
Method Article
Transcriptional Analysis by Nascent RNA FISH of In Vivo Trophoblast Giant Cells or In Vitro Short-term Cultures of Ectoplacental Cone Explants
In This Article
Summary
Trophoblast giant cells (TGCs) play a key role in the placenta to ensure a healthy pregnancy. We present a protocol for assessing the transcriptional status of genes in TGCs by nascent fluorescent in situ hybridization on cryostat sections of post-implantation embryos or short-term cultures of embryonic day 7 ectoplacental cones.
Abstract
The placenta derives from one extra-embryonic lineage, the trophectoderm. In the peri-implantation murine blastocyst, mural trophectoderm cells differentiate into primary trophoblast giant cells (TGCs) while the polar trophectoderm overlying the inner cell mass continues to proliferate later differentiating into secondary TGCs. TGCs play a key role in developing placenta and are essential for a successful pregnancy. Investigation of transcriptional regulation of specific genes during post-implantation development can give insights into TGCs development. Cells of the ectoplacental cone (EPC) from embryos at 7-7.5 days of gestation (E7-7.5), derived from the polar trophectoderm, differentiate into secondary TGCs1. TGCs can be studied in situ, on cryostat sections of embryos at E7 although the number of TGCs is very low at this stage. An alternative means of analyzing secondary TGCs is to use short-term cultures of individual EPCs from E7 embryos. We propose a technique to investigate the transcriptional status of genes of interest both in vivo and in vitro at the single-cell level using fluorescent in situ hybridization (RNA FISH) to visualize nascent transcripts. This technique provides a direct readout of gene expression and enables assessment of the chromosomal status of TGCs, which are large endoreplicating cells. Indeed, a key feature of terminal differentiation of TGCs is that they exit the cell cycle and undergo multiple rounds of endoreplication.This approach can be applied to detect expression of any gene expressed from autosomes and/or sex chromosomes and can provide important information into developmental mechanisms as well as placental diseases.
Introduction
Mammalian trophoblast giant cells (TGCs) form a barrier between maternal and embryonic tissues. They mediate implantation and invasion of the conceptus into the uterus and play critical roles in development for the formation of the placenta. They produce several growth factors (cytokines) and hormones of the Prolactin/Placental Lactogen family and steroids, necessary for embryonic growth and survival. TGCs are large, mononucleated and polyploid cells with a cell cycle, the endocycle, that consists of alternating S and G phases. Indeed, TGCs are endoreplicating cells, able to undergo multiple rounds of DNA synthesis without any division2. In order to investigate the transcriptional status of genes in vivo, in TGCs compared to other embryonic and extraembryonic cell types, nascent RNA FISH can be performed in vivo on cryostat sections3 at specific post-implantation stages. TGCs are easily recognizable on sections due to their large size and their transcriptional gene activity can be recorded but their number at early post-implantation stages is low. Paucity of TGCs on E7 embryonic sections, led us to perform short-term cultures of embryonic trophectodermal tissues to obtain differentiated TGCs in order to study the regulation of gene expression during trophectoderm development. Moreover, in order to remain as physiological as possible, established cell lines ie trophectoderm stem cells (TS) are not always suitable to investigate developmental mechanisms Generation of secondary TGCs provide a valuable tool to study pathological pregnancies associated with defects in TGCs due to abnormal gene regulation in mice.
Trophoblast cells of the ectoplacental cone (EPC) are precursors of secondary TGCs4. Spontaneous differentiation of cultured EPCs to secondary TGCs has previously been reported5. However, in contrast to primary TGCs, studies on secondary TGC differentiation have remained limited, presumablydue to the difficulties of isolating EPC explants free of any maternal or embryonic tissues. We adapted these methods, in order to perform RNA FISH on secondary TGCs derived from individual embryo at E7, a developmental post-implantation stage where TGCs are very few but could be generated from EPC precursors. RNA FISH to analyze nuclear primary transcripts has never been done at the level of the single cell level on secondary TGCs. This allows precise analysis of transcription and was used to show the epigenetic instability of TGCs at post-implantation stages3.
A classic example of epigenetics in mammals, the X-chromosome inactivation (XCI) is studied in the Heard lab6. In this process one of the 2 X chromosomes in female is inactivated. The non-coding Xist transcript coats the X chromosome from which it is expressed in female cells and triggers silencing of most genes. Using RNA FISH, nascent transcripts of X-linked genes can be investigated as can the accumulation of Xist RNA on the inactive X chromosome (Xi). We describe here a procedure to perform RNA FISH on sections of post-implantation embryos and on short-term cultures of EPC. This protocol adapted from those that were used to study XCI in differentiating female embryonic stem cells and preimplantation embryos7-11. We provide examples of XCI in female embryos in vivo, as well as in vitro TGCs.
Protocol
Animal procedures were performed in accordance with the approved institutional animal care and use committee of the Institut Curie (CEEA-IC) protocols (C 75-05-18). The work has also been conducted under the approval from the French Ministry of Higher Education and Research for the use of Genetically Modified Organisms (agreement number 5549CA-I).
1. Preparation of Cryostat Sections
- Collect embryos from naturally ovulating F1 C57BL/6 x DBA/2J mice as described in Shea et al.,12. On day 7 of gestation (E7), sacrifice 8-12 week old mouse by cervical dislocation. Collect the entire conceptus i.e decidua12.
- Isolate E7 conceptuses, as described in Shea et al.,12. Place them in a 60 mm Petri dish containing PBS.
- Freeze the E7 mouse conceptus for cryostat sections.
- Prepare adapted aluminum foil wells 1cm in height ('home-made' using a glass Pasteur pipet). Deposit a drop of tissue freezing medium on the bottom of this small container. Deposit the conceptus in the correct orientation (i.e., for longitudinal sections the conceptus should maintained in its horizontal orientation).
- Fill the well containing the conceptus with tissue freezing medium. Suspend it with forceps in vapor above liquid N2 in order to allow it to freeze slowly – then immerse the block in the liquid N2 for a few sec until it turns white. Subsequently, transfer the block into a freezing vial and store at -80 °C (can store for several months).
- Before cryo-sectioning, place the frozen block containing the conceptus at -20 °C in the cryostat for 30 min. Perform cryosections of 8 µm thickness. Deposit 4 sections on a slide to enable efficient attachment. Place sections close enough to fit under an 18 x 18 mm2 coverslip.
- Check the quality of the sections (intact and without scratches) and the orientation of the embryo (longitudinal sections) with a stereomicroscope and choose the sections suitable for further analysis. As quickly as possible deposit them in a coplin jar and perform RNA FISH (See 3.3.2).
2. Preparation of Secondary TGCs
- Isolate the Conceptus (see 1.1 and 1.2).
- Dissect the E7 Ectoplacental Cone (EPC)
- Use a stereomicroscope and Petri dishes containing sterile PBS. Dissect the decidua with forceps. Pierce the sample and open forceps to tear the two sides of the decidua apart.
- Shell out the embryo. Use tips of closed fine forceps (Dumont No. 5) in a scissor-like action to separate the EPC from the embryo proper. Use special care to obtain a perfectly clean sample, separate from maternal tissue as well as from chorion and yolk sac. Wash out EPC explants in PBS.
- With a little spoon, transfer individual dissected EPC to a 4-well plate containing a sterile coverslip in medium.
- Derive TGCs from EPCs Short-term Cultures.
- Prepare 4-well plates containing coverslips. Sterilize 12 mm diameter round glass coverslips by immersing in ethanol, followed by flame-drying.
- Add 0.5 ml EPC medium to each well (EPC medium: RPMI 1640 supplemented with 15% FCS, 0.1mM 2-mercaptoethanol and antibiotics).
- Deposit single, dissected-EPCs at the center of the coverslip in the well along with culture medium. Use fine forceps to apply the EPC on the glass coverslip. Culturefor 3-5 days at 37 °C, 5% CO2. Individual explant forms an outgrowth that spreads as a monolayer of flattened TGCs (Figure 2A).
3. RNA FISH
NOTE: The protocols are based on those described for embryonic stem cells (ESCs) in Chaumeil et al.,8 and Pollex and Heard13.
- Advanced Preparation of Stock Solutions
- Prepare a 3M sodium acetate buffer pH 5.2.
- Prepare 2x hybridization buffer containing 40 % (w/v) sodium dextran sulfate, 20x BSA, 400 mM Vanadyl Ribonucleoside Complex (VRC) in 4x Saline Sodium Citrate (SSC).
- Prepare mounting medium containing 90% (v/v) glycerol, 0.1% (w/v) p-phenylenediamine, pH 9 in PBS.
- Freshly Prepared Solutions
- Prepare fixative solution consisting of 3% freshly prepared paraformaldehyde (PFA) in PBS. Prepare permeabilization solution containing 0.5 % Triton-X-100 in PBS supplemented with 2 mM VRC. Prepare wash buffer by mixing 50% formamide (FA) and 2x SSC and adjust the pH to 7.2-7.4.
- Prepare DNA counter-staining solution consisting of 1 µg/ml DAPI (4′,6-diamidino-2-phenylindole dihydrochloride) in 2x SSC.
- Fixation and Permeabilization in Preparation for FISH
- For cells on coverslips, wash cells in 1x PBS for 5 min. Fix cells on coverslips, or embryonic sections on slides, for 10 min in fixative (3% PFA) solution at RT.
- Rinse cells three times with 1x PBS. Permeabilize cells for 5 min in ice-cold permeabilization solution on ice. Wash cells three times with 70% ethanol. Store coverslips in 4-well plates and slides in 4-slide transport box in 70% ethanol at -20 °C.
NOTE: Coverslips and slides can be stored for several months at -20 °C. Plates and boxes containing them have to be sealed with parafilm to avoid ethanol evaporation.
- DNA Probe Labeling
- Label DNA probes by nick translation using fluorescent nucleotides. Follow the manufacturer's instructions (see Table 1)8.
- For a 50 µl reaction mix, add 1-2 µg of plasmid, bacterial artificial chromosomes (BAC) or multiple displacement amplification (MDA) (for DNA amplification; see 3.4.4) DNA to 17.5 µl water, 2.5 µl 0.2 mM SR-, SG-, Cy5-dUTP, 10 µl 10 mM each dNTP mix (dGTP, dATP, dCTP), 5 µl 10 mM dTTP, 5 µl 10x nick translation buffer and 8 µl nick translation enzyme.
- Incubate for 16 hr at 15 °C in the dark. Inactivate the reaction by freezing at -20 °C. Store probes for months at -20 °C.
- MDA
NOTE: For BAC DNA, quantity of DNA obtained after classical preparation is low, therefore one can use a step of DNA amplification before labeling by using MDA. Use a commercial kit and follow the manufacturer's instructions (see Table 1). - Mix 0.5 µl BAC DNA with 9.5 µl sample buffer. Heat 3 min at 95 °C. Immediately quench on ice; leave on ice 10 min. Prepare reaction buffer/enzyme mix: (9.5 µl + 0.5 µl enzyme mix) and keep on ice.
- Mix 10 µl DNA mix + 10 µl reaction buffer/enzyme mix. Incubate at 30 °C at least 20 hr. Inactivate enzyme by heating sample 10 min at 65 °C. Cool sample to 4 °C before storing at -20 °C.
- Check MDA by digestion. Add 1 µl MDA, 2 µl 10x buffer, 2 µl Hind III and 15 µl H2O. Incubate at 37 °C overnight. Run slowly on a 0.8-1% agarose gel to confirm accurate DNA amplification. Clear DNA fragments of high molecular weight should be observed on the gel.
- Probe Preparation
- Use 0.1 or 1 µg probe per coverslip or slide.
NOTE: Add 2-5 µg of Cot-1 DNA if competition is required e.g., most probes as they can contain repeat sequences which otherwise cross hybridize and increase background.- For precipitation, add 5 µg salmon sperm DNA, 1/10 volume 3 M sodium acetate pH 5.2 and 3 volumes ethanol. Spin at 16,000 x g and 4 °C for 25 min. Wash pellets with 70% ethanol and spin down again for 5 min.
- Dry pellet for 2 min in a concentrator/speed vac. Resuspend in 100% formamide in half the volume required for hybridization (e.g., 2.5 µl for coverslip or 7 µl for embryonic sections on slide). Place 30 min at 37 °C with shaking in a thermomixer. Denature for 7 min at 75 °C.
- Quench on ice, or if competition is required put directly at 37 °C for 30-60 min. Mix the probe solution with an equal volume of 2x hybridization solution.
- Use 0.1 or 1 µg probe per coverslip or slide.
- Hybridization and Washes
- Dehydrate; coverslips in wells, and slides in a Coplin jar, by sequential washing in 1x 80%, 1x 95% and 2x 100% ethanol for 5 min each. Dry coverslips and slides completely.
- For hybridization, apply the 5 µl probe hybridization mix (see 3.5) onto a slide and lower the coverslip, with cells facing into the hybridization mix. For sections on slide, apply 14 µl probe hybridization mix (see 3.5) and cover with an 18 x 18 mm2 coverslip.
- Place slides in a humid chamber (tissue paper soaked in 50% FA/2x SSC) and incubate at 37 °C overnight, in the dark.
- Post-hybridization Washes:
- Add 1 ml of 50% FA/2x SSC onto the coverslip on the slide to loosen it; remove the coverslip carefully from the slide (taking care not to scrape the cells) and place it cell-side up into a 4-well plate containing 50% FA/2x SSC. For sections on slide, remove the coverslip in a similar manner and place the slide in a Coplin jar containing 50% FA/2x SSC.
- Perform 3 washes, each for 7 min with pre-warmed 50% FA/2x SSC at 42 °C or 44 °C for coverslip or slide, respectively.
- Perform 3 washes with pre-warmed 2x SSC for 5 min each at 42 °C or 44 °C for coverslip or slide, respectively.
- Counterstain nuclei by washing in 2x SSC with 1 µg/ml DAPI for 3 min at RT. Rinse twice with 2x SSC.
- Mounting of Coverslips and Slides
- For small coverslip from cultured TGCs, apply 5 µl mounting medium on a slide. Place the coverslip cell-side down on top of the drop.
- For slide with sections, apply 15 µl mounting medium on the slide. Place a 22 x 22 mm2 coverslip on top of the drop.
- Avoid bubbles. Wipe off excess mounting solution. Seal the coverslip with a small amount of nail polish. If possible, image slides immediately or store for up to several months at -20 °C.
4. Microscopy and Analysis
- Acquire 3D sequential z axis images in 0.3 µm using a fluorescence microscope (63X objective) and perform the analysis of 3D image stacks using the imageJ software14.
Results
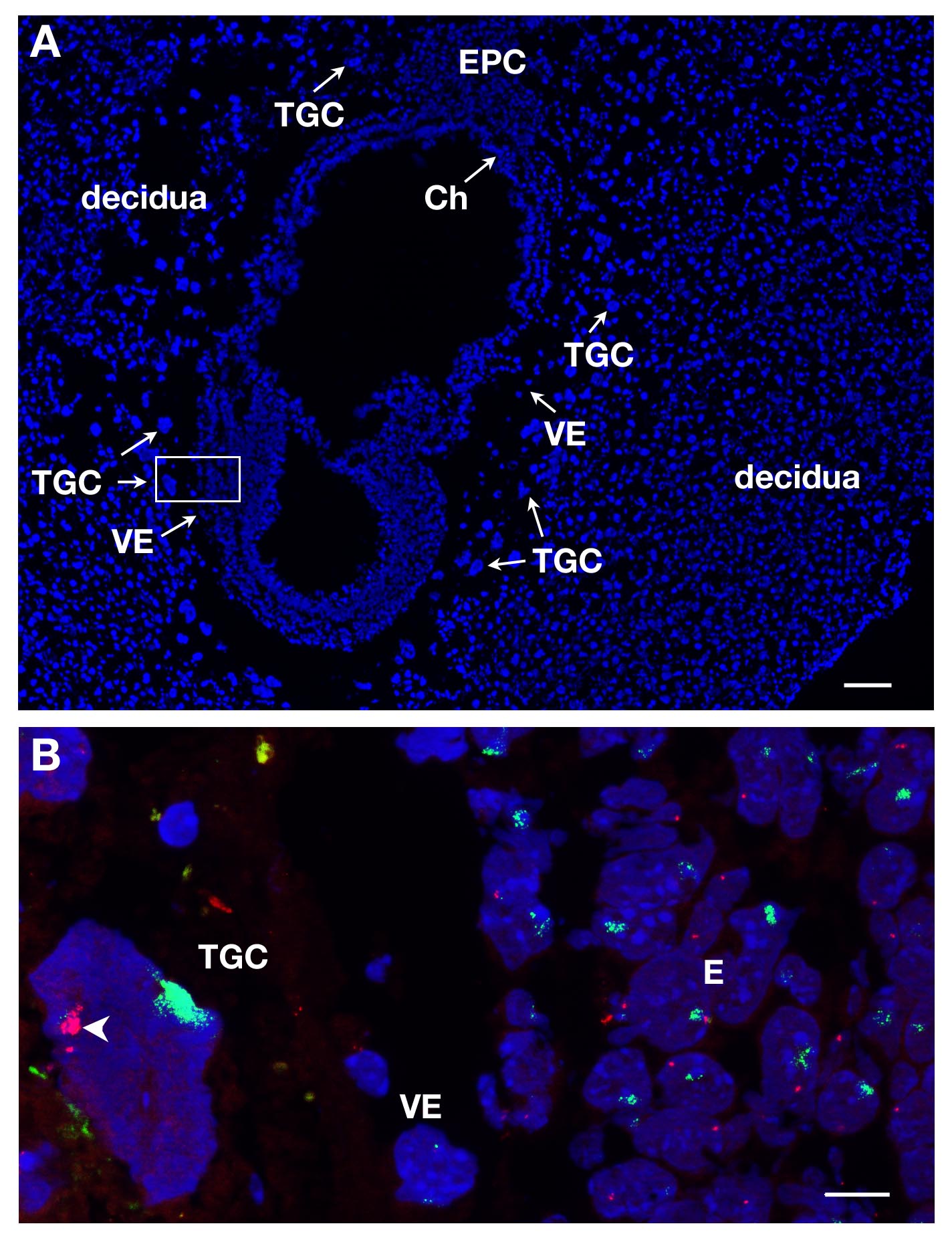
TGCs can be identified on E7 sections upon DAPI staining due to their localization in the conceptus and their large size. This is illustrated in Figure 1A on a longitudinal section. RNA FISH was performed on such embryonic sections in order to study X chromosome inactivation in this extra-embryonic lineage.
Several slides or coverslips can be processed at the same time. By using different dyes to label RNA of different genes, one can detect different primary transcripts in the same nucleus. At least 2 probes can be mixed together, for instance Xist coupled to SG (green signal) and Atrx coupled to SR (red signal). An example of a female TGC is shown in Figure 1B where 2 other lineages are represented, the embryo proper (E) and the visceral endoderm (VE). A TGC nucleus is shown with Xist RNA covering the X chromosome which is inactivated (Xi) while the other X chromosome which is active (Xa) displays a few pinpoints. X-linked gene Atrx primary transcripts are expressed from the active X chromosome (not decorated by Xist) in several copies due to endoreplication. This illustrates monoallelic expression e.g., inactivation of Atrx on one X chromosome.
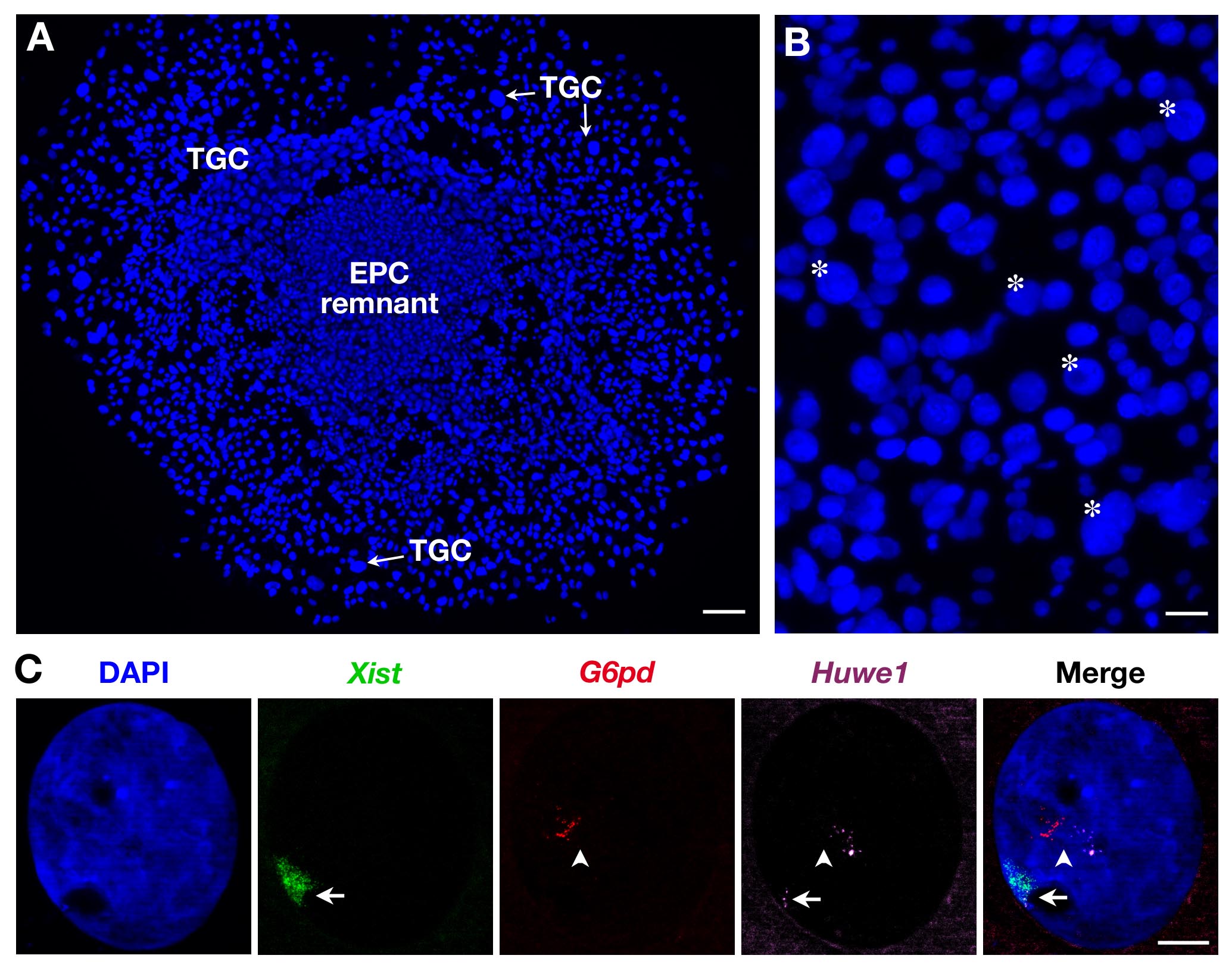
Since TGCs are heterogeneous in size as illustrated in Figure 2B, only the largest ones are recorded. Three probes can be also used as illustrated in Figure 2C for secondary TGCs. In this case, Xist-SG (green), two X-linked genes: G6pd-SR (red) and Huwe1-Cy (magenta) were analyzed at the same time in the same nucleus. While G6pd is monoallelically expressed as shown here (and previously shown in secondary TGCs3) Huwe1 is biallelically expressed demonstrating its property to escape XCI. Endoreplication, with several pinpoints i.e., nascent transcripts copies, is also evident.

Figure 1. Primary transcript expression in TGCs from E7 female embryonic section. (A) Longitudinal section of the E7 conceptus stained with DAPI. The embryo and extra-embryonic tissues are surrounded by the mother tissue, the decidua. The localization of the different lineages is done upon DAPI staining with 5X objective. TGCs are identified due to their large size. Ch, chorion, E, embryo, EPC, ectoplacental cone, VE, visceral endoderm, TGC, trophoblast giant cells. Scale bar = 100 µm. (B) Higher magnification of the boxed-in area in A (63X objective) RNA FISH. Atrx primary transcript in red and Xist RNA in green are visualized on the 3 lineages = E, VE, TGC. Scale bar = 10 µm. Example of an in vivo TGC where Atrx is monoallelically expressed (Xa, arrowhead) and silent on the Xist-coated X chromosome (Xi); several Atrx signals seen on the Xa are due to endoreplication. Atrx = BAC clone RP23-260I15. Please click here to view a larger version of this figure.
{kind=link}

Figure 2. Primary transcript expression in secondary TGCs derived from EPC. (A) General view of growing secondary TGCs (x5 objective). Scale bar = 100 µm. (B) Asterisk indicate example of TGCs according to their size (10X objective). Scale bar = 60 µm. (C) Example of a secondary female TGC RNA FISH with the use of 3 probes (Xist domain in green, covering the inactivated X chromosome: Xi) showing endoreplication of G6pd and Huwe1 primary transcripts (several pinpoints, in red and magenta). While G6pd is expressed monoallelically, Huwe1 expression is biallelic in this nucleus. Huwe1 = BAC clone RP24-157H12 ; G6pd = BAC clone RP23-13D21. Xi = arrow; Xa = arrowhead (63X objective). Scale bar = 10 µm. Please click here to view a larger version of this figure.
{kind=link}
Discussion
Nascent RNA FISH represents an easy and sensitive method for the single cell analysis of transcriptional activity in embryonic tissues at different developmental stages. The power of this approach is the capacity to identify different embryonic lineages at any particular stage according to morphological criteria. However this also requires that minimal background fluorescence is present. Any such background renders the identification of different embryonic regions and cell types challenging. To ensure minimal background, there are two critical steps in this protocol. The first is the quality of the cryosection and the second is the efficiency (signal to noise ratio) of the RNA FISH, which depends on the level of gene expression and the quality of the probe. For the latter, probes are therefore always tested on cultured cells on coverslips (embryonic stem cells or somatic cells) prior to use on sections.
In this protocol, we provide the techniques required to perform RNA FISH analysis on TGCs from two different types of sample preparation (cryostat sections and primary cultures of embryonic explants). Such single cell analysis enables dynamic changes in gene expression at different post-implantation stages to be assessed.
The methods we describe to generate secondary TGCs (at E7-7.5 post-implantation stages) were used in our lab to study the X-chromosome inactivation patterns of an extra-embryonic lineage which is constituted of TGCs at post-implantation stages. Extra-embryonic development in rodents depends on the differentiation of TGCs. Indeed, TGCs are essential for placental and thus embryonic development. Defects in TGC differentiation cause embryonic lethality (for review see reference15). Transcriptional activity of TGCs using RNA FISH allowed us to demonstrate an unusual X chromosome inactivation status of such cell type during mouse development3.
The methods presented here to obtain and study secondary TGCs can be applied to study molecular pathways in development of the important extra-embryonic tissues they are part of, both in normal and mutant mice. These approaches could be adapted to investigate TGC development in other mammals. Our analysis involved the use of wild type mouse embryos allowing us to assess the transcriptional activity of various genes in in vivo TGCs and in in vitro TGCs derived from EPC explant. This method could be extended to the analysis of transgenic mice and/or by the addition of inhibitor molecules in the culture medium. We have successfully used this method of RNA FISH on another type of TGCs, such as primary TGCs which appear at an earlier stage of mouse development, E3.0 blastocyts, which can be individually cultured for 4-5 days during which developed outgrowth, the ICM being surrounded by large primary TGCs16,17. Nascent transcripts for different genes as well as Xist could be visualized and quantified using the same RNA FISH approach3.
Combined immunofluorescence and RNA FISH can also be performed on cryostat sections as well as in vitro cultured TGCs3. This demonstrates that the method is quite robust, as primary transcripts are highly susceptible to degradation and there is an absolute requirement of RNase free compounds. IF/RNA FISH provides additional information about transcription levels, cellular localization and protein expression, simultaneously in a given cell. Although, sections of paraffin-embedded tissue have previously been used to detect nascent RNA in human tumors while maintaining tissue morphology18, in our hands, cryostat sections are more appropriate for preserving both the nascent RNA and the epitopes required for detection by antibodies during immunofluorescence.
In addition to RNA FISH, following immunofluorescence, chromosomal DNA FISH can also be performed on secondary TGCs using probes labeled with fluorochromes, similar to those used for RNA FISH3. Fluorescent probes can be either plasmids/fosmids or BACs, labeled with fluorescent dUTPs as used here, and described in Chaumeil et al.,8 . Alternatively, fluorescently labeled oligonucleotides can be used19. Since DNA FISH does not require strand-specificity, oligonucleotide probes can be designed to target either of the two complementary strands in the target region. Branched DNA probes, where signal amplification is achieved by two sequential rounds of specific and amplifier probes can also be used to enhance the signal-to-noise ratio of FISH signals20. Alternatively, RNA probes such as riboprobes can be used although in our DNA-based probes guarantee a better tradeoff between signal quality, specificity and ease of use.
Finally, 3D image acquisition is essential in order to obtain the required spatial information in these large cells, and can be performed using a variety of fluorescence microscopes, such as the Apotome microscope, or epifluorescence microscopes with deconvolution such as the Deltavision (GEH), or other microscopes suitable for imaging tissue sections, and large (>20 µm) TGCs. It should be noted that for single copy DNA loci, the punctate signal detected by nascent RNA FISH or DNA FISH may not be readily detected by confocal microscopy.
In conclusion, the methods we describe here should be useful both for the detailed analysis of TGS in a developmental context, but also in other disease situations. Many genes that are involved in TGC development and function in rodents are conserved between rodents and humans, such as transcription factors, proteases and cell adhesion molecules21. Mouse TGCs are a cell model for studying genes that regulate placental development and therefore give insights into human placental diseases. Furthermore, due to the fact that these cells are endoreplicating and since some cancer cells engage endocycle programs, in addition to gene amplification processes, the methods we describe should also be helpful in single cell investigations required to explore mechanisms leading to genome instability in cancer cells.
Disclosures
The authors have nothing to disclose.
Acknowledgements
We thank Sophie Gournet for help with illustrations, Julie Chaumeil for reading the manuscript, the animal housing facility and the imaging platform of the Unit. This work has received support under the program «Investissements d'Avenir» launched by the French Government and implemented by ANR with the references ANR-10-LABX-0044 and ANR-10-IDEX-0001-02 PSL, the EpiGeneSys FP7 no. 257082 Network of Excellence to E.H., an ERC Advanced Investigator award no. 250367 and EU FP7 SYBOSS grant no. 242129 to E.H. The authors would like to acknowledge the Cell and Tissue Imaging Platform of the Genetics and Developmental Biology Department (UMR3215/U934) of Institut Curie, member of France-Bioimaging (ANR-10-INSB-04), for help with light microscopy.
Materials
| Name | Company | Catalog Number | Comments |
| Stereomicroscope | Nikon | SMZ 1500 | |
| Stereomicroscope | Zeiss | Stemi SV6 | |
| Scissors Pascheff-Wolff | Moria | MC19 | |
| Dumont #5 forceps | Roth | PK78.1 | |
| 4-well tissue culture dishes | Nunc | 176740 | |
| 60 mm Petri dishes Falcon | Dutsher, France | 353004 | |
| 100 mm Petri dishes Falcon | Dutsher, France | 353003 | |
| Coverslips 18 mm x 18 mm | VWR | 631-1331 | |
| Coverslips 22 mm x 22 mm | VWR | 631-0125 | |
| 12 mm glass round coverslips | Harvard apparatus | 64-0712 | |
| Slides Superfrost plus | VWR | 631-9483 | |
| 4-slide Transport box Lockmailer | Dutsher, France | 40684 | |
| Cryotubes 1.8 ml Corning | Fisher Science | 10418571 | |
| Glass Coplin staining jars | Fischer Scientific | W1561L | |
| TissueTek O.C.T compound | VWR | 4583 | |
| RPMI 1640 medium | Invitrogen | 61870 | |
| Phosphate buffered saline (PBS) | Sigma-Aldrich | D1408 | 10x is used |
| Water | Sigma-Aldrich | W3500 | |
| Paraformaldehyde | Panreac Quimica, Spain | 141451 | 3% in PBS |
| Triton-X-100 | Euromedex | 2000-A | 0.5% final |
| Vanadyl ribonucleoside complex (VRC) | New England Biolabs, USA | S1402S | |
| Sodium dextran sulfate | Sigma-Aldrich | D8906 | |
| Bovine serum albumin (BSA) | New England Biolabs, USA | B9001S | |
| Formamide | Sigma-Aldrich | 47671-1L-F | aliquots kept at -20 °C |
| Illustra TempliPhi Kit Construct (Kit MDA) | Dutsher, France | 25-6400-80 | |
| Nick translation kit | Abbott, USA | 07J00-001 | |
| 20x SSC buffer concentrate | Sigma-Aldrich | S6639 | |
| Spectrum green dUTP | Abbott, USA | 02N32-050 | |
| Spectrum red dUTP | Abbott, USA | 02N34-050 | |
| Cy-5 dUTP | Dutsher, France | PA55022 | |
| Mouse Cot-1 DNA | Invitrogen | 18440016 | |
| DNA, MB grade | Invitrogen | Roche | DNA from fish sperm |
| 4′,6-diamidino-2-phenylindole dihydrochloride | Sigma-Aldrich | D9564 | DAPI |
| Glycerol | Sigma-Aldrich | G9012 | |
| p-phenylenediamine | Sigma-Aldrich | 695106 | |
| Centrifuge 5417R | Eppendorf, Germany | molecular biology grade | |
| Eppendorf concentrator plus | Eppendorf | ||
| Eppendorf Thermomixer comfort | Eppendorf | ||
| Liquiport Liquid pump | KNF Neuberger, Trenton, USA | ||
| Shake'N'Bake Hybridization oven | Boekel Scientific, USA | ||
| Cryostat | Leica | CM3050 |
References
- Cross, J. C. Genetic insights into trophoblast differentiation and placental morphogenesis. Sem. Cell Dev. Biol. 11, 105-113 (2000).
- Zybina, E. V., Zybina, T. G. Polytene chromosomes in mammalian cells. Int. Rev. Cytol. 165, 53-119 (1996).
- Corbel, C., Diabangouaya, P., Gendrel, A. -. V., Chow, J. C., Heard, E. Unusual chromatin status and organization of the inactive X chromosome in murine trophoblast giant cells. Development. 140, 861-887 (2013).
- Rossant, J., Tamura-Lis, W. Effect of culture conditions on diploid to giant-cell transformation in postimplantation mouse trophoblast. J. Embryol. Exp. Morphol. 62, 217-227 (1981).
- El-Hashash, A. H., Kimber, S. J. Trophoblast differentiation in vitro: establishment and characterization of a serum-free culture model for murine secondary trophoblast giant cells. Reproduction. 128, 53-71 (2004).
- Chow, J. C., Heard, E. X inactivation and the complexities of silencing a sex chromosome. Curr Opin Cell Biol. 3, 359-366 (2009).
- Chaumeil, J., Okamoto, I., Heard, E. X-chromosome inactivation in mouse embryonic stem cells: analysis of histone modifications and transcriptional activity using immunofluorescence and FISH. Methods Enzymol. 376, 405-419 (2004).
- Chaumeil, J., Augui, S., Chow, J. C., Heard, E. Combined immunofluorescence, RNA fluorescent in situ hybridization, and DNA fluorescent in situ hybridization to study chromatin changes, transcriptional activity, nuclear organization, and X-chromosome inactivation. Methods Mol. Biol. 463, 297-308 (2008).
- Okamoto, I., Otto, A. P., Allis, C. D., Reinberg, D., Heard, E. Epigenetic dynamics of imprinted XCI during early mouse development. Science. 303, 644-664 (2004).
- Okamoto, I., Arnaud, D., Le Baccon, P., Otte, A. P., Disteche, C. M., Avner, P., Heard, E. Evidence for de novo imprinted X-chromosome inactivation independent of meiotic inactivation in mice. Nature. 438, 369-373 (2005).
- Patrat, C., Okamoto, I., Diabangouaya, P., Vialon, V., Le Baccon, P., Chow, J., Heard, E. Dynamic changes in paternal X-chromosome activity during imprinted X-chromosome inactivation in mice. Proc. Natl. Acad. Sci. 106, 5198-5203 (2009).
- Geijsen Shea, K., N, Dissection of 6.5 dpc mouse embryos. J. Vis. Exp. (2), e160 (2007).
- Pollex, T., Piolot, T., Heard, E. Live-cell imaging combined with immunofluorescence, RNA, or DNA FISH to study the nuclear dynamics and expression of the X-inactivation center. Methods Mol. Biol. 1042, 13-31 (2013).
- Schneider, C. A., Rasband, W. S., Eliceiri, K. W. NIH Image to ImageJ: 25 years of image analysis. Nature Methods. 9, 671-675 (2012).
- Hemberger, M. IFPA award in placentology lecture - characteristics and significance of trophoblast giant cells. Placenta. 29, 4-9 (2008).
- Carney, E. W., Prideaux, V., Lye, S. J., Rossant, J. Progressive expression of trophoblast-specific genes during formation of mouse trophoblast giant cells in vitro. Mol. Reprod. Dev. 34, 357-368 (1993).
- Shin, J., et al. Maternal Rnf12/RLIM is required for imprinted X-chromosome inactivation in mice. Nature. 467, 977-981 (2010).
- Capodieci, P., Donovan, M., Buchinsky, H., Jeffers, Y., Cordon-Cardo, C., Gerald, W., Edelson, J., Shenoy, S. M., Singer, R. H. Gene expression profiling in single cells within tissue. Nat Methods. 9, 663-665 (2005).
- Beliveau, B. J., Joyce, E. F., Apostolopoulos, N., Yilmaz, F., Fonseka, C. Y., McCole, R. B., Chang, Y., Li, J. B., Senaratne, T. N., Williams, B. R., et al. Versatile design and synthesis platform for visualizing genomes with Oligopaint FISH probes. Proc. Natl. Acad. Sci. 109, 21301-21306 (2012).
- Kenny, D., Shen, L., Kolberg, J. A. Detection of viral infection and gene expression in clinical tissue specimens using branched DNA (bDNA) in situ hybridization. J. Histochem. Cytochem. 50, 1219-1227 (2002).
- Cross, J. C., Baczyk, D., Hemberger, M., Hugues, M., Simmons, D. G., Yamamoto, H., Kingdom, J. C. Genes, development and evolution of the placenta. Placenta. 24, 123-130 (2003).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved