
Method Article
Investigation of the Transcriptional Role of a RUNX1 Intronic Silencer by CRISPR/Cas9 Ribonucleoprotein in Acute Myeloid Leukemia Cells
In This Article
Summary
Direct delivery of preassembled Cas9/guide RNA ribonucleoprotein complexes is a fast and efficient means for genome editing in hematopoietic cells. Here, we utilize this approach to delete a RUNX1 intronic silencer and examine the transcriptional responses in OCI-AML3 leukemic cells.
Abstract
The bulk of the human genome (~98%) is comprised of non-coding sequences. Cis-regulatory elements (CREs) are non-coding DNA sequences that contain binding sites for transcriptional regulators to modulate gene expression. Alterations of CREs have been implicated in various diseases including cancer. While promoters and enhancers have been the primary CREs for studying gene regulation, very little is known about the role of silencer, which is another type of CRE that mediates gene repression. Originally identified as an adaptive immunity system in prokaryotes, CRISPR/Cas9 has been exploited to be a powerful tool for eukaryotic genome editing. Here, we present the use of this technique to delete an intronic silencer in the human RUNX1 gene and investigate the impacts on gene expression in OCI-AML3 leukemic cells. Our approach relies on electroporation-mediated delivery of two preassembled Cas9/guide RNA (gRNA) ribonucleoprotein (RNP) complexes to create two double-strand breaks (DSBs) that flank the silencer. Deletions can be readily screened by fragment analysis. Expression analyses of different mRNAs transcribed from alternative promoters help evaluate promoter-dependent effects. This strategy can be used to study other CREs and is particularly suitable for hematopoietic cells, which are often difficult to transfect with plasmid-based methods. The use of a plasmid- and virus-free strategy allows simple and fast assessments of gene regulatory functions.
Introduction
Cis-regulatory elements (CREs) are non-coding DNA sequences that contain binding sites for transcriptional regulators to control gene expression1,2. These elements are typically 100 to 1,000 base pairs (bp) long. Promoters and enhancers are the two most characterized types of CREs. Promoters are present in close proximity to the transcription start sites and constitute the basic unit of transcription. Many genes have more than one promoter and their alternative use contributes to transcriptome diversity and tissue specificity3,4. On the other hand, enhancers activate transcription and can be located upstream, downstream or within introns of the target genes. Enhancers can act from far distance (over a megabase) and independent of orientation1,2. CREs also include silencers and insulators5,6. The former acts oppositely to enhancers to inhibit gene expression by binding to transcriptional repressors, whereas the latter partitions the genome into discrete topologically domains to insulate genes from other CREs from neighboring domains. These elements act in concert with each other through short- and/or long-range chromatin interactions and are organized into regulatory hubs to direct proper spatiotemporal gene expression. Recent advancements in high-throughput sequencing techniques have accelerated the identification and functional annotation of many CREs that have greatly facilitated our understandings of the transcriptional networks that dictate lineage-specific gene expression in different cell and tissue types7,8,9,10,11,12.
Given the fundamental roles of CREs in regulating transcription, their alterations can lead to aberrant gene expression. It has been shown that CREs are frequently disrupted by genetic and epigenetic changes in different types of human cancers, thereby contributing to tumor initiation, progression and aggressiveness13,14. In addition, CRE-binding factors are often mutated and/or misexpressed in various cancer types, further highlighting the significance of CRE deregulation in oncogenesis15. CREs can also be affected by structural aberrations, as exemplified by frequent chromosomal rearrangements of the immunoglobulin heavy (IgH) gene enhancer that result in abnormal activation of the neighboring oncogenes in B-cell lymphomas16. In acute myeloid leukemia (AML), repositioning of a single enhancer by chromosome 3q rearrangements causes concomitant GATA2 downregulation and EVI1 activation, which can be potentially targeted by BET inhibition of enhancer functions17. Recently, we characterized a novel chromosomal translocation involving perturbation of a RUNX1 intronic silencer that might contribute to AML progression in a pediatric patient18. Thus, deciphering the non-coding cancer genome provides fruitful avenues for elucidating disease pathogenesis, biomarker discovery and therapeutic interventions, which ultimately improve patient outcomes.
The CRISPR/Cas9 nuclease pathway, originally identified as an adaptive immune system in prokaryotic cells, has been exploited as a rapid and cost-effective means for site-specific genomic editing in living cells and organisms19,20,21,22. The CRISPR/Cas9 system involves two principal components: the gRNA and Streptococcus pyogenes-derived Cas9 nuclease. The gRNA contains a specific sequence called protospacer that recognizes the target region and directs Cas9 for editing. The gRNA is composed of two parts: CRISPR RNA (crRNA), typically a 20-mer nucleotide sequence complementary to the target DNA, and a trans-activating crRNA (tracrRNA), which serves as a binding scaffold for the nuclease. A protospacer adjacent motif (PAM) (5'-NGG) immediately adjacent to the target site is required for Cas9 cleavage and the cleavage site is located 3 nucleotides upstream of the PAM. CRISPR/Cas9-mediated gene editing is commonly performed by transfecting cells with a plasmid that encodes Cas9 and the cloned gRNA23. However, this approach is challenging for hematopoietic cells, which are often hard to transfect and require lengthy virus-based transduction methods. An alternative approach is direct cellular delivery of preassembled Cas9/gRNA RNP complexes24. A common method for the RNP delivery is electroporation, which generates temporary pores in the cell membrane, thus allowing entry of the RNP complexes into the cells25,26. The advantages of this approach include ease of use, reduced off-target effects and stability of the RNP complexes. Here, we describe a protocol of using the RNP delivery method to investigate the transcriptional role of a RUNX1 intronic silencer in the OCI-AML3 leukemia cell line18, which was established from the peripheral blood of an AML patient diagnosed with the French-American-British M4 subtype27. The protocol includes the design of crRNA, preparation of RNP complexes, electroporation as well as screening and subsequent characterization of the desired clones.
Protocol
1. Design of crRNA
- Design two crRNAs, one 5' and the other 3' of the target CRE using a web-based CRISPR design tool28,29,30,31. Ensure that a PAM of NGG is located immediately downstream of the target sequence for Cas9 recognition. The design of the two crRNAs (crRNA-1 and crRNA-2) for the deletion of the RUNX1 silencer18 is shown in Figure 1.
NOTE: A crRNA typically contains a 20-mer protospacer that is complementary to the target sequence. - Check the presence of single nucleotide polymorphisms (SNPs)/indels in the target and adjacent PAM sequences by entering the genomic locations of the sequences into the search box of an online genome browser (e.g., NCBI 1000 Genomes or UCSC Genome Browser).
NOTE: Common SNPs/indels have a minor allele frequency of at least 1% in the general population. - Submit the selected crRNA sequences for synthesis with a commercial vendor. Also, purchase the tracrRNA for gRNA duplex formation.
2. Design of Deletion Screening Primers
- Design a pair of primers that flank the intended deletion region. Ensure that the primers are at least 50 bp from the Cas9 cleavage sites so that PCR amplification is minimally affected by indels formed at the cleavage sites.
- Ensure that the amplicon is smaller than 1,200 bp (preferably smaller than 600 bp for better size resolution). Also, label one of the primers with a fluorescent dye (e.g., 6-carboxyfluorescein (6-FAM)) at the 5'-end for the detection. The primers used for screening of the RUNX1 silencer deletion18 are shown in Figure 1.
3. Preparation of Cas9/gRNA RNP Complexes
- Resuspend the crRNAs and tracrRNA in 1x TE buffer (10 mM Tris, 0.1 mM EDTA, pH 7.5) to a final concentration of 200 µM.
- For each crRNA, mix 2.2 µL of 200 µM crRNA, 2.2 µL of 200 µM tracrRNA and 5.6 µL of 1x TE buffer (total 10 µL) in a 0.2 mL tube to obtain a final duplex concentration of 44 µM.
- Incubate at 95 °C for 5 min in a thermocycler. Allow the tubes to cool to room temperature for gRNA complex formation.
- Dilute 10.4 µL of 62 µM recombinant Cas9 nuclease with 7.6 µL of 1x phosphate-buffered saline (PBS) to obtain a final nuclease concentration of 36 µM.
NOTE: This amount is sufficient for the preparation of two Cas9/gRNA complexes. - Mix equal volumes (8.5 µL) of the diluted nuclease with each of the gRNA duplexes obtained from step 3.3.
- Incubate at room temperature for 20 min to allow RNP complex formation. Keep the mixtures on ice until electroporation.
4. Electroporation of the RNP Complexes into OCI-AML3 Cells
- Culture OCI-AML3 cells in RPMI 1640 medium supplemented with 10 % heat-inactivated fetal bovine serum (FBS), 1x GlutaMAX, 100 units/mL of penicillin and 100 µg/mL of streptomycin (hereafter referred to as complete RPMI 1640 medium) at 37 °C with 5 % CO2.
- Count cells using trypan blue staining. Ensure that the cell viability at the time of electroporation is over 90%.
- Centrifuge 2.5 x 106 cells in a 1.5 mL tube at 500 x g for 5 min at room temperature. Remove the supernatant and wash the cells with 1 mL of 1x PBS. Spin down the cells again and remove all residual supernatant.
- Resuspend the cells in 163 µL of RPMI 1640 medium without phenol red. Add 16.7 µL of each RNP complex (from step 3.6) and 3.6 µL of 100 µM Electroporation Enhancer to the cells (total volume = 200 µL). Mix gently by pipetting.
NOTE: The Electroporation Enhancer is a carrier DNA for enhancing the editing efficiency. - Transfer the mixture to a 0.2 cm-gap electroporation cuvette without any bubbles. Perform electroporation with an electroporation system (Mode: exponential; Voltage =: 150 V; Capacitance = 700 µF; Resistance = 50 Ω).
- Transfer the cells to a T-25 tissue culture flask containing 6 mL of complete RPMI 1640 medium and incubate at 37 °C with 5 % CO2.
5. Screening and Selection of Cell Clones with Biallelic Deletions
- Dilute the cells to 5 x 103/mL in complete RPMI 1640 medium one day after electroporation. Add 100 µL of the diluted cell suspension into each well of 96-well tissue culture plates and allow the cells to grow for 7-14 days.
- Extract genomic DNA from the cells using a high-throughput purification system.
- Add 100 µL of plate binding and lysis buffer to each well of a 96-well extraction plate. Add 5 x 104 cells resuspended in 10 µL of 1x PBS to the buffer and mix them by pipetting.
- Incubate at room temperature for 30 min to allow binding of genomic DNA to the wells.
- Aspirate the solution from the wells without scraping the well surfaces. Wash the wells with 120 µL of wash buffer.
- Air dry the wells containing the bound DNA.
- Prepare 20 µL of PCR mix (2 µL of 10x High Fidelity buffer, 0.8 µL of 50 mM MgSO4, 0.4 µL of 10 mM dNTPs, 0.4 µL of 10 µM FAM-labeled forward primer (step 2.2), 0.4 µL of 10 µM unlabeled reverse primer and 0.4 U of Taq DNA polymerase for each sample).
NOTE: Prepare a master mix to ensure the addition of standardized amounts of reagents in each sample. - Add the PCR mix to each well of the extraction plate and run the reactions in a thermocycler (Conditions: initial 94 °C for 2 min, followed by 35 cycles of 94 °C for 15 s, 56 °C for 30 s and 68 °C for 1 min).
- Estimate the amount of the products by measuring their concentrations in a selected number of samples using a fluorometer. Dilute all samples with nuclease-free H2O to 0.5 ng/µL.
- Mix 1 µL of the diluted PCR products with 8.5 µL of deionized formamide and 0.5 µL of fluorescent dye-labeled size standard in a 96-well plate compatible with the genetic analyzer.
NOTE: Prepare a master mix containing deionized formamide and the size standard. - Cover the plate with a plate septa and denature the samples at 95 °C for 3 min in a thermocycler. Do not close the lid of the machine.
- Perform capillary gel electrophoresis to separate the labeled PCR products as previously described32.
- After electrophoresis, open the analysis software to analyze the results.
- Click New Project and select Microsatellite. Then click OK.
- Click Add Samples to Project and select the result files (contain the .fsa extension). Then click Add to list to import the files.
- In the table showing the selected result files, choose Microsatellite Default in the Analysis Method column. Also, select the size standard used in the Size Standard column. Then click the Analyze icon, enter the experiment name and save the experiment.
- Click Display Plots to view the results and choose Fragment Analysis in the plot setting.
- Choose the appropriate colored channels for analysis. Check the orange icon to view the labeled fragments in the size standard to assess the quality of size calling.
- Check the blue icon to view the labeled PCR products. Identify the peaks that correspond to the wild-type and mutant (i.e., bearing the expected deletions) products. Estimate the mutant level in each sample by dividing the area under the mutant peak by the sum of the area under the wild-type and mutant peaks.
- Select multiple cell pools with high levels of the expected deletions for further serial dilutions.
- Repeat the DNA extraction, fluorescent PCR and capillary electrophoresis steps. Select cell clones with mutant levels >95% representing biallelic deletions for subsequent analyses.
- Verify the identity of the deletions in the selected clones by Sanger sequencing.
6. Functional Analyses of the Silencer Deletion by Real-time Quantitative RT-PCR Analysis
- Extract total RNA from the selected clones and perform complementary DNA (cDNA) synthesis.
- Mix 1 µg of RNA with 1 µL of 50 µM oligo(dT)20 primer and 1 µL of 10 mM dNTP mix in a total volume of 10 µL. Incubate at 65 °C for 5 min and then place on ice for at least 1 min.
- Add 10 µL of cDNA synthesis mix containing 2 µL of 10x RT buffer, 4 µL of 25 mM MgCl2, 2 µL of 0.1 M DTT, 1 µL of RNase inhibitor (40 U/µL) and 1 µL of reverse transcriptase (200 U/µL). Incubate at 50 °C for 50 min and then 85 °C for 5 min in a thermocycler.
NOTE: Prepare a master mix for the reverse transcription. - Chill the samples on ice. Add 1 µL of RNase H and incubate at 37 °C for 20 min. Store the cDNA at -20 °C.
- Design primers and TaqMan probes that specifically recognize individual transcript variants generated from alternative promoters.
NOTE: Pre-designed transcript-specific primer/probe sets are commercially available. - Clone DNA fragments containing the specific transcript sequences into plasmid DNA. Prepare a 10-fold dilution series (106 to 10 copies) of the recombinant plasmids as standard curves for transcript quantification.
- Prepare 20 µL of PCR mix (0.5 µL of DNA template, 1 µL of 20x pre-designed TaqMan probe/primer assay and 10 µL of 2x TaqMan PCR Master Mix) for each sample (both cDNA and plasmid standards). Measure each sample in triplicate.
NOTE: Prepare a master mix for the real-time PCR. - Run the reactions in a real-time PCR machine (Conditions: initial 50 °C for 2 min and 95 °C for 10 min, followed by 40 cycles of 94 °C for 15 s and 60 °C for 1 min).
- After the amplification, click the Analyze icon in the software to analyze the data. Check the slope and the correlation coefficient of the standard curves to evaluate the efficiency and linearity of the reactions. Ensure that the slopes are between -3.1 and -3.6 and correlation coefficients are greater than 0.99.
- Normalize the copy number of the target transcripts in each sample with a housekeeping gene (e.g., GAPDH).
Results
The aim of this experiment is to delete an intronic silencer in the RUNX1 gene and examine the impacts on RUNX1 transcription in OCI-AML3 cells. The silencer was identified by a combinatorial molecular approach and found to contain a 209 bp core element18. To enable more accurate evaluation of this core element in controlling RUNX1 expression, the crRNAs (crRNA-1 and crRNA-2) were designed to target closely to this region18 (Figure 1). The predicted Cas9 cleavage sites brought by crRNA-1 and crRNA-2 were 29 bp and 35 bp from the core element, respectively (Figure 1). It should be noted that while the PAM sites of the two crRNAs reside on opposite strands, DSBs will occur independently of PAM sequence location. Thus, the concomitant introduction of two Cas9/gRNA RNP complexes guided by crRNA-1 and crRNA-2 is expected to excise the silencer element from the RUNX1 locus.
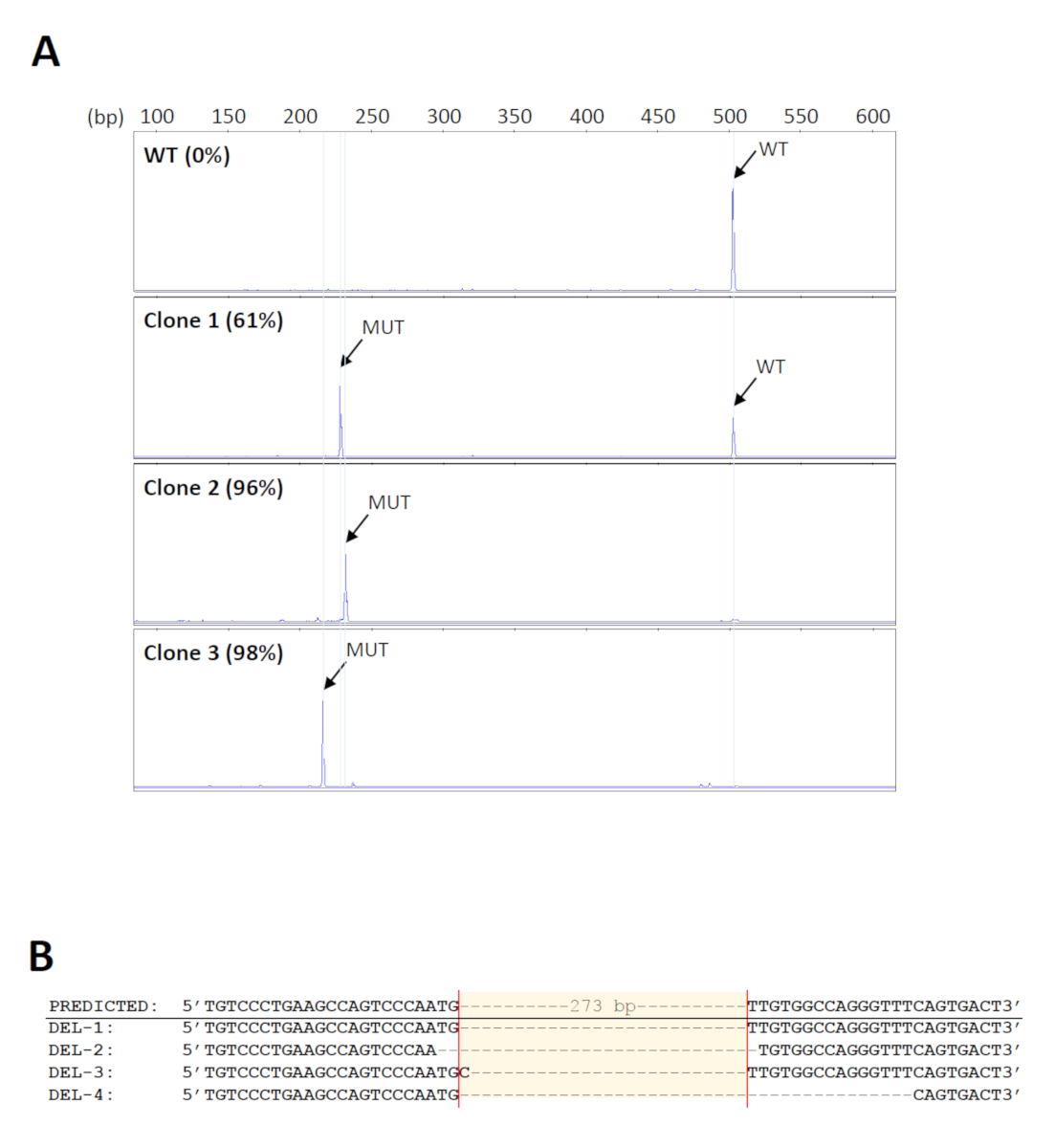
To screen for the desired deletions in a large number of samples, a 96-well format of genomic DNA extraction kit was used for high-throughput purification. Also, DNA bound on the wells of the extraction plates can be subjected to direct amplification, thus minimizing errors or contamination due to repeated sample transfer. The primers used for screening of the deletions18 are shown in Figure 1. The expected size of the wild-type PCR product is about 500 bp. Since the intended deletion spans 273 bp, the mutant product is expected to be about 230 bp. This size range enables simple and rapid fragment analysis by capillary gel electrophoresis. A total of 160 initial cell pools were screened and 14 were found to carry the expected deletions with mutant levels of at least 70%. Five pools were then selected for further serial dilutions to identify clones bearing biallelic deletions. Representative electropherograms from cell clones with different levels of mutant products are shown in Figure 2A. The identity of the deletions was verified by Sanger sequencing (Figure 2B). As expected, indels formed by non-homologous end joining repair of DSBs33 were observed at the predicted cleavage sites in the deletion clones. These resulted in the amplification of mutant products of varying sizes, which could also be detected by capillary electrophoresis (Figure 2A).
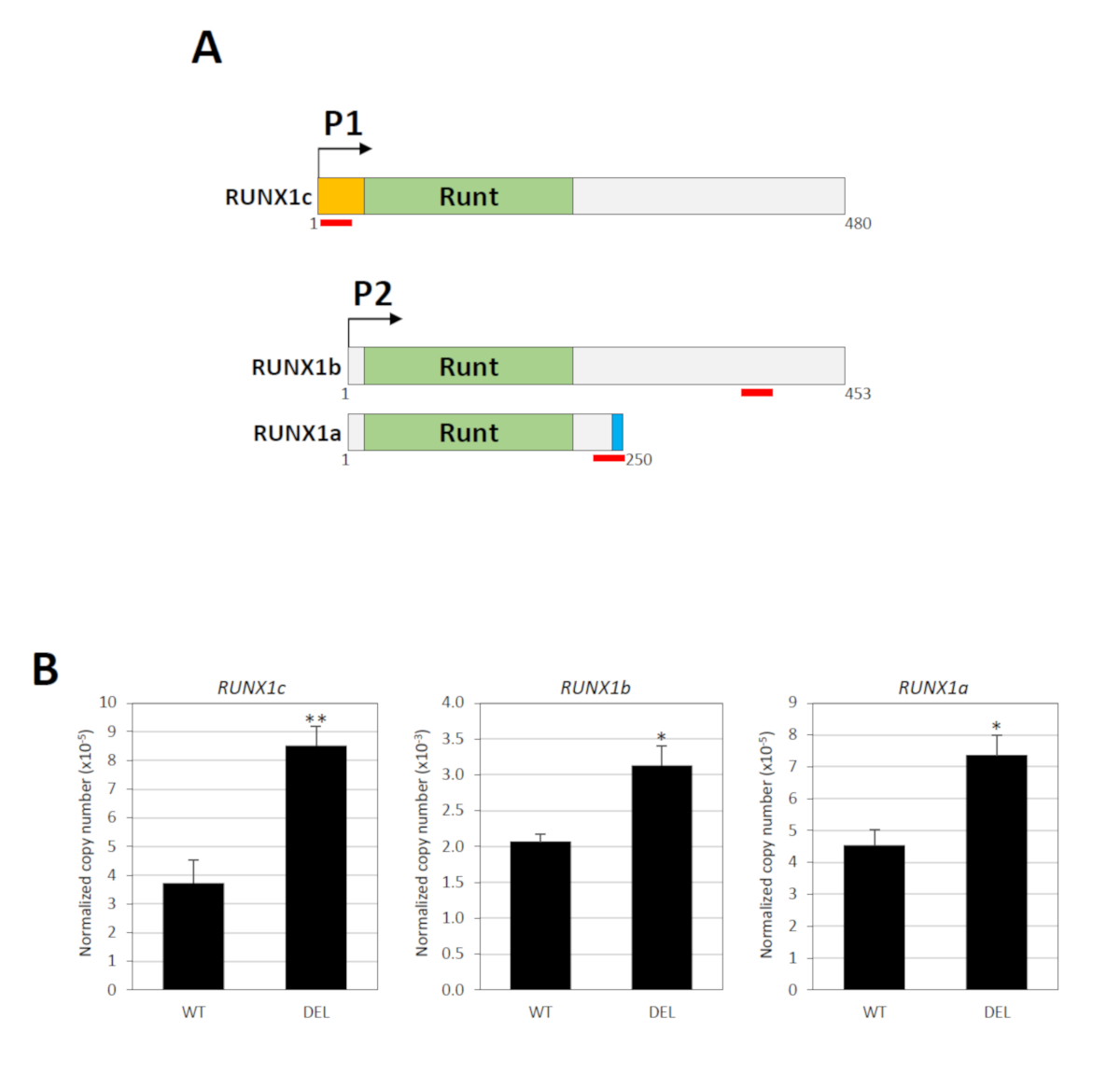
The RUNX1 gene contains two promoters namely the distal P1 and proximal P2, which are separated by a large intron harboring the silencer element34. Three major mRNA transcripts are produced by these promoters: RUNX1c by P1 and RUNX1a and RUNX1b by P234. The nucleotide sequence of RUNX1c and RUNX1b are identical except the former has a unique N-terminus, from which a specific TaqMan gene expression assay can be designed (Figure 3A). To measure RUNX1b, a TaqMan assay recognizing both RUNX1b and RUNX1c was used (Figure 3A). RUNX1b levels were then determined by subtracting total RUNX1b/RUNX1c from RUNX1c. RUNX1a is a distinctly shorter isoform due to alternative splicing and a specific TaqMan assay is available for this variant (Figure 3A). Thus, the activity of the P1 and P2 promoters can be determined individually. Real-time quantitative RT-PCR showed that deletion of the silencer element significantly upregulated the expression levels of both P1- and P2-derived transcripts (Figure 3B).

Figure 1: Strategy to delete the RUNX1 intronic silencer. The silencer (red box) is located in the first intron of the RUNX1 gene separating the two promoters P1 and P2 (hg19 coordinates are shown). Two crRNAs (crRNA-1 and crRNA-2) were designed to introduce DSBs flanking the silencer element. The predicted Cas9 cleavage sites are indicated by vertical red lines and the PAM sites (NGG) are in purple. The two primers used for screening of the deletions are represented by open arrows. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Identification of the deletion clones. (A) Representative electropherograms of cell clones showing different levels of mutant PCR products (MUT). The size of the mutant products varies among the clones because of indels formed at the cleavage sites. WT = wild-type. (B) Verification of the deletions from representative clones by Sanger sequencing. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Functional consequences of the silencer deletion. (A) The three major RUNX1 isoforms (RUNX1a, RUNX1b and RUNX1c) are shown. These variants contain the same Runt DNA binding domain but different N- (orange) or C-terminus (blue). The location of the TaqMan probe/primer pairs is indicated by red lines. Numbers indicate the amino acid residues. (B) Real-time quantitative RT-PCR analysis of RUNX1 P1- and P2-derived transcripts in cell populations with (DEL) or without (WT) the biallelic deletions18. GAPDH was used for normalization. * and ** indicate P < 0.05 and P < 0.01, respectively by the Mann-Whitney test. This figure has been modified from Cheng et al.18. Please click here to view a larger version of this figure.
{kind=link}
Discussion
The CRISPR/Cas9 system has been used in a wide range of genome editing applications such as gene knockout and knock-in studies35,36, transcriptional regulation37,38, genetic engineering of various model organisms39,40,41,42,43,44 and gene therapy45,46. Here, we demonstrate the use of CRISPR/Cas9 to investigate the functional consequences of deleting an intronic silencer on the RUNX1 gene. The delivery of the CRISPR components in our approach did not rely on plasmid DNA, cloning of gRNA or virus but electroporation of preassembled Cas9/gRNA RNP complexes. It has been shown that the use of exogenous DNA can be associated with undesirable integration of foreign vector sequences into the host genome, increased toxicity and low efficiency25,47,48, whereas virus transduction methods are time-consuming. In addition, prolonged expression of Cas9 from plasmid DNA can augment off-target effects48. On the contrary, the direct RNP-based delivery approach has been established as the preferred method as it is fast and straightforward with improved editing efficiency, selectivity and cell viability. Indeed, a variety of methods such as lipofection49,50, electroporation25,51, nanoparticles52, cell-penetrating peptides53, iTOP54 and TRIAMF55 have been developed for efficient CRISPR/Cas9 delivery into diverse cell types as well as animal and plant species24,25,26,56,57,58,59,60,61,62,63. Since non-coding DNA sequences are hotspots of genetic variations64, checking the presence of common SNPs/indels in the target and neighboring PAM sequences is particularly relevant when designing gRNA that targets regulatory elements.
A bottleneck in CRISPR/Cas9 genome editing involves screening of desired mutant clones in a large number of samples. We employed fluorescent PCR coupled with capillary gel electrophoresis for the screening as the target mutation is a small genomic deletion of about 300 bp. This method is rapid and sensitive and can be performed in a high-throughput fashion. Also, this method allows accurate estimation of mutant levels and deletion sizes simultaneously. In addition, multiplex analysis of PCR fragments labeled with different fluorescent dyes is supported. We have been routinely using this technique to genotype small insertions/deletions in myeloid neoplasms65,66. In our experience, we can consistently detect fragment sizes that are differ by 4 bp with high precision and mutant burden down to ~3 %. However, it should be noted that this method has a fragment size limit of 1,200 bp, and thus it is not suitable for screening of large deletions. Also, base substitutions (resulting in unchanged fragment size) and potential off-target events in other genomic regions cannot be detected. For the latter, the costly whole genome sequencing is required to comprehensively profile global undesirable changes in the target clones. To adopt our current approach for investigation of large non-coding regulatory sequences (>1,000 bp), a detailed deletion and mutagenesis analyses of putative transcription factor binding sites using in vitro reporter gene assays can be performed beforehand to delineate the minimal functional region for CRISPR/Cas9 editing18.
As many genes contain more than one promoters3,4, it is important to be aware of the existence of alternative promoters in the target gene locus as manipulating regulatory elements may affect the promoters differentially. Thus, transcript variants derived from different promoters need to be measured individually to evaluate any promoter-specific responses. The use of TaqMan probe-based assays is preferred over SYBR Green because of better specificity and reproducibility. If the more advanced digital PCR system is available, transcript quantification can be performed more precisely without the need of standard curve construction.
An important consideration in performing CRISPR/Cas9 experiments in cancer cell lines is the ploidy and target gene copy number in the cells used as virtually all cancer cell lines harbor genetic alterations including structural and copy number variations. In our case, OCI-AML3 has a hyperdiploid karyotype with 45 to 50 chromosomes. Also, the cell line was found to carry a normal RUNX1 copy number as revealed from the Cancer Cell Line Encyclopedia67 and fluorescence in situ hybridization studies18. When targeting a gene with copy number gain, the delivery method may need to be optimized to provide sufficient levels of the CRISPR components for the editing. Also, more clones may need to be screened in order to identify the complete knockouts. Importantly, it has been shown that targeting at amplified genomic regions, particularly those caused by structural rearrangements, can trigger gene-independent antiproliferative responses in cancer cells, leading to false-positive results in gene functional studies68,69,70. In this regard, alternative approaches like RNA interference (RNAi) knockdown and/or cDNA overexpression should be employed to verify the CRISPR findings. Also, multiple cell lines should be used to avoid misinterpretation of cell line-specific but gene-independent CRISPR editing effects.
The CRISPR/Cas9 system has revolutionized basic and translational research by providing a simple and efficient means to genome editing. Here we demonstrate the ease of using CRISPR/Cas9 to disrupt an intronic silencer for transcriptional studies in a cancer cell line. This technique allows for the study of CREs at the DNA level and offers the opportunities to examine CRE functions in the endogenous context rather than the traditional heterologous reporter genes. Recently, a CRISPR-based RNA editing system has also been identified71 and may serve as a novel tool to study CREs by targeting RNA transcribed from the regulatory elements. By combining with chromosome conformation capture techniques, CRISPR/Cas9 will certainly help decipher the involvements of CREs in altered genome organization and gene expression linked to various health problems.
Disclosures
The authors have nothing to disclose.
Acknowledgements
The authors would like to thank Prof. M.D. Minden (Princess Margaret Cancer Centre, University Health Network, Toronto, Canada) for providing the OCI-AML3 cell line. Also, the authors would like to thank the Core Utilities of Cancer Genomics and Pathobiology (The Chinese University of Hong Kong) for providing the facilities and assistance in support of this research.
Materials
| Name | Company | Catalog Number | Comments |
| 0.2 cm gap electroporation cuvette | Bio-Rad | 1652086 | |
| 1×TE buffer, pH 7.5 | Integrated DNA Technologies | 11-01-02-02 | |
| 10 mM dNTP mix | Thermo Fisher Scientific | 18427013 | |
| 3500 Genetic Analyzer | Thermo Fisher Scientific | 4405673 | |
| 6-FAM-labeled fluorescent PCR forward primer | Thermo Fisher Scientific | None | |
| 7300 Real-Time PCR System | Thermo Fisher Scientific | None | |
| 7300 System SDS Software | Thermo Fisher Scientific | None | Version 1.3.1 |
| Bio-Rad Gene Pulser Xcell system | Bio-Rad | 1652660 | |
| ChargeSwitch Direct gDNA Purification Kit, 96-well | Thermo Fisher Scientific | CS11205 | |
| crRNA-1 | Integrated DNA Technologies | Alt-R CRISPR-Cas9 crRNA | Components of the Cas9/gRNA complex |
| crRNA-2 | Integrated DNA Technologies | Alt-R CRISPR-Cas9 crRNA | Components of the Cas9/gRNA complex |
| Deionized formamide | Thermo Fisher Scientific | 4311320 | |
| Electroporation Enhancer | Integrated DNA Technologies | 1075915 | |
| Fetal bovine serum | Thermo Fisher Scientific | 10270098 | |
| Fluorometer | Thermo Fisher Scientific | Q32857 | |
| GeneMapper Software 5 | Thermo Fisher Scientific | 4475073 | |
| GeneScan 600 LIZ Size Standard | Thermo Fisher Scientific | 4408399 | |
| GlutaMAX | Thermo Fisher Scientific | 35050061 | |
| PBS, 10x Solution, pH 7.4 | Affymetrix | 75889 | |
| Penicillin and streptomycin | Thermo Fisher Scientific | 15140122 | |
| Platinum Taq DNA Polymerase High Fidelity | Thermo Fisher Scientific | 11304029 | |
| Qubit dsDNA HS Assay Kit | Thermo Fisher Scientific | Q32854 | |
| Recombinant S. pyogenes Cas9 nuclease | Integrated DNA Technologies | 1081058 | Components of the Cas9/gRNA complex |
| RPMI 1640 medium | Thermo Fisher Scientific | 31800-022 | |
| RPMI 1640 medium without phenol red | Thermo Fisher Scientific | 11835030 | |
| RUNX1a TaqMan gene expression assays | Thermo Fisher Scientific | 4331182 | Hs04186042_m1 |
| RUNX1b/c TaqMan gene expression assays | Thermo Fisher Scientific | 4331182 | Hs00231079_m1 |
| RUNX1c TaqMan gene expression assays | Thermo Fisher Scientific | 4331182 | Hs01021966_m1 |
| SuperScript III First-Strand Synthesis System | Thermo Fisher Scientific | 18080051 | |
| TaqMan Universal PCR Master Mix | Thermo Fisher Scientific | 4304437 | |
| tracrRNA | Integrated DNA Technologies | 1072533 | Components of the Cas9/gRNA complex |
| TRIzol Reagent | Thermo Fisher Scientific | 15596018 | |
| Unlabeled PCR reverse primer | Thermo Fisher Scientific | None |
References
- Maston, G. A., Evans, S. K., Green, M. R. Transcriptional regulatory elements in the human genome. Annual Review of Genomics and Human Genetics. 7, 29-59 (2006).
- Shlyueva, D., Stampfel, G., Stark, A. Transcriptional enhancers: from properties to genome-wide predictions. Nature Reviews Genetics. 15 (4), 272-286 (2014).
- Kimura, K., et al. Diversification of transcriptional modulation: large-scale identification and characterization of putative alternative promoters of human genes. Genome Research. 16 (1), 55-65 (2006).
- Landry, J. R., Mager, D. L., Wilhelm, B. T. Complex controls: the role of alternative promoters in mammalian genomes. Trends in Genetics. 19 (11), 640-648 (2003).
- West, A. G., Gaszner, M., Felsenfeld, G. Insulators: many functions, many mechanisms. Genes & Development. 16 (3), 271-288 (2002).
- Riethoven, J. J. Regulatory regions in DNA: promoters, enhancers, silencers, and insulators. Methods in Molecular Biology. 674, 33-42 (2010).
- Thurman, R. E., et al. The accessible chromatin landscape of the human genome. Nature. 489 (7414), 75-82 (2012).
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 489 (7414), 57-74 (2012).
- Roadmap Epigenomics Consortium. Integrative analysis of 111 reference human epigenomes. Nature. 518 (7539), 317-330 (2015).
- Andersson, R., et al. An atlas of active enhancers across human cell types and tissues. Nature. 507 (7493), 455-461 (2014).
- Carninci, P., et al. The transcriptional landscape of the mammalian genome. Science. 309 (5740), 1559-1563 (2005).
- Bernstein, B. E., et al. The NIH roadmap epigenomics mapping consortium. Nature Biotechnology. 28 (10), 1045-1048 (2010).
- Zhou, S., Treloar, A. E., Lupien, M. Emergence of the Noncoding Cancer Genome: A Target of Genetic and Epigenetic Alterations. Cancer Discovery. 6 (11), 1215-1229 (2016).
- Khurana, E., et al. Role of non-coding sequence variants in cancer. Nature Reviews Genetics. 17 (2), 93-108 (2016).
- Herz, H. M., Hu, D., Shilatifard, A. Enhancer malfunction in cancer. Molecular Cell. 53 (6), 859-866 (2014).
- Willis, T. G., Dyer, M. J. The role of immunoglobulin translocations in the pathogenesis of B-cell malignancies. Blood. 96 (3), 808-822 (2000).
- Gröschel, S., et al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell. 157 (2), 369-381 (2014).
- Cheng, C. K., et al. RUNX1 upregulation via disruption of long-range transcriptional control by a novel t(5;21)(q13;q22) translocation in acute myeloid leukemia. Molecular Cancer. 17 (1), 133 (2018).
- Wiedenheft, B., Sternberg, S. H., Doudna, J. A. RNA-guided genetic silencing systems in bacteria and archaea. Nature. 482 (7385), 331-338 (2012).
- Bhaya, D., Davison, M., Barrangou, R. CRISPR-Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation. Annual Review of Genetics. 45, 273-297 (2011).
- Mali, P., et al. RNA-guided human genome engineering via Cas9. Science. 339 (6121), 823-826 (2013).
- Jinek, M., et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337 (6096), 816-821 (2012).
- Ran, F. A., et al. Genome engineering using the CRISPR-Cas9 system. Nature Protocols. 8 (11), 2281-2308 (2013).
- Cho, S. W., Lee, J., Carroll, D., Kim, J. S., Lee, J. Heritable gene knockout in Caenorhabditis elegans by direct injection of Cas9-sgRNA ribonucleoproteins. Genetics. 195 (3), 1177-1180 (2013).
- Kim, S., Kim, D., Cho, S. W., Kim, J., Kim, J. S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Research. 24 (6), 1012-1019 (2014).
- Lin, S., Staahl, B. T., Alla, R. K., Doudna, J. A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife. 3, 04766 (2014).
- Quentmeier, H., et al. Cell line OCI/AML3 bears exon-12 NPM gene mutation-A and cytoplasmic expression of nucleophosmin. Leukemia. 19 (10), 1760-1767 (2005).
- Heigwer, F., Kerr, G., Boutros, M. E-CRISP: fast CRISPR target site identification. Nature Methods. 11 (2), 122-123 (2014).
- Montague, T. G., Cruz, J. M., Gagnon, J. A., Church, G. M., Valen, E. CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Research. 42, 401-407 (2014).
- Doench, J. G., et al. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nature Biotechnology. 32 (12), 1262-1267 (2014).
- Haeussler, M., et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biology. 17 (1), 148 (2016).
- Ramlee, M. K., Wang, J., Cheung, A. M., Li, S. Using a Fluorescent PCR-capillary Gel Electrophoresis Technique to Genotype CRISPR/Cas9-mediated Knockout Mutants in a High-throughput Format. Journal of Visualized Experiments. (122), e55586 (2017).
- Canver, M. C., et al. Characterization of genomic deletion efficiency mediated by clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 nuclease system in mammalian cells. Journal of Biological Chemistry. 289 (31), 21312-21324 (2014).
- Sood, R., Kamikubo, Y., Liu, P. Role of RUNX1 in hematological malignancies. Blood. 129 (15), 2070-2082 (2017).
- Wang, H., et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 153 (4), 910-918 (2013).
- Yang, H., et al. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell. 154 (6), 1370-1379 (2013).
- Wright, J. B., Sanjana, N. E. CRISPR Screens to Discover Functional Noncoding Elements. Trends in Genetics. 32 (9), 526-529 (2016).
- Korkmaz, G., et al. Functional genetic screens for enhancer elements in the human genome using CRISPR-Cas9. Nature Biotechnology. 34 (2), 192-198 (2016).
- Cong, L., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Hwang, W. Y., et al. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nature Biotechnology. 31 (3), 227-229 (2013).
- Gratz, S. J., et al. Genome engineering of Drosophila with the CRISPR RNA-guided Cas9 nuclease. Genetics. 194 (4), 1029-1035 (2013).
- Chen, C., Fenk, L. A., de Bono, M. Efficient genome editing in Caenorhabditis elegans by CRISPR-targeted homologous recombination. Nucleic Acids Research. 41 (20), 193 (2013).
- Jacobs, T. B., LaFayette, P. R., Schmitz, R. J., Parrott, W. A. Targeted genome modifications in soybean with CRISPR/Cas9. BMC Biotechnology. 15, 16 (2015).
- Svitashev, S., et al. Targeted Mutagenesis, Precise Gene Editing, and Site-Specific Gene Insertion in Maize Using Cas9 and Guide RNA. Plant Physiology. 169 (2), 931-945 (2015).
- Schwank, G., et al. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell. 13 (6), 653-658 (2013).
- Ye, L., et al. Genome editing using CRISPR-Cas9 to create the HPFH genotype in HSPCs: An approach for treating sickle cell disease and β-thalassemia. Proceedings of the National Academy of Sciences of the United States of America. 113 (38), 10661-10665 (2016).
- Gabriel, R. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nature Biotechnology. 29 (9), 816-823 (2011).
- Gaj, T., Guo, J., Kato, Y., Sirk, S. J., Barbas, C. F. Targeted gene knockout by direct delivery of zinc-finger nuclease proteins. Nature Methods. 9 (8), 805-807 (2012).
- Zuris, J. A., et al. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nature Biotechnology. 33 (1), 73-80 (2015).
- Wang, M., et al. Efficient delivery of genome-editing proteins using bioreducible lipid nanoparticles. Proceedings of the National Academy of Sciences of the United States of America. 113 (11), 2868-2873 (2016).
- Gundry, M. C., et al. Highly Efficient Genome Editing of Murine and Human Hematopoietic Progenitor Cells by CRISPR/Cas9. Cell Reports. 17 (5), 1453-1461 (2016).
- Mout, R., et al. Direct Cytosolic Delivery of CRISPR/Cas9-Ribonucleoprotein for Efficient Gene Editing. ACS Nano. 11 (3), 2452-2458 (2017).
- Ramakrishna, S., et al. Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Research. 24 (6), 1020-1027 (2014).
- D'Astolfo, D. S., et al. Efficient intracellular delivery of native proteins. Cell. 161 (3), 674-690 (2015).
- Yen, J., et al. TRIAMF: A New Method for Delivery of Cas9 Ribonucleoprotein Complex to Human Hematopoietic Stem Cells. Scientific Reports. 8 (1), 16304 (2018).
- Sung, Y. H., et al. Highly efficient gene knockout in mice and zebrafish with RNA-guided endonucleases. Genome Research. 24 (1), 125-131 (2014).
- Martin, A., et al. CRISPR/Cas9 mutagenesis reveals versatile roles of Hox genes in crustacean limb specification and evolution. Current Biology. 26 (1), 14-26 (2016).
- Menoret, S., et al. Homology-directed repair in rodent zygotes using Cas9 and TALEN engineered proteins. Scientific Reports. 5, 14410 (2015).
- Woo, J. W., et al. DNA-free genome editing in plants with preassembled CRISPR-Cas9 ribonucleoproteins. Nature Biotechnology. 33 (11), 1162-1164 (2015).
- Malnoy, M., et al. DNA-free genetically edited grapevine and apple protoplast using CRISPR/Cas9 ribonucleoproteins. Frontiers in Plant Science. 7, 1904 (2016).
- Liang, Z., et al. Efficient DNA-free genome editing of bread wheat using CRISPR/Cas9 ribonucleoprotein complexes. Nature Communications. 8, 14261 (2017).
- Svitashev, S., Schwartz, C., Lenderts, B., Young, J. K., Mark Cigan, A. Genome editing in maize directed by CRISPR-Cas9 ribonucleoprotein complexes. Nature Communications. 7, 13274 (2016).
- Shin, S. E., et al. CRISPR/Cas9-induced knockout and knock-in mutations in Chlamydomonas reinhardtii. Scientific Reports. 6, 27810 (2016).
- 1000 Genomes Project Consortium et al. A global reference for human genetic variation. Nature. 526 (7571), 68-74 (2015).
- Klampfl, T., et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. The New England Journal of Medicine. 369 (25), 2379-2390 (2013).
- Döhner, H., et al. Diagnosis and management of AML in adults: 2017 ELN recommendations fro an international expert panel. Blood. 129 (4), 424-447 (2017).
- Barretina, J., et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 483 (7391), 603-607 (2012).
- Aguirre, A. J., et al. Genomic Copy Number Dictates a Gene-Independent Cell Response to CRISPR/Cas9 Targeting. Cancer Discovery. 6 (8), 914-929 (2016).
- Munoz, D. M., et al. CRISPR Screens Provide a Comprehensive Assessment of Cancer Vulnerabilities but Generate False-Positive Hits for Highly Amplified Genomic Regions. Cancer Discovery. 6 (8), 900-913 (2016).
- Gonçalves, E., et al. Structural rearrangements generate cell-specific, gene-independent CRISPR-Cas9 loss of fitness effects. Genome Biology. 20 (1), 27 (2019).
- Abudayyeh, O. O., et al. RNA targeting with CRISPR-Cas13. Nature. 550 (7675), 280-284 (2017).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved