
Method Article
In Vitro Tumor Cell Rechallenge For Predictive Evaluation of Chimeric Antigen Receptor T Cell Antitumor Function
* These authors contributed equally
In This Article
Summary
Here, we describe an in vitro co-culture method to recursively challenge tumor-targeted T cells, which allows for phenotypic and functional analysis of antitumor T cell activity.
Abstract
The field of chimeric antigen receptor (CAR) T cell therapy is rapidly advancing with improvements in CAR design, gene-engineering approaches and manufacturing optimizations. One challenge for these development efforts, however, has been the establishment of in vitro assays that can robustly inform selection of the optimal CAR T cell products for in vivo therapeutic success. Standard in vitro tumor-lysis assays often fail to reflect the true antitumor potential of the CAR T cells due to the relatively short co-culture time and high T cell to tumor ratio. Here, we describe an in vitro co-culture method to evaluate CAR T cell recursive killing potential at high tumor cell loads. In this assay, long-term cytotoxic function and proliferative capacity of CAR T cells is examined in vitro over 7 days with additional tumor targets administered to the co-culture every other day. This assay can be coupled with profiling T cell activation, exhaustion and memory phenotypes. Using this assay, we have successfully distinguished the functional and phenotypic differences between CD4+ and CD8+ CAR T cells against glioblastoma (GBM) cells, reflecting their differential in vivo antitumor activity in orthotopic xenograft models. This method provides a facile approach to assess CAR T cell potency and to elucidate the functional variations across different CAR T cell products.
Introduction
Immunotherapy using chimeric antigen receptor (CAR)-engineered T cells has seen promising outcomes against B cell malignancies1,2,3,4, while the potential of targeting other tumors continues to be under rigorous investigation5,6,7. Great progress has been made to optimize the CAR construct, manufacturing process and patient pre-infusion regimens6,7,8, and novel synthetic biology approaches are expected to increase their persistence, safety and tumor specificity9,10. However, it has been difficult and labor-intensive to appropriately assess the therapeutic potential of CAR T cells in order to guide the best choice for clinical translation. The most established model thus far to evaluate CAR T cell function is in immunodeficient mice bearing human tumor xenografts, whereby CAR T cells are examined for antitumor efficacy and compared at titrated cell doses11,12,13,14,15. These in vivo murine studies are labor-intensive and time-consuming, especially when screening large numbers of parameters. Further, in vivo studies can be restrained by the accessibility of mouse strains, animal care facilities and animal-handling techniques. Therefore, there is a need to develop more convenient in vitro assays allowing for quick readouts of effector activity, which also faithfully reflect the in vivo antitumor function of these T cells.
Conventional methods to determine the cytotoxicity of T cells in vitro have focused on the detection of degranulation, cytokine production and the ability to lyse radioisotope-labeled target cells (i.e., chromium release assays). While these assays are informative for defining CAR T cell specificity and redirected target recognition, they often fail to reflect in vivo antitumor potential of engineered T cells12,13,16. In certain cases, in vitro killing activity in short term assays showed an inverse correlation with in vivo antitumor function16. Such inconsistency is likely the result of high effector:target (E:T) ratios used in these in vitro assays, and therefore the inability to differentiate CAR T cell products that are prone to exhaustion17. By contrast, during in vivo tumor eradication T cells usually respond against large tumor burdens, thereby requiring multiple rounds of killing and subsequently driving T cell differentiation and exhaustion18,19,20, which is one of the major barriers against effective tumor clearance by CAR T cells12,13. Meanwhile, most short-term in vitro killing assays also do not readout differences in T cell proliferation, whereas in CAR T cell treated patients the capacity for CAR T cell expansion is strongly correlated with clinical responses4. Thus, the appropriate in vitro assay would need to recapitulate conditions of high tumor burden, induction of T cell exhaustion, and allows for the readout of T cell expansion.
Here we describe a strategy to evaluate CAR T cells for repetitive tumor killing potential, with a simple in vitro co-culture assay. Different T cell effector activity parameters can be simultaneously examined, including target cell killing, CAR T cell expansion and memory- or exhaustion-associated phenotypes. The results generated from this assay correlate well with the in vivo antitumor effect of CAR T cells, and can be exploited to assess the potency of CAR T cell products. While we describe an assay to evaluate IL13Ra2-targeted CAR T cells against primary GBM lines21, it can be readily adapted to any CAR T cell platform.
Protocol
Our IRB does not require review of this protocol. We receive discarded fresh glioblastoma tumor samples from the City of Hope Pathology Department that are coded, and our laboratory cannot gain access to the key. We receive the specimens with data only on prior treatment and disease state at the time of biopsy/resection, with no identifying data.
1. Media Preparation
- Prepare neural stem cell media for culturing primary GBM cell lines: DMEM:F12, 1:50 B27, 5 µg/mL heparin, and 2 mmol/L L-glutamine; supplemented with 20 ng/mL epidermal growth factor (EGF) and 20 ng/mL basic fibroblast growth factor (FGF) twice a week (see Table of Materials).
- Prepare T cell media: X-VIVO 15 containing 10% fetal calf serum (FCS); supplemented with 70 IU/mL rhIL-2 and 0.5 ng/mL rhIL-15 every 48 h (see Table of Materials).
- Prepare co-culture media: take neural stem cell media without EGF and FGF supplement, and add 10% FCS.
- Prepare FACS staining solution (FSS): HBSS, 2% FCS, NaN3 (0.5 g/500 mL).
2. Preparation of GBM Tumor Cells
- Harvest low-passage GBM tumor spheres (TSs) by centrifugation at 300 x g for 4 min and discard supernatant.
NOTE: GBM tumor spheres (TSs) are generated from resected tumors as described previously22,23,24, and maintained in neural stem cell media, in incubators with 5% CO2 at 37 °C. - Pre-warm co-culture media in a 37 °C water bath.
- Add 1 mL of cold accutase to GBM TSs, dissociate TSs by pipetting for 30-60 s, and stop dissociation by adding 5 mL of warm co-culture media.
NOTE: GBM TSs should not be kept on accutase for more than 5 min. - Harvest GBM cells by centrifugation at 300 x g for 4 min, discard supernatant, and resuspend cells in 2 mL of co-culture media.
- Determine cell concentration using a cell viability counter.
NOTE: Cell viability must be >70%.
3. Preparation of CAR T Cells
- Less than 24 h before the assay, take 100 µL of cultured CAR T cells into a flow cytometry tube and add 2 mL of FSS.
NOTE: CAR T cells were generated as described previously11,21 and cultured in T cell media. - Centrifugation at 300 x g for 4 min and discard supernatant.
- Add 2 mL of FSS to wash the cells, centrifuge at 300 x g for 4 min, and discard supernatant.
- Stain the cells with appropriate antibody to indicate CAR expression at 4 °C for 30 min.
NOTE: For example, if an IL13Rα2-targeted CAR described previously25 is used, then stain with anti-IL13 antibody. - Wash twice with 2 mL of FSS and analyze CAR exn using a flow cytometer.
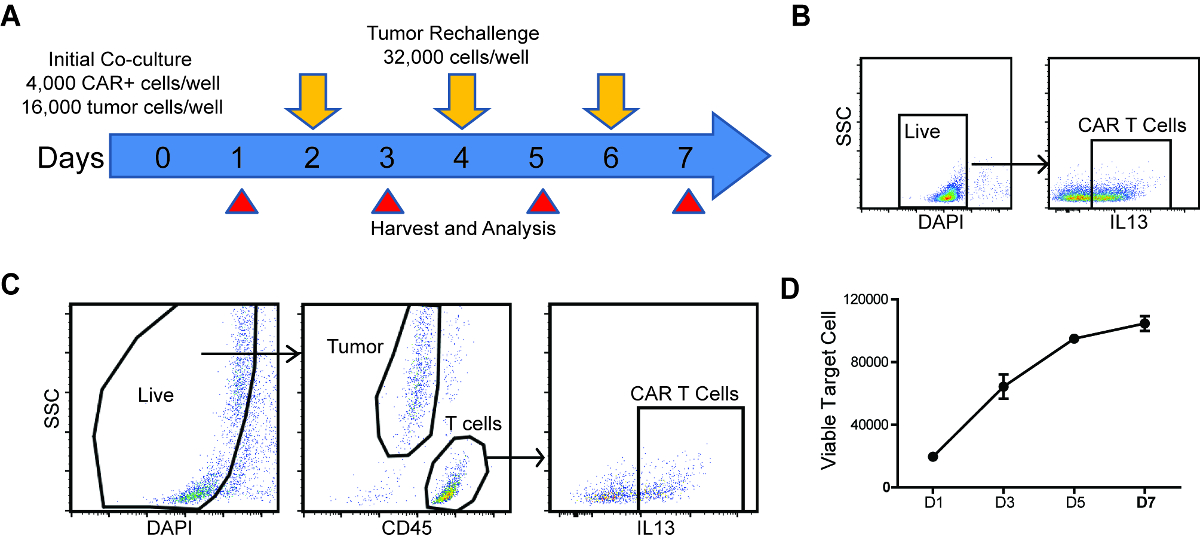
- Determine the CAR% on T cells using the gating strategy shown in Figure 1B.
- At the day of co-culture, harvest all CAR T cells by centrifugation at 300 x g for 4 min, discard supernatant, and resuspend cells in 2 mL of co-culture media.
- Determine cell concentration using a cell viability counter.
NOTE: Cell viability must be >70%.
4. Set up tumor — T Cell Co-culture
- Dilute tumor cells to a concentration of 0.16 million/mL with co-culture media.
- Based on CAR%, dilute CAR T cells to a concentration of 0.04 million CAR+ cells/mL with co-culture media.
NOTE: For example, if the CAR is 50% and T cell concentration is 0.4 million/mL, then make a 1:5 dilution to get the final concentration of 0.04 million CAR+ cells/mL. - Pipette 100 µL of diluted tumor cells into each well of a 96-well flat-bottom tissue culture plate.
- Pipette 100 µL of diluted CAR T cells into each well that contains tumor cells and mix well.
NOTE: Each tumor-T cell co-culture will be analyzed at 4 time points. 4-6 replicates might be required at every time point based on different analysis, but <3 replicates per time point is not recommended; see Table 1 for a representative platemap. - Maintain the plate in a 37 °C, 5% CO2 incubator.
5. Tumor Cell Rechallenge
NOTE: Rechallenge takes place at 2, 4 and 6 days post the initial co-culture setup (Figure 1A).
- Harvest and dissociate GBM TSs as described above (Steps 2.1-2.6).
- Resuspend GBM cells at a concentration of 0.64 million/mL.
- Determine the co-culture wells that need rechallenge (see Table 1) and carefully remove 50 µL media from the top of each well.
- Add 50 µL of GBM cell suspension into each well and mix well, then put the plate back into a 37 °C, 5% CO2 incubator.
6. Harvest Samples and Flow Cytometric Analysis
NOTE: Samples will be harvested at 1, 3, 5 and 7 days post the initial co-culture setup, with 1, 2, 3 and 4 rounds of tumor challenge, respectively.
- Pre-warm 0.05% trypsin-EDTA solution in 37 °C waterbath.
- Determine the wells that need harvesting and transfer the media into a new round-bottom 96-well plate.
- Pipette 50 µL of trypsin-EDTA into the wells to digest remaining tumor cells at 37 °C for 5 min.
- Under a microscope, confirm the cells have detached from the bottom.
- Pipette around the well bottom to resuspend detached cells, then transfer trypsin-EDTA containing detached cells to the corresponding wells of the round-bottom 96-well plate.
- Centrifuge the round-bottom 96-well plate at 300 x g, 4 °C for 4 min, then discard supernatant.
- Add 200 µL/well of FSS to wash the cells, centrifuge at 300 x g, 4 °C for 4 min, then discard supernatant.
- Resuspend cells in 100 µL/well FSS containing antibodies (see Table of Materials), and stain cells at 4 °C for 30 min.
- Add 100 µL/well FSS to cells, centrifuge at 300 x g, 4 °C for 4 min, then discard supernatant.
- Add 200 µL/well of FSS to wash the cells, centrifuge at 300 x g, 4 °C for 4 min, then discard supernatant.
- Resuspend cells with 100-200 µL/well of FSS with 500 ng/mL DAPI, then analyze samples by flow cytometry.
7. Functional and Phen Otypic Analysis of CAR T Cells
- Retrieve the data files from flow cytometer, and gate all live (DAPI-) cells (Figure 1C).
- Quantify tumor cells by gating the CD45- population and CAR T cells by gating the CD45+, CAR+ population (Figure 1C).
NOTE: If the tumor cells express CD45 (e.g. Raji lymphoma cells), anti-CD3 staining can be used to differentiate T cells from tumor cells. - Plot tumor cell and CAR T cell number through the time course.
- Identify T cell activation by the co-expression of 4-1BB and CD69.
NOTE: These surface markers can be used to analyze T cell activation 6-24 h post initial co-culture. - Identify T cell exhaustion by the expression of PD-1, LAG-3 and TIM-3.
- Identify T cell memory status by the expression of CD45RO and CD62L.
Results
Using the assay described above, we have distinguished the differential effector potency and killing dynamics mediated by CD4+ and CD8+ CAR T cells. The results presented here illustrate the difference between CD4+ and CD8+ CAR T cells, generated from a healthy donor independent of our previous publication21.
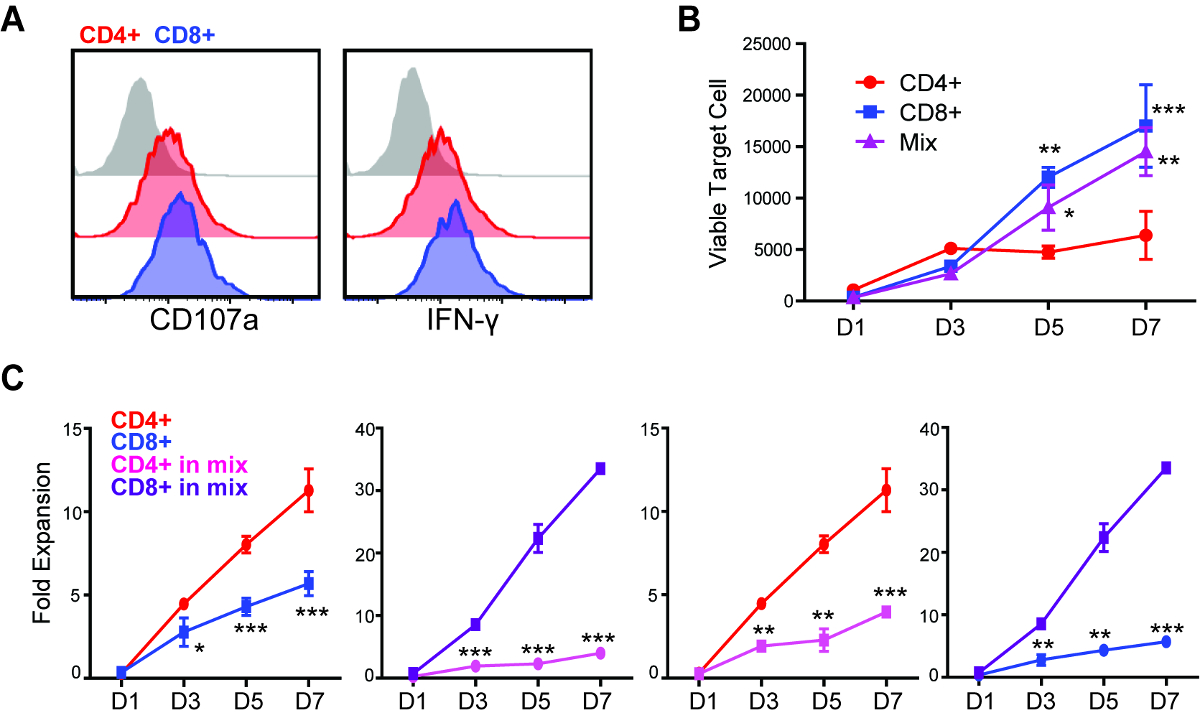
Through a standard degranulation assay, we were able to observe that both CD4+ and CD8+ CAR T cells became equally activated against GBM cells that express the targeted antigen, as indicated by the expression of CD107a and intracellular cytokine (Figure 2A). However, using the repetitive tumor challenge assay, we found that CD4+ but not CD8+ CAR T cells exhibited the capability of multiple-round killing (Figure 2B). CD4+ CAR T cells also achieved better expansion in comparison to CD8+ cells during this assay (Figure 2C). The difference of expansion between CAR T cell subsets were only observed from D3 after two rounds of tumor challenge (1:12 E:T), while the difference of cytotoxicity was observed beginning at D5 after three rounds of tumor challenge (1:20 E:T), indicating that short-term assays failed to elucidate differences in the potency of different CAR T cell products. When the 1:1 mixed CD4+ and CD8+ CAR T cells were applied to this assay, they were found to outperform CD8+ but not CD4+ CAR T cells on long-term cytotoxicity (Figure 2B). The expansion of CD8+ CAR T cells was induced by the co-applied CD4+ cells, while CD4+ CAR T cell expansion was inhibited in the presence of CD8+ cells (Figure 2C).
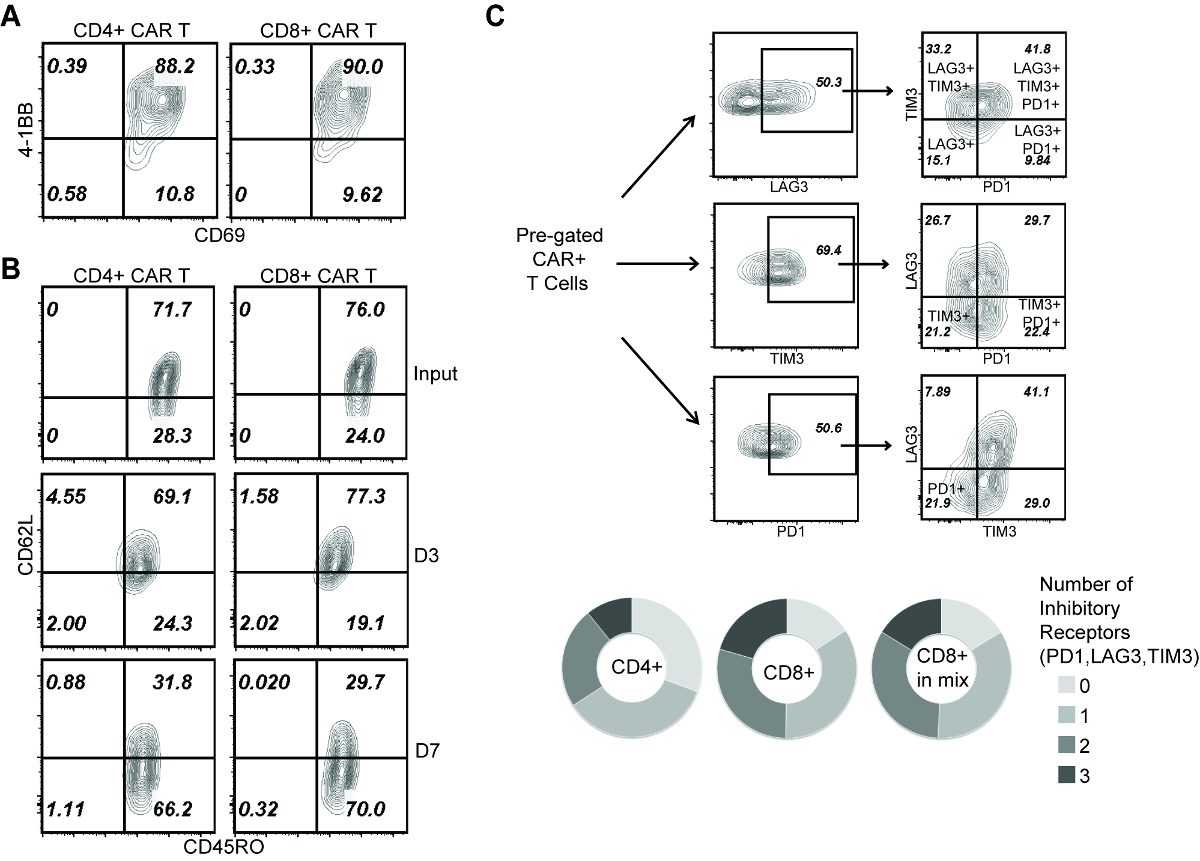
To explain the mechanism that distinguishes the effector activity between CD4+ and CD8+ CAR T cells, we first confirm that 24 h after initial co-culture, both CD4+ and CD8+ CAR T cells were comparably activated (Figure 3A). Meanwhile, as indicated by CD45RO and CD62L expression, both CD4+ and CD8+ CAR T cells showed a transition from central memory (CD45RO+, CD62L+) to effector memory (CD45RO+, CD62L-) phenotype during repetitive tumor challenge (Figure 3B). Then we characterized the cells at D3 of this assay, a time before the CAR T cell subsets started to display functional difference. T cell exhaustion was marked by the co-expression of inhibitory receptors PD-1, LAG-3 and TIM-315,21, which showed that CD8+ CAR T cells were more prone to exhaustion compared with CD4+ CAR T cells (Figure 3C). Further, no difference was seen on CD8+ CAR T cell exhaustion in the presence/absence of co-applied CD4+ cells (Figure 3C), indicating that CD4-induced CD8+ CAR T cell expansion is not associated with a better effector function.
Together, the results from this assay identified that CD4+ CAR T cells outperformed CD8+ cells specifically in long-term cytotoxicity. Despite the similar short-term cytotoxicity (1-3 days, once rechallenged), CD4+ CAR T cells sustained effector function upon repetitive tumor challenge, while CD8+ cells became exhausted and failed to control tumor cell growth. When the two subsets were mixed, CD4+ CAR T cells facilitated the expansion of CD8+ CAR T cells, but the exhaustion status of CD8+ cells were not ameliorated, resulting in the lack of synergistic effect between the two groups. Similar difference between CD4+ and CD8+ CAR T cells was seen in an in vivo model as previously described21. Indeed, CD8+ CAR T cells were able to mediate short-term tumor clearance but followed with antigen-positive recurrence; in contrast, CD4+ CAR T cell treatment resulted in long-term tumor eradication21, which is reminiscent of their repetitive killing potential using the in vitro rechallenge assay.

Figure 1: Schema and analysis strategy of repetitive challenge assay. (A) Schema and timeline of repetitive tumor challenge assay. For each well, CAR T cells were first co-cultured with PBT030-2 GBM cells (4,000 CAR+ cells, 16,000 tumor cells) and re-challenged with 32,000 tumor cells every other day (D2, D4 and D6). Analysis of tumor cell and CAR T cell number, as well as CAR T cell phenotype is carried out at D1, D3, D5 and D7. (B) Gating strategy to determine CAR% in T cells before setting up the co-culture. (C) Gating strategy of live cells, tumor cells and CAR T cells from the repetitive challenge assay. (D) Tumor cells number at different times of the rechallenge assay, co-cultured with untransduced T cells. Error bars = ±SEM. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Repetitive challenge assay reveals differences in killing and proliferative potency between CD4+ and CD8+ CAR T cells. (A) Degranulation and intracellular cytokine staining of CD4+ and CD8+ CAR T cells after 5 h of co-culture with GBM cells (E:T=1:1). (B) CD4+, CD8+ or mixed (CD4:CD8=1:1) CAR T cells were applied to repetitive challenge assay, and viable tumor cells were quantified. *p < 0.05, **p < 0.01, ***p < 0.001 compared with CD4+, using one-way ANOVA with Bonferroni's Multiple Comparison Tests. (C) CD4+ and CD8+ CAR T cell expansion during repetitive tumor challenge assay. Comparison of (left to right) CD4+ vs CD8+, single subset; CD4+ vs CD8+, within the "mixed" group; CD4+, single vs mixed; CD8+, single vs mixed. *p < 0.05, **p < 0.01, ***p < 0.001 using an unpaired Student's t test. Error bars = ±SEM. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Analysis of T cell phenotype during repetitive challenge assay. (A) 4-1BB and CD69 staining on CAR T cells at D1 of the assay. (B) CD45RO and CD62L staining of CAR T cells before applying to rechallenge assay (input), and at D3 and D7 of the assay. (C) Co-expression of PD-1, LAG-3 and TIM-3 on CAR T cells at D3 of the assay. (Top) Gating strategies to identify T cells expressing 1, 2 or 3 inhibitory receptors. (Bottom) Comparison of: CD4+ cells (single subset), CD8+ cells (single subset), CD8+ cells (within the "mixed" group). Please click here to view a larger version of this figure.
{kind=link}

Table 1: Representative plate map of rechallenge setup.
Discussion
This repetitive tumor cell challenge assay provides a convenient approach to evaluate CAR T cell functional potency, using an in vitro setup that recapitulates the high tumor burden associated with in vivo tumor models. During this 7-day assay, CAR T cells undergo four rounds of tumor challenge (initial co-culture and 3x rechallenge), creating an environment where exhausted T cells may lose their capability to respond to tumor challenge despite a potent initial response. Tumor cell elimination and CAR T cell expansion represent two fundamental readouts that can be acquired from this assay, while the phenotypes of CAR T cells can be easily analyzed by staining surface or intracellular markers that indicate T cell activation, exhaustion and/or memory. This assay is also convenient to combine with non-biased analysis of CAR T cells transcriptome, proteome or phosphor-proteome21,26, and T cell polyfunctionality which has been adopted to predict clinical responses27. Furthermore, tumor cells can also be tested for their adaptive response against T cell immunity such as cytokine secretion28. Since samples are harvested at multiple time points, this assay allows for not only static, but also dynamic analysis of CAR T cell behavior when responding to excess number of tumor cells.
The assay is particularly powerful in comparing the effector potency of CAR T cells targeting the same antigen but with different designs and/or manufacturing processes. We have used this assay to identify that CD4+ CAR T cells were able to mediate superior long-term effector function than CD8+ cells21. It was also shown that in vivo CAR efficacy was correlated with the cytotoxicity at later time points (D5-D7) during this assay, further highlighting the necessity of using repetitive tumor challenge for in vitro CAR T cell evaluation. Although GBM cells were used as the example of target cells here, this assay could be easily adopted to a different T cell-tumor combination. Notably, CAR T cell behavior can also vary upon different antigen densities and engineering cancer cells to express various levels of targeted antigens provided the tool to investigate sequential molecular events upon CAR recognition29. Therefore, this assay can also be exploited to examine the activation pattern of certain CAR T cells against different targets.
Since the target cell viability and CAR T cell expansion are two upfront readouts of this assay, it is critical that all the co-cultures start with viable (>70%) tumor and T cells. When performing the rechallenge processes, the action of aspirating media (step 5.3) should not disturb tumor and T cells in the bottom of the wells, in order not to introduce unnecessary variations of cells counts. When performing this assay on suspension target cell lines, it is recommended to harvest remaining cells by centrifugation before tumor cell rechallenge.
One limitation of this assay is that the setup parameters (E:T ratios of initial co-culture and rechallenge) needs to be adjusted based on different CAR-tumor combinations. For example, if the tumor cells express a considerable level of immuno-inhibitory molecules (e.g. PD-L1), a higher E:T ratio is recommended given that the overall killing efficiency is impaired compared with PD-L1-low tumor cells. Before applying the assay to new CAR-tumor combinations, a pilot study is recommended to determine the optimal conditions, which usually allows the most potent CAR T cells tested in the assay to eliminate >80% of tumor cells at the end point. Since direct cell-cell contact is needed for CAR T cell-mediated killing, if the tumor cells were fluorescence-labeled, then the panel of flow cytometric analysis must be adjusted accordingly (e.g. if the tumor cells were GFP-labeled, then any FITC-conjugated antibodies should not be used).
The clinical activity of CAR T cells against B cell malignancies has led to two FDA-approved drugs. However, the response has shown substantial variation across different patients27,30,31, which was elucidated to correlate with the phenotypic properties of the CAR products30. This in vitro repetitive tumor challenge assay further provides an approach to functionally test the CAR effector potency in addition to the phenotypic characterizations and polyfunctionality cytokine production, and allows for higher-throughput screening of clinical products in comparison to in vivo tumor models while retaining the predictive fidelity. Overall, this assay could be used for T cell functional test for CAR T cell design, pre-clinical and clinical development.
Disclosures
The CAR construct described in this work have been licensed by Mustang Bio., Inc., for which S.J.F. and C.E.B. receive royalty payments. All other authors declare no potential conflicts of interest.
Acknowledgements
This work was supported by grants from the California Institute for Regenerative Medicine (CIRM) grant TR3-05641 and NIH grants P30 CA33572 (cores). D.W. is supported by NCI fellowship 1F99CA234923-01.
Materials
| Name | Company | Catalog Number | Comments |
| 0.05% Trypsin/0.53mM EDTA in HBSS without Calcium and Magnesium | Corning | MT25051CI | |
| 1 M Hepes | Irvine Scientific | 9319 | |
| 200 mM L-Glutamine | Cambrex Bio Science | 17-605E | |
| 4',6-diamidino-2-phenylindole, dihydrochloride (DAPI) - FluoroPure grade | Invitrogen | D21490 | |
| Accutase | Innovative Cell Technologies | AT104 | |
| Aldesleukin Proleukin (rhIL-2) | Novartis Oncology | NDC 0078-0495-61 | |
| Anti-CD137, PE | BD Biosciences | 555956 | 4B4-1 |
| Anti-CD19, PE-Cy7 | BD Biosciences | 557835 | SJ25C1 |
| Anti-CD3, PERCP | BD Biosciences | 340663 | SK7 |
| Anti-CD4, FITC | BD Biosciences | 340133 | SK3 |
| Anti-CD45, PERCP | BD Biosciences | 340665 | 2D1 |
| Anti-CD45RO, PE | BD Biosciences | 561137 | UCHL1 |
| Anti-CD62L, APC | BD Biosciences | 559772 | DREG-56 |
| Anti-CD69, APC | BD Biosciences | 340560 | L78 |
| Anti-CD8, APC-Cy7 | BD Biosciences | 348793 | SK1 |
| Anti-IL-13, PE | BD Biosciences | 340508 | JES10-5A2 |
| Anti-LAG-3, PE | eBiosciences | 12-2239-41 | 3DS223H |
| Anti-PD-1, APC-Cy7 | BioLegend | 329921 | EH12.2H7 |
| Anti-TIM-3, APC | eBiosciences | 17-3109-42 | F38-2E2 |
| B-27 Serum-Free Supplement (50x) | Invitrogen | 17504-044 | |
| Corning 96 Well Clear Flat Bottom Polystyrene TC-Treated Microplates, Individually Wrapped, with Lid, Sterile (Product #3596) | Corning Life Sciences | 3596 | |
| Defined fetal bovine serum,HI IR | Hyclone Labs | SH30070.03IH | |
| DMEM F-12 50:50 (1x) without Glutamine | MediaTech, Inc. | 15-090-CV | |
| DMEM High Gluc w/o L-Glu, Na Pyr 1 L | Invitrogen | 11960051 | |
| DMEM-Ham's F12 50:50 Mixture with L-glutamine and 15 mM HEPES | Fisher Scientific | MT10092CV | |
| HBSS | Irvine Scientific | 9224 | |
| Heat-Inactivated FCS | Hyclone | SH30070.03 | |
| Heparin Sodium(1,000 U/mL) | American Pharmaceutical | 401811B | |
| MACSQuant | Milteni Biotech Inc. | BIS013220 | |
| PBS 1x W/CA & MG | Irvine Scientific | 9236 | |
| PBS with 1 mM EDTA (No Ca2+ or Mg2+) | VWR Scientific Products | PB15009A | |
| Recombinant human EGF | R&D Systems | 236-EG-200 | |
| Recombinant human FGF | R&D Systems | 233-FB | |
| rhIL-15 Working Dilution (10 ng/µL) | CellGenix, US Operations | tF0297 | |
| Sodium Azide (NaN3) | Sigma | S8032 | |
| Sorvall ST40R | Thermo Scientific | 41693700 | |
| TC-HEPES BUFFER (1 M) SOLN 100ML | IRVINE SCIENTIFIC CORP | 6472 | |
| Tecnol Procedure Mask | Kimberly-Clark | 47117 | |
| X-VIVO 15 | Lonza | BW04-744Q |
References
- Porter, D. L., et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Science Translational Medicine. 7 (303), (2015).
- Brentjens, R. J., et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Science Translational Medicine. 5 (177), (2013).
- Grupp, S. A., et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. The New England Journal of Medicine. 368 (16), 1509-1518 (2013).
- Lee, D. W., et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. The Lancet. 385 (9967), 517-528 (2015).
- Yong, C. S. M., et al. CAR T-cell therapy of solid tumors. Immunology and Cell Biology. 95 (4), 356-363 (2017).
- Barrett, D. M., Singh, N., Porter, D. L., Grupp, S. A., June, C. H. Chimeric antigen receptor therapy for cancer. Annual Review of Medicine. 65, 333-347 (2014).
- Priceman, S. J., Forman, S. J., Brown, C. E. Smart CARs engineered for cancer immunotherapy. Current Opinion in Oncology. 27 (6), 466-474 (2015).
- Fesnak, A. D., June, C. H., Levine, B. L. Engineered T cells: the promise and challenges of cancer immunotherapy. Nature Reviews Cancer. 16 (9), 566-581 (2016).
- Esensten, J. H., Bluestone, J. A., Lim, W. A. Engineering Therapeutic T Cells: From Synthetic Biology to Clinical Trials. Annual Review of Pathology. 12, 305-330 (2017).
- Lim, W. A., June, C. H. The Principles of Engineering Immune Cells to Treat Cancer. Cell. 168 (4), 724-740 (2017).
- Brown, C. E., et al. Optimization of IL13Ralpha2-Targeted Chimeric Antigen Receptor T Cells for Improved Anti-tumor Efficacy against Glioblastoma. Molecular Therapy. , (2017).
- Long, A. H., et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nature Medicine. 21 (6), 581-590 (2015).
- Cherkassky, L., et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. The Journal of Clinical Investigation. 126 (8), 3130-3144 (2016).
- Priceman, S. J., et al. Co-stimulatory signaling determines tumor antigen sensitivity and persistence of CAR T cells targeting PSCA+ metastatic prostate cancer. Oncoimmunology. 7 (2), 1380764(2018).
- Eyquem, J., et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 543 (7643), 113-117 (2017).
- Gattinoni, L., et al. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. The Journal of Clinical Investigation. 115 (6), 1616-1626 (2005).
- Malandro, N., et al. Clonal Abundance of Tumor-Specific CD4(+) T Cells Potentiates Efficacy and Alters Susceptibility to Exhaustion. Immunity. 44 (1), 179-193 (2016).
- Gattinoni, L., et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nature Medicine. 15 (7), 808-813 (2009).
- Klebanoff, C. A., et al. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proceedings of the National Academy of Sciences of the United States of America. 102 (27), 9571-9576 (2005).
- Gattinoni, L., Klebanoff, C. A., Restifo, N. P. Paths to stemness: building the ultimate antitumour T cell. Nature Reviews Cancer. 12 (10), 671-684 (2012).
- Wang, D., et al. Glioblastoma-targeted CD4+ CAR T cells mediate superior antitumor activity. JCI Insight. 3 (10), (2018).
- Kahlon, K. S., et al. Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Research. 64 (24), 9160-9166 (2004).
- Brown, C. E., et al. Recognition and killing of brain tumor stem-like initiating cells by CD8+ cytolytic T cells. Cancer Research. 69 (23), 8886-8893 (2009).
- Lee, J., et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 9 (5), 391-403 (2006).
- Brown, C. E., et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. The New England Journal of Medicine. 375 (26), 2561-2569 (2016).
- Salter, A. I., et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Science Signaling. 11 (544), (2018).
- Rossi, J., et al. Preinfusion polyfunctional anti-CD19 chimeric antigen receptor T cells are associated with clinical outcomes in NHL. Blood. 132 (8), 804-814 (2018).
- Garcia-Diaz, A., et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Reports. 19 (6), 1189-1201 (2017).
- Walker, A. J., et al. Tumor Antigen and Receptor Densities Regulate Efficacy of a Chimeric Antigen Receptor Targeting Anaplastic Lymphoma Kinase. Molecular Therapy. 25 (9), 2189-2201 (2017).
- Fraietta, J. A., et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nature Medicine. 24 (5), 563-571 (2018).
- Fraietta, J. A., et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature. 558 (7709), 307-312 (2018).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved