
Method Article
2 in 1: One-step Affinity Purification for the Parallel Analysis of Protein-Protein and Protein-Metabolite Complexes
* These authors contributed equally
In This Article
Summary
Protein-protein and protein-metabolite interactions are crucial for all cellular functions. Herein, we describe a protocol that allows parallel analysis of these interactions with a protein of choice. Our protocol was optimized for plant cell cultures and combines affinity purification with mass spectrometry-based protein and metabolite detection.
Abstract
Cellular processes are regulated by interactions between biological molecules such as proteins, metabolites, and nucleic acids. While the investigation of protein-protein interactions (PPI) is no novelty, experimental approaches aiming to characterize endogenous protein-metabolite interactions (PMI) constitute a rather recent development. Herein, we present a protocol that allows simultaneous characterization of the PPI and PMI of a protein of choice, referred to as bait. Our protocol was optimized for Arabidopsis cell cultures and combines affinity purification (AP) with mass spectrometry (MS)-based protein and metabolite detection. In short, transgenic Arabidopsis lines, expressing bait protein fused to an affinity tag, are first lysed to obtain a native cellular extract. Anti-tag antibodies are used to pull down protein and metabolite partners of the bait protein. The affinity-purified complexes are extracted using a one-step methyl tert-butyl ether (MTBE)/methanol/water method. Whilst metabolites separate into either the polar or the hydrophobic phase, proteins can be found in the pellet. Both metabolites and proteins are then analyzed by ultra-performance liquid chromatography-mass spectrometry (UPLC-MS or UPLC-MS/MS). Empty-vector (EV) control lines are used to exclude false positives. The major advantage of our protocol is that it enables identification of protein and metabolite partners of a target protein in parallel in near-physiological conditions (cellular lysate). The presented method is straightforward, fast, and can be easily adapted to biological systems other than plant cell cultures.
Introduction
The method described here aims at the identification of metabolite and protein partners of a protein of choice in near-in-vivo cellular lysate conditions. It has been speculated that many more metabolites than characterized today have an important regulatory function1. Metabolites can act as biological switches, changing the activity, functionality, and/or localization of their receptor proteins2,3,4. In the last decade several breakthrough methods, enabling identification of PMI in vivo or in near-in-vivo conditions, have been developed5. Available approaches can be separated into two groups. The first group comprises techniques that start with a known-metabolite bait in order to trap novel protein partners. Methods include affinity chromatography6, drug affinity responsive target-stability assay7, chemo-proteomics8, and thermal proteome profiling9. The second group consists of a single method that starts with a known protein in order to identify small-molecule ligands10,11.
AP coupled with MS-based lipidomics was used to analyze protein-lipid complexes in Saccharomyces cerevisiae12. As a starting point, the authors used yeast strains expressing 21 enzymes involved in ergosterol biosynthesis and 103 kinases fused to a tandem-affinity purification (TAP) tag. 70% of the enzymes and 20% of the kinases were found to bind different hydrophobic ligands, shedding light into the intricate protein-lipid interaction network.
Previously, we could demonstrate that, similarly to lipids, polar and semi-polar compounds also remain bound to protein complexes isolated from the cellular lysates13. Based on these findings, we decided to optimize the AP method published previously10,11 for plant cells and hydrophilic compounds14. For this purpose, we used TAP vectors described by Van Leene et al. 2010, successfully used in plant PPI studies15. To shorten the time required to obtain transgenic lines, we decided on Arabidopsis cell cultures. We employed a one-step methyl tert-butyl ether, (MTBE)/methanol/water extraction method, allowing the characterization of proteins (pellet), lipids (organic phase), and hydrophilic metabolites (aqueous phase)16 in a single affinity-purification experiment. EV control lines were introduced to exclude false positives, e.g. proteins binding to the tag alone. As proof of concept we tagged three (of the five) nucleoside diphosphate kinases present in the Arabidopsis genome (NDPK1-NDPK3). Among other findings, we could demonstrate that NDPK1 interacts with glutathione S-transferase and glutathione. Consequently we could prove that NDPK1 is subjected to glutathionylation14.
To sum up, the presented protocol is an important tool for characterizing protein-protein and protein-small-molecule interaction networks and constitutes a major advance over existing methods.
Protocol
Preparation of transgenic Arabidopsis cell culture lines, including cloning, transformation, selection, and growth conditions can be found in17. Note that EV control lines are recommended to correct for false positives. Prior to the experiment, confirm the overexpression of the bait protein by western blot analysis, e.g. using IgG antibodies against the G-protein part of the tandem affinity tag. It is important to separate the growth media from plant cell culture material.
1. Preparing Plant Cell Material Prior to the Experiment
- Grow a PSB-L A. thaliana cell culture line overexpressing the protein of interest18.
- Prepare MSMO medium, which contains 4.43 g/L MSMO mixed with 30 g/L sucrose. Adjust the pH of the buffer to 5.7 with 1 M KOH and autoclave the solution. Prior to the experiment, supplement the medium with 0.5 mg/L α-naphthaleneacetic acid, 0.05 mg/L kinetin and 50 μg/mL kanamycin.
- Cultivate transformed plant cell cultures in 50 mL of MSMO medium in a 100 mL flask on an orbital platform shaker with gentle agitation (130 rpm). Grow cells in a culture room at 20 °C and light intensity equal to 80 μmol m-2 s-1.
- Subculture cells into fresh media every 7 days, diluting them 1:10.
- Collect cells at the logarithmic growth phase using a glass funnel combined with a vacuum pump, using a nylon mesh as filter. Wrap the infiltrate in aluminum foil and freeze in liquid nitrogen.
Caution: Remember that liquid nitrogen is extremely cold. Incorrect handling can cause burns. Wear appropriate personal protective equipment, including thermally insulated gloves, protective glasses, and a lab coat.
2. TAP Protocol
Note: The following step is adapted from Maeda et al. 201411 and Van Leene et al. 201117.
- Homogenize harvested and frozen plant cell culture material using a mixer mill (2 min at 20 Hz) or mortar and pestle to obtain fine powder. Aliquot 3 g of the ground material (corresponding to around 90 mg of total protein) per sample. Avoid thawing of the sample during this step by using liquid nitrogen pre-chilled equipment.
Note: Store ground plant material in 50 mL tube at –80 °C to the beginning of an AP procedure. - Triturate the sample in a liquid-nitrogen-precooled mortar with 3 mL of ice-cold lysis buffer (0.025 M Tris–HCl pH 7.5; 0.5 M NaCl; 1.5 mM MgCl2; 0.5 mM DTT; 1 mM NaF; 1 mM Na3VO4; 100x diluted commercial protease inhibitor cocktail; 1 mM PMSF) until the material thaws. Once the sample thaws, immediately proceed to the next step.
Note: Prepare the lysis buffer fresh. Introduce blank samples at this step. Detergents are not recommended as they can cause problems in MS detection. - To remove cellular debris, divide the material into 2 mL microcentrifuge tubes and centrifuge at 20,817 x g for 10 min at 4 °C. Collect 3 mL of the clear lysate in a 15-mL conical centrifuge tube.
- During centrifugation step, equilibrate IgG-Sepharose beads. Aliquot 100 µL of the beads per sample and wash them with 1 mL of lysis buffer. Vortex to resuspend beads and flash-spin. Discard the lysis buffer and repeat the step twice. Resuspend beads in 400 µL of lysis buffer.
- Add beads to the collected plant lysate and incubate the mixture on a rotating wheel for 1 h at 4 °C.
- Transfer the mixture into a syringe combined via Luer-lock cap with a spin column with filter pore size 35 µm. Apply pressure to pass the lysate through. Beads with attached complexes will remain on the filter, while the lysate will go through.
Note: Optionally, use a vacuum manifold system. Make certain to apply gentle pressure so as not to harm the beads. - Wash beads at first with 10 mL of washing buffer (0.025 M Tris–HCl pH 7.5; 0.5 M NaCl), and then with 1 mL of elution buffer (10 mM Tris–HCl pH 7.5; 150 mM NaCl; 0.5 mM EDTA; 1000x diluted E64 and 1 mM PMSF). Perform the washing using a syringe attached to the column or vacuum manifold system.
Note: When using a vacuum manifold system make certain to apply gentle pressure so as not to harm the beads. - Incubate beads with the 400 µL of elution buffer containing 50 U of an improved version of the tobacco etch virus (AcTEV) protease. Use table shaker at 1,000 rpm for 30 min at 16 °C.
Note: Remember to use a plug to close the column at the bottom adding the elution buffer. - Add an extra portion (50 U) of the enzyme into the column and incubate the mixture for the next 30 min under the same, described above, conditions.
- Collect the eluate in a 2 mL microcentrifuge tube either by centrifugation (1 min, 20,817 x g) or by vacuum manifold. To remove remaining complexes, introduce an additional elution step using 200 µL of elution buffer.
Note: Store the sample at –20 °C or –80 °C, or immediately proceed with the protein and metabolite extraction step. Thaw frozen samples on ice.
3. Western Blot Analysis
- To confirm the presence of the bait protein in the collected eluate use 10 µL of the protein–metabolite-containing eluate to perform SDS–PAGE and western blot analysis. To identify the protein of interest, use mouse primary antibodies against streptavidin-binding protein (1:200), a part of the TAP tag remaining after TEV protease cleavage, as described in Van Leene et al. 201117. Next, use secondary goat anti-mouse antibodies coupled with HRP.
4. Metabolite and Protein Extraction
Note: This protocol is adapted from Giavalisco et al. 201116.
Note: From this step onwards use UPLC–MS–grade solutions.
- Add 1 mL of methyl tert-butyl ether (MTBE)/methanol/water solvent (3:1:1) to the collected eluate and mix the sample by inversion. Make sure that the solvent is cooled to –20 °C prior to the extraction step.
CAUTION: MTBE and methanol are harmful substances. Perform the extraction step under the fume hood and wear appropriate personal protection equipment, e.g., gloves. - Add 0.4 mL of methanol:water 1:3 solution to each sample and mix the content of the sample by inversion.
Note: Supplementation of the mixture with methanol:water solution results in phase separation. The upper phase contains lipids, lower phase contains polar and semi-polar metabolites, and proteins can be found in the pellet. - To separate phases, centrifuge the sample at 20,817 x g for 2 min at room temperature, then collect the upper phase for lipid measurements (not done in this protocol) using a manual liquid handling pipette with 1 mL volume capacity.
- Add 0.2 mL of methanol and mix by inversion.
- Centrifuge the sample at 20,817 x g for 2 min at RT, then collect the polar phase for metabolite measurements (polar and semi-polar compounds). To avoid disturbing the protein pellet, leave around 50 µL of the liquid phase at the bottom of the tube.
- Dry collected samples for metabolite measurements overnight in a centrifugal evaporator. Avoid over-drying the protein pellets by removing samples from the evaporator after 30–60 min.
Note: Store samples at –20 °C or –80 °C, or immediately proceed with the preparation of proteins for LC–MS/MS analysis.
5. Preparing Samples for Proteomic Analysis
Note: This step is adapted from Olsen et al. 200419 and the technical manual of the Trypsin/Lys-C Mix (see Table of Materials).
- Perform enzymatic digestion of the sample.
Caution: Solvents used during enzymatic digestion and desalting of the sample are harmful. Work under the fume hood and wear appropriate personal protection equipment, e.g., gloves.- Dissolve protein pellet in 30 µL of freshly prepared denaturation buffer (40 mM ammonium bicarbonate containing 2 M thiourea/6 M urea, pH 8). To achieve better protein solubility, perform a 15-min sonication step. Repeat the step until the pellet dissolves.
- Centrifuge the sample at 20,817 x g for 10 min at 4 °C, then transfer the supernatant to a new microcentrifuge tube.
- Determine protein concentration using the Bradford protein assay.
- For further analysis, aliquot a volume equivalent to 100 µg of protein and fill the sample up to 46 µL with denaturation buffer.
- Add to the sample 2 µL of freshly prepared reduction buffer (50 mM DTT dissolved in H2O) and incubate for 30 min at room temperature.
- Treat the sample with 2 µL of freshly prepared alkylation buffer (150 mM iodoacetamide dissolved in 40 mM ammonium bicarbonate buffer) and incubate the mixture in the dark for 20 min at room temperature.
- Dilute the sample with 30 µL of 40 mM ammonium bicarbonate buffer and add 20 µL of LysC/Trypsin Mix.
- After 4 h incubation at 37 °C, dilute the sample with 300 µL of 40 mM ammonium bicarbonate buffer.
- Continue with overnight incubation at 37 °C.
- Acidify the sample with approximately 20 µL of 10% trifluoroacetic acid (TFA) to obtain pH < 2. Check sample pH using a pH strip.
Note: Store the sample at –20 °C or proceed to the next step.
- Desalt the digested proteins.
Note: Preferably, use a vacuum manifold system. Avoid over-drying of the column.- Rinse the C18 SPE column (see Table of Materials) with 1 mL of 100% MeOH and then with 1 mL of 80% acetonitrile (ACN) containing 0.1% TFA diluted in water. Use, here and in further protein-desalting steps, a vacuum manifold system to speed up the process. Avoid over-drying of the column.
- Equilibrate the column by washing it twice with 1 mL of 0.1% TFA diluted in water.
- Load the sample onto the column. Rinse tube with additional 200 µL of 0.1% TFA and transfer the solution on the column. Run the solutions through the column.
- Wash the column twice with 1 mL of 0.1% TFA.
- Elute desalted peptides from the column with 800 µL of 60% ACN, 0.1% TFA solution. Dry the collected fraction in a centrifugal evaporator, avoiding over-drying of the protein fraction by removing samples from the evaporator after 30–60 min.
Note: Store samples at –20 °C or –80 °C or immediately proceed to the next step. In subsequent steps, keep samples on ice.
6. Measurement Prepared Protein Samples Using UPLC–MS/MS.
Note: Prior to proteomic and metabolomic measurements, filter (0.2 µm pore size) and degas all buffers using a vacuum pump for 1 h.
- Resuspend dried peptide pellet stored in 2 mL microcentrifuge tube in 50 µL of buffer C (3% v/v ACN, 0.1% v/v formic acid) using manual liquid handling pipette with 200 µL volume capacity. Sonicate samples for a 15 min in an ultrasonic bath with 35 kHz ultrasonic frequency.
Caution: ACN and formic acid are harmful substances. Work under the fume hood and wear appropriate personal protection equipment, e.g., gloves. - Centrifuge the sample at 20,817 x g for 10 min at 4 °C, then transfer 20 µL of the supernatant to a glass vial.
- Separate digested peptides using a C18 reversed-phase column connected to a liquid chromatography and acquire mass spectra using a mass spectrometer.
- Separate on the column 3 µL of the sample using a 300-nl/min flow rate. For a mobile phase, use buffer C and D (63% v/v ACN, 0.1% v/v formic acid), forming a gradient ramping from 3% ACN to 15% ACN over 20 min and then to 30% ACN over the next 10 min.
Note: Store the rest of the sample at –20 °C or –80 °C up to few months. Prior to proteomic measurement, refreeze the sample on ice. - Wash contaminants out for 10 min using 60% ACN and equilibrate the column with 5 µL of buffer C before measurement of the next sample.
- Gain mass spectra using data-dependent MS/MS method with resolution set at 70,000, AGC target of 3e6 ions, maximum injection time of 100 ms, and an m/z ranging from 300 to 1600. Acquire maximum of 15 MS/MS scans at a resolution of 17,500, AGC target of 1e5, maximum injection time of 100 ms, underfill ratio of 20%, with an isolation window of 1.6 m/z and m/z ranging from 200 to 2000. Enable apex trigger (6–20 s), set dynamic exclusion to 15 s, and exclude charges of 1 and > 5.
- Separate on the column 3 µL of the sample using a 300-nl/min flow rate. For a mobile phase, use buffer C and D (63% v/v ACN, 0.1% v/v formic acid), forming a gradient ramping from 3% ACN to 15% ACN over 20 min and then to 30% ACN over the next 10 min.
7. Processing of Proteomic Data
- Download the newest Arabidopsis thaliana proteome database from http://www.uniprot.org/ and include contaminant database. Analyze raw data obtained from the LC–MS runs using MaxQuant with the integrated Andromeda peptide search engine using default setting with enabled LFQ normalization20,21,22. Find detailed information about parameters used in Table S1.
- Open the “protein groups.txt” output file. For further analysis, filter for protein groups identified with at least two unique peptides. Remove protein groups defined by MaxQuant as potential contaminants and filter for A. thaliana proteins (ARATH in Fasta headers column) present in database.
- To test the significance of protein enrichment between samples, use LFQ normalized intensities and perform unpaired, two-tailed Student's t-test followed by multiple comparison correction (e.g. Benjamini & Hochberg false discovery rate (FDR) correction or Bonferroni correction).
- Calculate p-value by comparing LFQ intensities obtained for EV control and NDPK1. Filter out all undetermined values. Sort p-values in ascending order and use R script or online calculator (e.g., https://www.sdmproject.com/utilities/?show=FDR) to compute FDR correction. Filter for FDR values below 0.1.
Note: Consider the form of data analysis suitable for the research. For quantitative studies (analysis of protein enrichment between samples) use the “LFQ intensity” value, whereas for qualitative research (presence or absence of particular protein) choose the “Intensity” value. - Filter for protein groups that are more abundant in NDPK1 comparing to EV control. Determine localization of potential protein partners using the SUBA database23 and correct for protein co-localized with NDPK1.
- Calculate p-value by comparing LFQ intensities obtained for EV control and NDPK1. Filter out all undetermined values. Sort p-values in ascending order and use R script or online calculator (e.g., https://www.sdmproject.com/utilities/?show=FDR) to compute FDR correction. Filter for FDR values below 0.1.
8. Measurement of Samples Containing Polar Phase Using UPLC–MS.
- Resuspend dried polar phase from step 4.5 in 200 µL of water and sonicate the sample for 5 min.
- Centrifuge the sample at 20,817 x g for 10 min at 4 °C, then transfer the supernatant to a glass vial.
Note: Store the rest of the sample at –20 °C or –80 °C for up to several months. Prior to metabolomic measurement, refreeze the sample on ice. - Perform a separation step using UPLC coupled to C18 reversed-phase column and acquire mass spectra with MS.
- Load onto the column 2 µL of the sample per injection for each ionization mode (positive and negative) and separate the fraction using 400 µL/min flow rate. To create the required gradient for metabolite measurement, prepare mobile phase solution as follows: buffer A (0.1% formic acid in H2O) and buffer B (0.1% formic acid in ACN).
- Separate metabolites at 400 µL/min and the following gradient: 1 min 99% of buffer A, 11-min linear gradient from 99% of buffer A to 60% of buffer A, 13-min linear gradient from 60% of buffer A to 30% of buffer A, 15-min linear gradient from 30% of buffer A to 1% of buffer A, hold 1% concentration until 16 min. Starting from 17 min, use linear gradient from 1% of buffer A to 99% of buffer A. Re-equilibrate the column for 3 min with 99% concentration of buffer A before measurement of the next sample.
- Acquire mass spectra covering mass range between 100 and 1500 m/z with resolution set to 25,000 and loading time restricted to 100 ms. Set AGC target to 1e6, capillary voltage to 3kV with a sheath gas flow and auxiliary gas value of 60 and 20, respectively. Set capillary temperature to 250 °C and skimmer voltage to 25V.
9. Processing of Metabolomics Data
- Process collected chromatograms acquired from both ionization modes. Use software to extract mass to charge ratio (m/z), retention time (RT) and intensity of associated peaks, e.g., commercial software (see Table of Materials) or alternative24.
- Start processing software by double-clicking the .exe file
- Create new workflow, search for activity “Load from File” and move this activity by “drag and drop” into the blank workflow space. Press the activity with the right mouse button and open settings of the activity.
- In the tab containing “General” settings, set a name of the experiment in “Name” field and next click “Select files and folders” and mark raw chromatograms.
- In the tab containing “Advanced” settings, set “Profile Data Cutoff” to 0 intensity. Click “Apply” and “OK”.
- Search for and add activity “Data Sweep”. Press the activity with the right mouse button and open settings of the activity.
- In the tab containing “General” settings, mark “Centroid data” and “MS/MS Data”. Remove all MS/MS data by selecting “Everything” in the selection panel.
- Search for and add the activity “Chromatogram Chemical Noise Subtraction”. Press the activity with the right mouse button and open settings of the activity.
- In the tab containing “General” settings, mark “Chromatogram Smoothing” and set number of scans to “3” and “Estimator” to “Moving average”. Set “RT Window” to 51 scans, “Quantile” to 50%, Subtraction “Method” and 750 Intensity “Threshold”.
- In the tab containing “Advanced” settings, mark “RT Structure Removal” and set “Minimum RT Length” to 5 scans.
- In the tab containing “Advanced” settings, mark “m/z Structure Removal” and set “Minimum m/z Length” to 3 points.
- Search for and add the activity “Chromatogram RT Alignment”. Press the activity with the right mouse button and open settings of the activity.
- In the tab containing “General” settings, set “Alignment Scheme” to “Pairwise Alignment Base Tree” and “RT Search Interval” to 0.5 min.
- In the tab containing “Advanced” settings, use default parameters.
- Search for and add the activity “Peak Detection” in “Chromatogram” group of activities. Press the activity with the right mouse button and open settings of the activity.
- In the tab containing “General” settings, set “Summation Window” to 0.09 min, “Minimum Peak Size” to 0.03 min, “Maximum Merge Distance” to 5 points and “Merge Strategy” to “Centers”. In the “Peak RT Splitting” box set “Gap/Peak Ratio” to 50%.
- In the tab containing “Advanced” settings, set “Smoothing Window” to 5 points, “Refinement Threshold” to 80% and “Consistency Threshold” to 1. Set “Center Computation” as “Intensity-weighted” with “Intensity Threshold” set at 70%.
- Search for and add the activity “Isotope Clustering” in “Chromatogram” group of activities. Press the activity with the right mouse button and open settings of the activity.
- In the tab containing “General” settings, set “RT tolerance” to 0.015 min and “m/z Tolerance” to 5 ppm.
- In the tab containing “Envelope fitting” settings, set “Method” as “No Shape Restriction” and “Ionization” as “Protonation (for positive mode) and “Deprotonation” (for negative mode). Set “Minimum and Maximum charge” to 1 and 4, respectively.
- In the tab containing “Advanced” settings, use default parameters.
- Search for and add activity “Singleton Filter”.
- To export results of data processing, search for and add the activity “Analyst” in “Export” group of activities.
- In the tab containing “General” settings, set “Type” as “Clusters” and “Observable” as “Summed Intensity”. Choose “Custom Destination” and specify directory of export file.
- In the tab containing “Advanced” settings, use default parameters.
- Annotate mass features using in-house reference-compound database.
- Analyze one or multiple MS grade reference-compound using UPLC-MS. Use the same LC-MS method for analysis of reference-compounds and metabolites co-purified with protein of interest.
Note: For this study, a set of almost 300 dipeptides was analyzed and used as reference-compound library. - Use analysis (see Table of Materials) software to open raw chromatogram file and search for specific m/z and RT associated with measured reference-compound (see User Guide).
Note: Secondary metabolites differ in types of ionization. Check for the presence of the common adducts by searching for ion mass equal to M-1.007276, M+1.007276, M+18.033823 and M+22.989218 for [M-H], [M+H], [M+NH4] and [M+Na], respectively. - Use spreadsheet to open exported “Analyst” file obtained after processing of chromatograms and search for specific ion mass. Compare RT of the mass feature measured in the experiment and RT of the reference compound. Allow a deviation of 0.005 Da for m/z and 0.1 min for RT.
- Analyze one or multiple MS grade reference-compound using UPLC-MS. Use the same LC-MS method for analysis of reference-compounds and metabolites co-purified with protein of interest.
- To test the significance of metabolite enrichment co-purified with particular protein between samples (line with overexpressed protein of interest vs EV control), compare peak values using two-tailed non-paired Student's t-test followed by multiple comparison correction (e.g. Benjamini & Hochberg false discovery rate correction or Bonferroni correction).
Results
In the original study, three A. thaliana NDPK genes were overexpressed in PSB-L cell suspension cultures under the control of the constitutive 35S promoter14 (Figure 1). Tandem affinity tag was fused to either carboxy- or amino-terminal end of a bait protein. The affinity-purified complexes were subjected to MTBE/methanol/water extraction16. Affinity-pulled proteins and small molecules were identified using MS (Tables S2 and S3).
To correct for false positives, blank samples were used to exclude small-molecule contaminants from the chemicals and laboratory consumables. Furthermore, metabolites and proteins that bind to either an affinity tag or resin alone were accounted for by using EV control lines.To retrieve true positives, two-tailed non-paired Student's t-test and Benjamini & Hochberg false discovery rate correction was applied to identify metabolites (Table S4) and proteins (Table S5) significantly enriched in the NDPKs AP experiments (N- and C-terminally tagged NDPKs) in comparison to the EV control lines (FDR < 0.1). Note that in the previous work, we used absence/presence criteria to delineate protein and small-molecule interactors.
Representative results are given for NDPK1, while metabolite data focus on dipeptides, a novel class of the small-molecule regulators studied in our group. Proteomic analysis revealed 26 putative protein partners of NDPK1. By further filtering for proteins co-localized in the same subcellular compartment as NDPK1 (cytosol), the list narrowed down to 13 putative protein interactors. Among the identified proteins were glutathione S-transferase, two elongation initiation factors, tubulin, and aconitate hydratase. Metabolomic analysis revealed four dipeptides Val-Leu, Ile-Glu, Leu-Ile, and Ile-Phe that specifically co-eluted with NDPK1 (Figure 2). Note that all four dipeptides share a hydrophobic residue in their N-terminus, suggesting shared binding specificity.
To look for known protein-protein and protein-metabolite complexes we queried 13 identified proteins and four dipeptides against the Stitch database25 (Figure 3). Several observations could be made: (i) None of the interactors was previously reported for NDPK1. (ii) APX1 ortholog was reported to interact with aldehyde dehydrogenase family member ALDH7B4, while translation initiation factor FBR12 with another translation initiation factor encoded by gene AT2G40290. (iii) The identified dipeptides have no reported protein partners. Co-eluted dipeptides were not reported earlier as associated to any retrieved plant protein. However, they play important roles in other organisms: Leu-Ile, e.g., has a neurotrophin-activating effect in a human cell line26. Note that the experiment does not allow identifying the exact topology of the system. For example, a dipeptide may interact directly with NDPK1 but may well be related to any of the co-purified proteins.
Taken together, our results show that the established procedure, employing AP together with mass spectrometry, facilitates identification of protein-protein and protein-small-molecule interactors and helps generate extensive information about the interactome of the target protein.

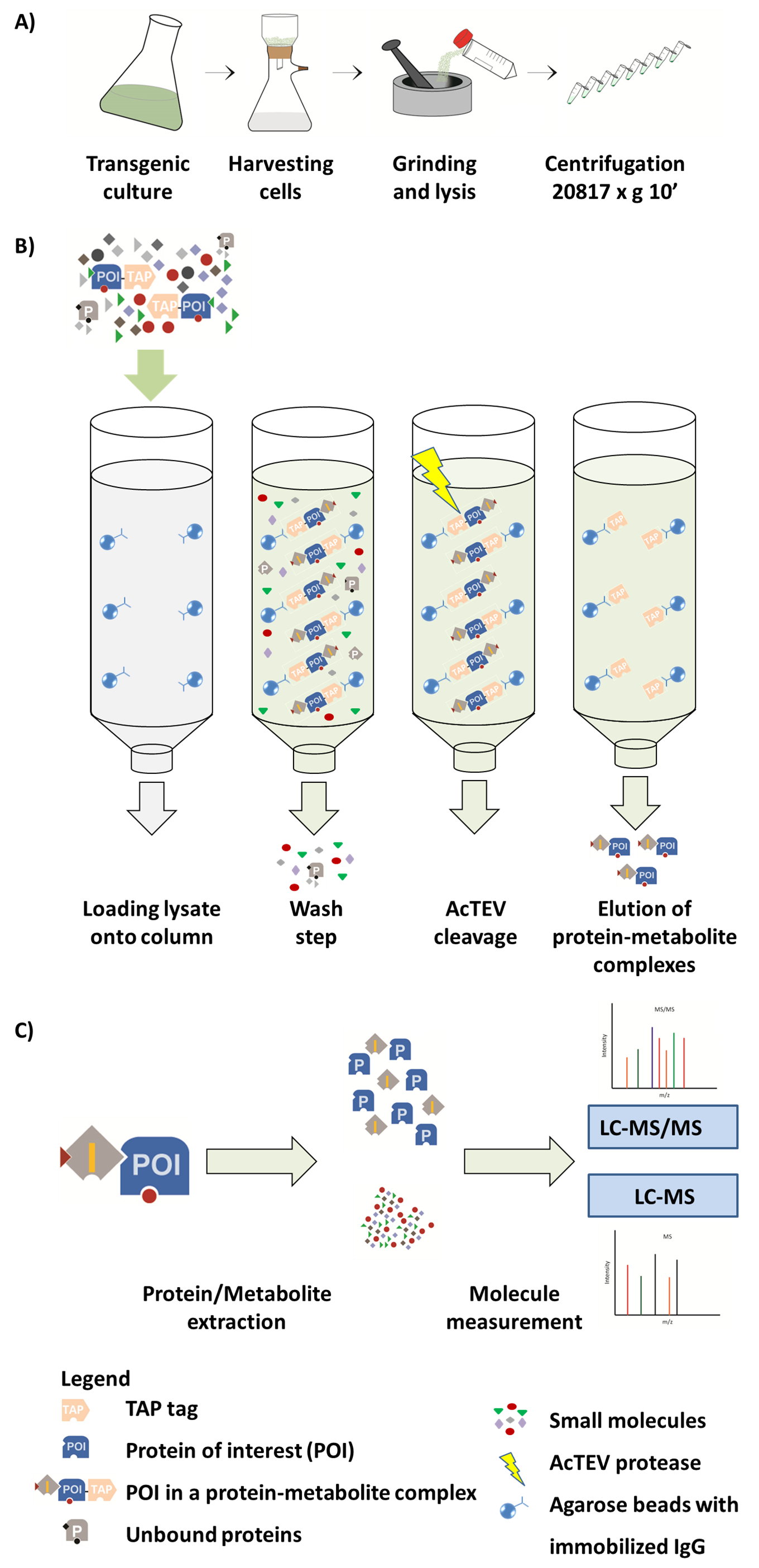
Figure 1. Scheme of AP-MS workflow. (A) Preparation of a native soluble fraction from plant cell culture. (B) Next steps in the AP procedure. After loading the sample onto the column, the protein of interest (POI) fused to a TAP tag binds to the IgG antibody immobilized on the agarose beads. Washing of the column facilitates removal of unbound proteins and metabolites. After performing AcTEV cleavage, POI protein-metabolite complexes are eluted. (C) Separation of complexes into protein and metabolite fraction followed by semi-quantitative MS analysis. Part of this Figure is reproduced from Luzarowski et al. 201714. Please click here to view a larger version of this figure.
{kind=link}

Figure 2. Dipeptides specifically co-eluting with NDPK1. Average intensities of four dipeptides Val-Leu (A), Ile-Glu (B), Leu-Ile (C), and Ile-Phe (D) measured in AP experiment were plotted. All four dipeptides show significant enrichment in NDPK1 samples compared to EV control (asterisks represent FDR < 0.1). Error bars represent standard error for 6 measurements (3 replicates of N- and 3 of C-terminally tagged proteins). Please click here to view a larger version of this figure.
{kind=link}

Figure 3. Interaction network of all molecules co-eluting with NDPK1, queried against STITCH database considering only previous experimental and database evidences (confidence > 0.2). Higher confidence indicates higher chances of interaction and is calculated based on the deposited data. Please click here to view a larger version of this figure.
{kind=link}
Table S1. MaxQuant output table "parameters.txt". Table includes threshold values for identification and quantification, as well as information about the databases used. Please click here to download this file.
Table S2. Information from MaxQuant output table "proteinGroups.txt". Table contains a list of all identified protein groups, intensities, and additional information such as number of unique peptides and score. Please click here to download this file.
Table S3. Output file containing analysis of polar metabolites. Table contains a list of all identified mass features characterized by specific m/z, RT and intensity. Please click here to download this file.
Table S4. Dipeptides found in AP samples in which NDPK1, NDPK2 or NDPK3 were used as bait. Dipeptides present in blank samples were excluded from the list. Two independent lines (tagged in either N- or C-terminus) for each NDPK were run in triplicate. Student's t-test and further correction of p-value using Benjamini & Hochberg method were used to determine significantly enriched interactor partners of NDPKs (FDR < 0.1). Given is ΔRT calculated in relation to the reference compounds and Δppm in relation to the monoisotopic mass given in Metlin27. Please click here to download this file.
Table S5. Proteins co-purified with NDPK1. Two independent lines (tagged in either N- or C-terminus) for each NDPK were run in triplicate. Student's t-test and further correction of p-value using Benjamini & Hochberg method were used to determine significantly enriched interactor partners of NDPKs (FDR < 0.1). Please click here to download this file.
Discussion
The presented protocol allows parallel identification of PP and PM complexes of a target protein. From cloning to final results, the experiment can be completed in as little as 8-12 weeks. Complete AP takes about 4-6 h for a set of 12 to 24 samples, rendering our protocol suitable for mid-throughput analysis.
The protocol, despite being overall straightforward, has a number of critical steps. (i) Sufficient amount of input protein and affinity beads is crucial to reach a dynamic range of metabolite detection. Efficient cell lysis is therefore a crucial step in the procedure. Poor protein yields can be a consequence of insufficient pulverization of the material or of suboptimal lysis-buffer/material ratio. (ii) Care should be taken that used reagents are MS-friendly. Strong detergents, glycerol, or excessive amounts of salt should be avoided as they interfere with MS detection. (iii) Agarose beads should not be over-dried during washing steps, and when using a vacuum manifold it is important to apply a slow flow rate so as not to destroy the beads or affect complex stability.
There are some important possible modifications to the presented protocol: (i) We use the constitutive CaMV35S promoter to maximize the amount of bait protein. Overexpression, while very useful, can have serious effects on cell homeostasis28 and lead to the formation of physiologically irrelevant interactions. Expression of tagged proteins using native promoters and where possible in a loss-of-function background is considered superior for retrieving true biological interactors. For proteins normally not expressed in plant cell cultures, a plant background may prove necessary to identify relevant interactors. (ii) When working with membrane proteins, the lysis buffer needs to be supplemented with an MS-compatible detergent. (iii) Introduction of a second affinity-purification step could improve false-positives to true-positives ratio and eliminate the need for EV controls29. A novel tandem tag with two independent protease-cleavage sites presents an attractive alternative to the size-exclusion chromatography step added by Maeda et al. 201411, which is both laborious and time consuming.
The most serious drawback of the AP is the high rate of false positives. The reasons are numerous. Constitutive overexpression was already mentioned. Another source of physiologically irrelevant interactions, unless working with isolated organelles, is preparation of whole-cell lysates containing mixtures of proteins and metabolites from different subcellular compartments. Subcellular localization should be used to filter for true interactors. Nevertheless, the majority of false positives result from unspecific binding between proteins and agarose resins. Introduction of a second purification step, as described above, offers the best solution to the problem, however comes at the cost of time and throughput. Moreover, weaker interaction may be lost as the protocol lengthens. Another caveat of AP is that despite the comprehensive information it provides about the interactome of a target protein, differentiating between direct and indirect targets of the baited protein is impossible. Targeted bimolecular approaches are needed to confirm interactions.
AP coupled with MS-based metabolomics was used to study protein- complexes in S. cerevisiae12. This work, together with our earlier observation13 that, similarly to lipids, polar and semi-polar compounds remain bound to protein complexes isolated from cellular lysates, provided conceptual groundwork for the presented protocol. Our protocol is characterized by three unique points: (i) In contrast to the yeast work12, it demonstrates that AP is suitable for retrieving not only hydrophobic but also hydrophilic protein ligands. (ii) By introducing a three-in-one extraction protocol, a single AP can be used to study protein and metabolite interactors of the bait protein. (iii) We adapted the protocol to plant cells.
Future efforts will focus on creating a novel tandem tag with two independent protease-cleavage sites. We would also like to explore suitability of the protocol to low-abundance small molecules such as plant hormones.
Disclosures
The authors have nothing to disclose.
Acknowledgements
We would like to kindly acknowledge Prof. Dr. Lothar Willmitzer for his involvement in the project, productive discussions, and great supervision. We are grateful to Dr. Daniel Veyel for helping with proteomic MS measurements. We appreciate Mrs. Änne Michaelis who provided us invaluable technical help with LC-MS measurements. Furthermore, we would like to thank Dr. Monika Kosmacz and Dr. Ewelina Sokołowska for their help and involvement in the work on the original manuscript, and to Weronika Jasińska for technical support.
Materials
| Name | Company | Catalog Number | Comments |
| Murashige and Skoog Basal Salts with minimal organics | Sigma-Aldrich | M6899 | |
| 1-Naphthylacetic acid | Sigma-Aldrich | N1641 | |
| Kinetin solution | Sigma-Aldrich | K3253 | |
| Tris base | Sigma-Aldrich | 10708976001 | |
| NaCl | Sigma-Aldrich | S7653 | |
| MgCl2 | Carl Roth | 2189.1 | |
| EDTA | Sigma-Aldrich | 3609 | |
| NaF | Sigma-Aldrich | S6776 | |
| DTT | Sigma-Aldrich | D0632 | |
| PMSF | Sigma-Aldrich | P7626 | |
| E-64 protease inhibitor | Sigma-Aldrich | E3132 | |
| Protease Inhibitor Cocktail | Sigma-Aldrich | P9599 | |
| Na3VO4 | Sigma-Aldrich | S6508 | |
| AcTEV Protease | Thermo Fischer Scientific | 12575015 | |
| Rotiphorese Gel 30 (37,5:1) | Carl Roth | 3029.2 | |
| TEMED | Carl Roth | 2367.3 | |
| PageRuler Prestained Protein Ladder | Thermo Fischer Scientific | 26616 | |
| SBP Tag Antibody (SB19-C4) | Santa Cruz Biotechnology | sc-101595 | |
| Goat anti-mouse IgG-HRP | Santa Cruz Biotechnology | sc-2005 | |
| Bradford Reagent | Sigma-Aldrich | B6916 | |
| Trypsin/Lys-C Mix, Mass Spec Grade | Promega | V5071 | |
| Urea | Sigma-Aldrich | U5128 | |
| Thiourea | Sigma-Aldrich | T8656 | |
| Ammonium bicarbonate | Sigma-Aldrich | 9830 | |
| Iodoacetamide | Sigma-Aldrich | I1149 | |
| MTBE | Biosolve | 138906 | |
| Methanol | Biosolve | 136806 | |
| Water | Biosolve | 232106 | |
| Acetonitrile | Biosolve | 12006 | |
| Trifluoroacetic acid | Biosolve | 202341 | |
| Formic acid | Biosolve | 69141 | |
| Unimax 2010 Platform Shaker | Heidolph | 5421002000 | |
| Nylon Mesh (Wire diameter 34 µM, thickness 55 µM, open area 14%) | Prosepa | Custom order | |
| Glass Funnel, 47 mm, 300 ml | Restek | KT953751-0000 | |
| Filter Bottle Top 500 mL 0,2 µM Pes St | VWR International GmbH | 514-0340 | |
| Mixer Mill MM 400 | Retsch GmbH | 207450001 | |
| IgG Sepharose 6 Fast Flow | GE Healthcare Life Sciences | 17-0969-02 | |
| Mobicol ""Classic"" with 2 different screw caps without filters | MoBiTec GmbH | M1002 | |
| Filter (small) 35 µM pore size, for Mobicol M 1002, M1003, M1050 & M1053 | MoBiTec GmbH | M513515 | |
| Variable Speed Tube Rotator SB 3 | Carl Roth | Y550.1 | |
| Rotary dishes for rotators SB 3 | Carl Roth | Y555.1 | |
| Resprep 24-Port SPE Manifolds | Restek | 26080 | |
| Finisterre C18/17% SPE Columns 100mg / 1ml | Teknokroma | TR-F034000 | |
| Autosampler Vials | Klaus Trott Chromatographie-Zubehör | 40 11 01 740 | |
| Acclaim PepMap 100 C18 LC Column | Thermo Fischer Scientific | 164534 | |
| EASY-nLC 1000 Liquid Chromatograph | Thermo Fischer Scientific | LC120 | |
| Q Exactive Plus Hybrid Quadrupole-Orbitrap Mass Spectrometer | Thermo Fischer Scientific | IQLAAEGAAPFALGMBDK | |
| Acquity UPLC system | Waters | Custom order | |
| ACQUITY UPLC HSS C18 Column, 100A, 1.8 µM, 2.1 mM X 100 mM, 1/pkg | Waters | 186003533 | |
| High-power ultrasonic cleaning baths for aqueous cleaning solutions | Bandelin | RK 31 | |
| Genedata Expressionist | Genedata | NaN | |
| Xcalibur Software | Thermo Fischer Scientific | NaN | |
| MaxQuant | NaN | NaN |
References
- Li, X., Snyder, M. Metabolites as global regulators: A new view of protein regulation. Bioessays. 33 (7), 485-489 (2011).
- Jacob, F., Monod, J. Genetic regulatory mechanisms in the synthesis of proteins. Journal of Molecular Biology. 3 (3), 318-356 (1961).
- Schlattner, U., et al. Dual Function of Mitochondrial Nm23-H4 Protein in Phosphotransfer and Intermembrane Transfer a cardiolipin-dependent switch. Journal of Biological Chemistry. 288 (1), 111-121 (2013).
- Ramírez, M. B., et al. GTP binding regulates cellular localization of Parkinson's disease-associated LRRK2. Human Molecular Genetics. , ddx161 (2017).
- Jung, H. J., Kwon, H. J. Target deconvolution of bioactive small molecules: the heart of chemical biology and drug discovery. Archives of Pharmacal Research. 38 (9), 1627-1641 (2015).
- Harding, M. W., Galat, A., Uehling, D. E., Schreiber, S. L. A receptor for the immunosuppressant FK506 is a cis-trans peptidyl-prolyl isomerase. Nature. 341 (6244), 758-760 (1989).
- Lomenick, B., et al. Target identification using drug affinity responsive target stability (DARTS). Proceedings of the National Academy of Sciences of the United States of America. 106 (51), 21984-21989 (2009).
- Manabe, Y., Mukai, M., Ito, S., Kato, N., Ueda, M. FLAG tagging by CuAAC and nanogram-scale purification of the target protein for a bioactive metabolite involved in circadian rhythmic leaf movement in Leguminosae. Chemical Communications. 46 (3), 469-471 (2010).
- Pantoliano, M. W., et al. High-density miniaturized thermal shift assays as a general strategy for drug discovery. Journal of Biomolecular Screening. 6 (6), 429-440 (2001).
- Li, X., Snyder, M. Analyzing In vivo Metabolite-Protein Interactions by Large-Scale Systematic Analyses. Current Protocols in Chemical Biology. , 181-196 (2010).
- Maeda, K., Poletto, M., Chiapparino, A., Gavin, A. -. C. A generic protocol for the purification and characterization of water-soluble complexes of affinity-tagged proteins and lipids. Nature Protocols. 9 (9), 2256-2266 (2014).
- Li, X., Gianoulis, T. A., Yip, K. Y., Gerstein, M., Snyder, M. Extensive in vivo metabolite-protein interactions revealed by large-scale systematic analyses. Cell. 143 (4), 639-650 (2010).
- Veyel, D., et al. System-wide detection of protein-small molecule complexes suggests extensive metabolite regulation in plants. Scientific Reports. 7, (2017).
- Luzarowski, M., et al. Affinity purification with metabolomic and proteomic analysis unravels diverse roles of nucleoside diphosphate kinases. Journal of Experimental Botany. , (2017).
- Van Leene, J., et al. Targeted interactomics reveals a complex core cell cycle machinery in Arabidopsis thaliana. Molecular systems biology. 6 (1), 397 (2010).
- Giavalisco, P., et al. Elemental formula annotation of polar and lipophilic metabolites using 13C, 15N and 34S isotope labelling, in combination with high-resolution mass spectrometry. The Plant Journal. 68 (2), 364-376 (2011).
- Van Leene, J., et al. Isolation of transcription factor complexes from Arabidopsis cell suspension cultures by tandem affinity purification. Plant Transcription Factors: Methods and Protocols. , 195-218 (2011).
- Van Leene, J., et al. A tandem affinity purification-based technology platform to study the cell cycle interactome in Arabidopsis thaliana. Molecular & Cellular Proteomics. 6 (7), 1226-1238 (2007).
- Olsen, J. V., Ong, S. -. E., Mann, M. Trypsin cleaves exclusively C-terminal to arginine and lysine residues. Molecular & Cellular Proteomics. 3 (6), 608-614 (2004).
- Cox, J., Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nature Biotechnology. 26 (12), 1367-1372 (2008).
- Cox, J., et al. Andromeda: A peptide search engine integrated into the MaxQuant environment. Journal of Proteome Research. 10 (4), 1794-1805 (2011).
- Tyanova, S., Temu, T., Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nature Protocols. 11 (12), 2301 (2016).
- Hooper, C. M., et al. SUBAcon: a consensus algorithm for unifying the subcellular localization data of the Arabidopsis proteome. Bioinformatics. 30 (23), 3356-3364 (2014).
- Katajamaa, M., Orešič, M. Data processing for mass spectrometry-based metabolomics. Journal of Chromatography A. 1158 (1-2), 318-328 (2007).
- Szklarczyk, D., et al. STITCH 5: augmenting protein-chemical interaction networks with tissue and affinity data. Nucleic Acids Research. 1277, (2015).
- Tanaka, K. -. i., et al. Dipeptidyl compounds ameliorate the serum-deprivation-induced reduction in cell viability via the neurotrophin-activating effect in SH-SY5Y cells. Neurological Research. 34 (6), 619-622 (2012).
- Smith, C. A., et al. METLIN: A metabolite mass spectral database. Therapeutic Drug Monitoring. 27, 747-751 (2005).
- Bhattacharyya, S., et al. Transient protein-protein interactions perturb E. coli metabolome and cause gene dosage toxicity. Elife. 5, (2016).
- Rigaut, G., et al. A generic protein purification method for protein complex characterization and proteome exploration. Nature Biotechnology. 17 (10), 1030-1032 (1999).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved