutagenic PCR.
| Step | Temp | Time |
| Initial denaturation | 98 °C | 30 sec |
| 30 cycles | 98 °C | 10 sec |
| 55 °C | 30 sec | |
| 72 °C | 120 sec | |
| Final Extension | 72 °C | 300 sec |
| Storage | 10 °C |
Table 5: Suggested Thermocycler Protocol for the Single-Primer Mutagenic PCR.
| Component | Volume |
| Template DNA | 1 μl |
| 10 µM Forward Primer | 0.5 μl |
| 10 µM Reverse Primer | 0.5 μl |
| Taq 2X Master Mix | 25 μl |
| Nuclease-free water | 23 μl |
| Total volume | 50 μl |
Table 6: Suggested Conditions for the Sequence Verification PCR.
| Step | Temp | Time |
| Initial denaturation | 95 °C | 30 sec |
| 30 cycles | 95 °C | 30 sec |
| 55 °C | 30 sec | |
| 68 °C | 30 sec | |
| Final Extension | 68 °C | 300 sec |
| Storage | 10 °C |
Table 7: Suggested Thermocycler Protocol for the Sequence Verification PCR.
Discussion
The present method achieved 80% reduction in mKate fluorescence levels compared to untargeted controls. This is in line with other RNA knockdown methods, where complete silencing is not observed and 50-90% efficiency is typical16,17. At the phenotypic level, CAT-targeted knockdowns were able to significantly attenuate chloramphenicol resistance, and eliminate it under some conditions.
The knockdown phenotype was detectable at the population level after only a few hours post-infection (Figure 3B). This illustrates an important feature of phage-based delivery: a high knockdown frequency can be obtained directly in batch culture without prior genetic modification. Unlike conventional genetic modifications using plasmid transformation or genomic integration, phage infection does not require that a population be re-grown from a single isolated colony. This allows the effects of phage infection to be explored in populations with complex spatial dynamics11, with pre-existing spatial structures like biofilms18, or in genetically mixed natural populations19.
A critical step in this method is the production of packaged phagemid at high titer. The metabolic burden associated with phage particle production may lead to high rates of mutation or plasmid loss in the phagemid production strain. It is recommended that the phagemid production strain be cultured directly from a single cotransformed colony and not refrigerated, frozen or sub-cultured prior to phage harvest. A low efficiency of co-transformation may also be observed when introducing the phagemid and helper plasmid to E. coli simultaneously. In this case, higher efficiencies may be obtained by transforming the helper plasmid first, then preparing competent cells bearing the helper plasmid for subsequent transformation with the phagemid.
Phagemid infection or sRNA expression also imposes a detectable metabolic burden on the target cells, and may result in some phenotypic perturbation. For example, a reduction in mKate2 fluorescence was observed even when cells were infected with a phagemid targeting CAT (Figure 3). Infection with M13 is not thought to trigger systemic stress responses in E. coli20, but may alter transcriptional patterns indirectly. Alternately, the GFP or ampicillin resistance markers included on the phagemid may compete for cellular resources, reducing mKate2 expression and growth9. Finally, the sRNA cassette itself may globally alter gene expression profiles by titrating the Hfq protein, or through off-target mRNA silencing. Off target effects are common in in vivo RNAi targeting21-23, but they have yet to be systematically investigated for this system.
One limitation of this method is that the infection efficiency is less than 100%, allowing some non-infected bacteria to persist in the population. The results of this work and earlier work10 suggest that noninfected cells represent 1-10% of the final population, and are responsible for most of the nonsilenced phenotypes observed. A variety of routes to M13-resistance are known, with the most common being mutational loss of pilus expression24. In light of these limitations, controls should be used to confirm high infection rates and knockdown efficiency.
Another potential limitation for some applications is the occasional transfer of contaminating helper phage. Although M13K07 contains a mutated packaging signal, it can be packaged into phage capsid at low frequency and transferred to infected populations, resulting in cells competent for phage production and the continued spread of phage beyond the initial infection event25. Modifications to the helper phage have proven effective at reducing nonspecific packaging, although sometimes at the cost of reduced phage production26.
Engineered bacteriophage have become an indispensable tool for E. coli synthetic biology, allowing quick delivery of new genes to a growing population. Recent work has produced intercellular communication circuits11 or expressed transcription factors to suppress antibiotic resistance pathways27. The protocol presented here adds to a growing collection of tools that allow control of bacterial physiology through programmed RNAs. CRISPR-Cas nucleases, when mutated to eliminate nuclease activity, have been shown to repress transcription at RNA-guided gene targets17,28. In contrast, sRNA silencing works at the translational level and does not require exogenous protein expression. Next-generation biotechnologies may unite transcriptional and translational control with phage-mediated delivery to program complex phenotypes in real time.
Disclosures
The authors have nothing to disclose.
Acknowledgements
Funding for this work was provided by the Fondation Bettencourt Schueller in support of the Paris Bettencourt iGEM team. We thank the INSERM U1001 research unit and Chantal Lotton for technical assistance and advice. Phagemid Litmus28i_J23115-B0032-GFP was provided by Monica Ortiz and Drew Endy of Stanford.
Materials
| Name | Company | Catalog Number | Comments |
| Plasmid Miniprep Kit | Qiagen | 27104 | |
| DpnI Enzyme | NEB | R0176S | |
| Phusion High Fidelity Polymerase | NEB | M0530S | |
| Taq 2x Master Mix | NEB | M0270L | |
| M13KO7 Helper Phage | NEB | N0315S | |
| DH5α Competent Cells | Life Technologies | 18265-017 | |
| TOP10F' Cells | Life Technologies | C3030-03 | |
| LB Broth | Sigma | L3022-250G | |
| Ampicillin | Sigma | A9393-5G | |
| Kanamycin | Sigma | 60615-5G | |
| Chloramphenicol | Sigma | C0378-5G | |
| Tetracycline | Sigma | 87128-25G |
References
- Ohkumo, T., Masutani, C., Eki, T., Hanaoka, F. Use of RNAi in C. elegans. RNAi. , 129-137 (2008).
- Fire, A., Xu, S., Montgomery, M. K., Kostas, S. A., Driver, S. E., Mello, C. C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 391 (6669), 806-811 (1998).
- Iordanou, E., Chandran, R. R., Blackstone, N., Jiang, L. RNAi interference by dsRNA injection into Drosophila embryos. J Vis Exp. (50), e2477 (2011).
- Ramadan, N., Flockhart, I., Booker, M., Perrimon, N., Mathey-Prevot, B. Design and implementation of high-throughput RNAi screens in cultured Drosophila cells. Nat Protoc. 2 (9), 2245-2264 (2007).
- Tsong, T. Y. Electroporation of cell membranes. Biophys J. 60 (2), 297-306 (1991).
- Kim, W. J., Chang, C. -. W., Lee, M., Kim, S. W. Efficient siRNA delivery using water soluble lipopolymer for anti-angiogenic gene therapy. J Control Release. 118 (3), 357-363 (2007).
- Shi, Y. Mammalian RNAi for the masses. Trends Genet. 19 (1), 9-12 (2003).
- Na, D., Yoo, S. M., Chung, H., Park, H., Park, J. H., Lee, S. Y. Metabolic engineering of Escherichia coli using synthetic small regulatory RNAs. Nat Biotechnol. 31 (2), 170-174 (2013).
- Ceroni, F., Algar, R., Stan, G. -. B., Ellis, T. Quantifying cellular capacity identifies gene expression designs with reduced burden. Nat Methods. 12 (5), 415-418 (2015).
- Libis, V. K., Bernheim, A. G., et al. Silencing of Antibiotic Resistance in E. coli with Engineered Phage Bearing Small Regulatory RNAs. ACS Synth Biol. 3 (12), 1003-1006 (2014).
- Ortiz, M. E., Endy, D. Engineered cell-cell communication via DNA messaging. J Biol Eng. 6 (1), 16 (2012).
- Edelheit, O., Hanukoglu, A., Hanukoglu, I. Simple and efficient site-directed mutagenesis using two single-primer reactions in parallel to generate mutants for protein structure-function studies. BMC Biotechnol. 9 (1), 61 (2009).
- Chung, C. T., Miller, R. H. Preparation and storage of competent Escherichia coli cells. Recombinant DNA Part I. , 621-627 (1993).
- Phornphisutthimas, S., Thamchaipenet, A., Panijpan, B. Conjugation in Escherichia coli. Biochem Mol Biol Educ. 35 (6), 440-445 (2007).
- Sezonov, G., Joseleau-Petit, D., D'Ari, R. Escherichia coli Physiology in Luria-Bertani Broth. J Bacteriol. 189 (23), 8746-8749 (2007).
- Mittal, V. Improving the efficiency of RNA interference in mammals. Nat Rev Genet. 5 (5), 355-365 (2004).
- Qi, L. S., Larson, M. H., et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 152 (5), 1173-1183 (2013).
- Lu, T. K., Collins, J. J. Dispersing biofilms with engineered enzymatic bacteriophage. Proc Natl Acad Sci USA. 104 (27), 11197-11202 (2007).
- Yosef, I., Manor, M., Kiro, R., Qimron, U. Temperate and lytic bacteriophages programmed to sensitize and kill antibiotic-resistant bacteria. Proc Natl Acad Sci USA. 112 (23), 7267-7272 (2015).
- Karlsson, F., Malmborg-Hager, A. -. C., Albrekt, A. -. S., Borrebaeck, C. A. K. Genome-wide comparison of phage M13-infected vs. uninfected Escherichia coli. Can J Microbiol. 51 (1), 29-35 (2005).
- Senthil-Kumar, M., Mysore, K. S. Caveat of RNAi in plants: the off-target effect. Methods in molecular biology. 744, 13-25 (2011).
- Jackson, A. L., Linsley, P. S. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat Rev Drug Discov. 9 (1), 57-67 (2010).
- Cho, S. W., Kim, S., et al. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 24 (1), 132-141 (2014).
- Hagens, S., Blasi, U. Genetically modified filamentous phage as bactericidal agents: a pilot study. Lett Appl Microbiol. 37 (4), 318-323 (2003).
- Kasman, L. M., Kasman, A., Westwater, C., Dolan, J., Schmidt, M. G., Norris, J. S. Overcoming the phage replication threshold: a mathematical model with implications for phage therapy. J Virol. 76 (11), 5557-5564 (2002).
- Chasteen, L., Ayriss, J., Pavlik, P., Bradbury, A. R. M. Eliminating helper phage from phage display. Nucleic Acids Res. 34 (21), e145 (2006).
- Lu, T. K., Collins, J. J. Engineered bacteriophage targeting gene networks as adjuvants for antibiotic therapy. Proc Natl Acad Sci USA. 106 (12), 4629-4634 (2009).
- Bikard, D., Jiang, W., Samai, P., Hochschild, A., Zhang, F., Marraffini, L. A. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res. 41 (15), 7429-7437 (2013).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved