
Method Article
Measuring Microbial Mutation Rates with the Fluctuation Assay
In This Article
Summary
Here, a protocol is presented to perform a fluctuation assay and estimate microbial mutation rate using phenotypic markers. This protocol will enable researchers to assay mutations in diverse microbes and environments, determining how genotype and ecological context affect spontaneous mutation rates.
Abstract
Fluctuation assays are widely used for estimating mutation rates in microbes growing in liquid environments. Many cultures are each inoculated with a few thousand cells, each sensitive to a selective marker that can be assayed phenotypically. These parallel cultures grow for many generations in the absence of the phenotypic marker. A subset of cultures is used to estimate the total number of cells at risk of mutations (i.e., the population size at the end of the growth period, or Nt). The remaining cultures are plated onto the selective agar. The distribution of observed resistant mutants among parallel cultures is then used to estimate the expected number of mutational events, m, using a mathematical model. Dividing m by Nt gives the estimate of the mutation rate per locus per generation. The assay has three critical aspects: the chosen phenotypic marker, the chosen volume of parallel cultures, and ensuring that the surface on the selective agar is completely dry before the incubation. The assay is relatively inexpensive and only needs standard laboratory equipment. It is also less laborious than alternative approaches, such as mutation accumulation and single-cell assays. The assay works on organisms that go through many generations rapidly and it depends on assumptions about the fitness effects of markers and cell death. However, recently developed tools and theoretical studies mean these issues can now be addressed analytically. The assay allows mutation rate estimation of different phenotypic markers in cells with different genotypes growing in isolation or in a community. By conducting multiple assays in parallel, assays can be used to study how an organism's environmental context affects spontaneous mutation rate, which is crucial for understanding antimicrobial resistance, carcinogenesis, aging, and evolution.
Introduction
In 1901 the Dutch botanist Hugo de Vries coined the term mutation1. Twenty-six years later, when Hermann Joseph Muller discovered the mutagenic action of X-rays2, mutations were already perceived as one of the driving forces of evolution. However, the nature of mutations was not clear. To answer the fundamental question of whether mutations emerge spontaneously (i.e., a spontaneous mutation) or in response to selection (i.e., an induced mutation), a method was needed to observe mutational events. Such a method would measure the expected number of mutations per cell division or what was already known as a mutation rate3,4.

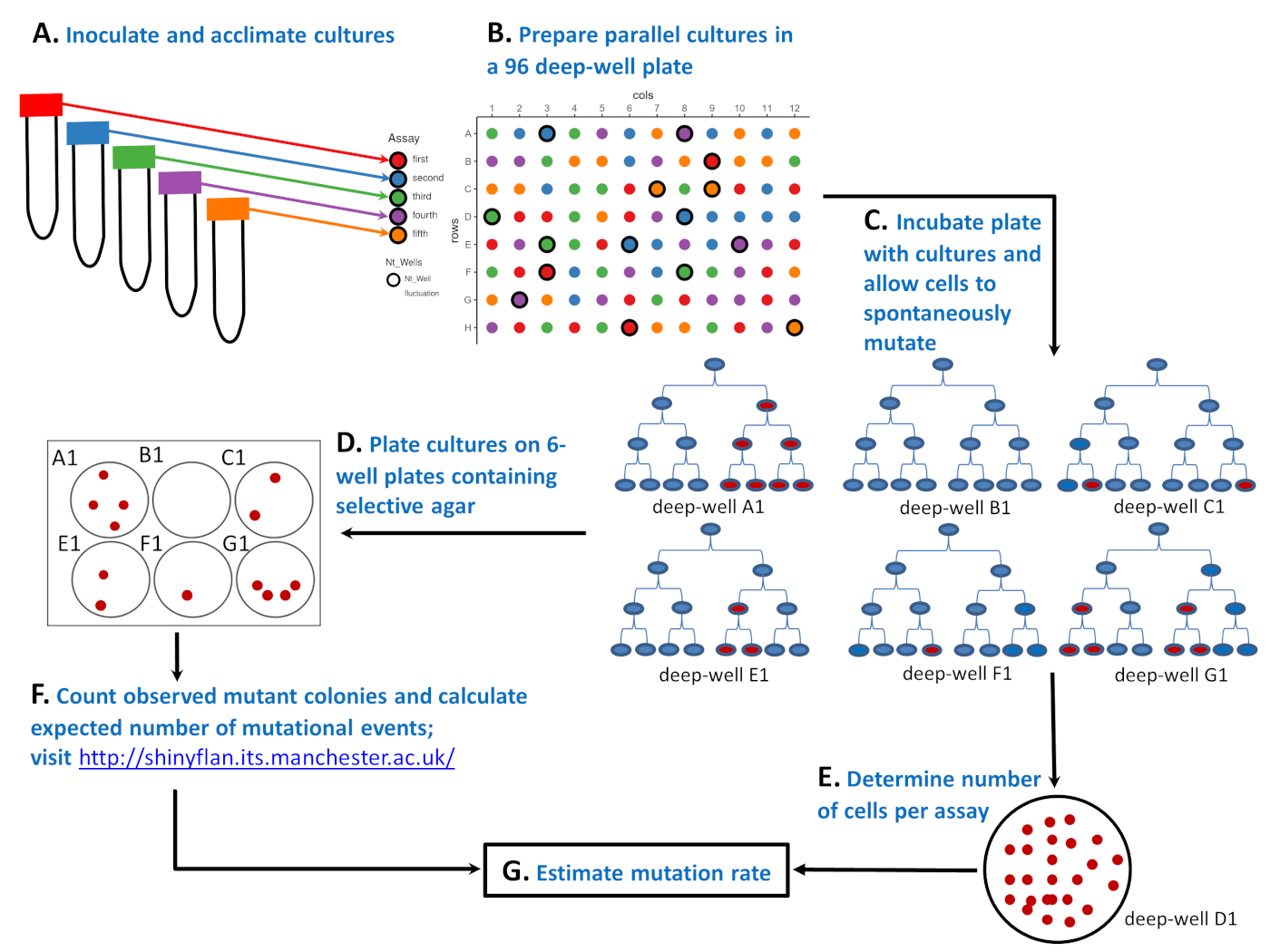
Figure 1: Schematic illustration of how to perform the fluctuation assay with a microbial strain in a 96 deep well plate. (A) Inoculate and acclimate cells in 50 mL tubes containing five different environments ('red', 'blue', 'green', 'purple', and 'orange' assays). (B) Prepare parallel cultures with a small number of sensitive cells in a 96 deep well plate. The 'red' assay has 20 parallel cultures, whereas the 'blue', 'green', 'purple', and 'orange' assays all have 19 parallel cultures. The positions of the parallel cultures on the 96 deep well plate are random. Randomization can be done with the Supplementary LayoutGenerator.R script or by some other tool. The layout on the top right is the randomization result. (C) Incubate the 96 deep well plate and allow the cells to divide and spontaneously mutate. Six cultures from deep wells A1, B1, C1, E1, F1, and G1 show how the number of mutants fluctuates: 4, 0, 2, 2, 1, and 4 red cells after the third cell division, respectively. The number of mutants differs not only because of the different number of spontaneous mutations (0, 1, or 2 as shown by the first red cell), but also because it is important when during a culture cycle a resistance mutation spontaneously emerges (cell division 1, 2, or 3). (D) After the incubation of the 96 deep well plate the number of mutants is determined by plating 81 parallel cultures. On the layout these are circles without bolded edges. The entire parallel culture is plated on one well of a 6 well plate containing a selective agar. (E) The remaining 15 cultures are diluted and plated on the non-selective agar to determine the average number of cells (Nt). On the layout these are labelled as Ntwells and have bolded edges. For each assay Nt is averaged over three parallel cultures. On the bottom right is a Petri dish containing a non-selective agar plate with 25 CFUs of a diluted culture grown in a deep well D1 (part of a 'green' assay). (F) After incubation of the selective 6 well plates the number of observed mutants were counted and the expected number of mutational events, m, was estimated using a Maximum Likelihood estimator. (G) Knowing both the number of mutations, m, and the number of cells per assay, Nt, the mutation rate was estimated as m/Nt. Please click here to view a larger version of this figure.
{kind=link}
Salvador Luria and Max Delbrück in 1943 provided an ingenious solution to this problem with the fluctuation assay5 (see Figure 1). The assay starts with multiple populations (named parallel cultures) that are initiated with a small number of microbial cells (Figure 1A,B). After growth in a benign, non-selective environment (Figure 1C), parallel cultures are transferred on plates containing a selective marker (phages, antibiotics, etc.), where only cells with a resistance mutation survive and can produce a colony (Figure 1D). The main expectation was that if resistance mutations are induced, the number of cells that carry a mutation should be distributed among different populations with the mean equal to the variance. What Luria and Delbrück found with the fluctuation assay is that the number of mutants fluctuated drastically and that the variance in the number of mutants among different populations was considerably greater than the mean. Luria and Delbrück thereby demonstrated that mutations are spontaneous. They showed that mutations spontaneously emerge whenever the DNA is replicated, and the number of mutants depends on when the mutation occurs during the growth of the population. See Figure 1C, where six populations, each initiated with a microbial cell (in blue), experience none, 1, or 2 single mutations. Populations A1, E1, and F1 experienced one single mutation (first red cell), but because a single mutation spontaneously emerges at various time points during a culture cycle, populations ended up with a very different number of observed mutants (four, two, and one, respectively). On the other hand, populations C1 and G1 ended up with the same number of observed mutants as E1 and A1, despite experiencing two mutational events rather than one. The fluctuation of observed mutants among populations not only gave the assay the name, but also showed that a mutant frequency (i.e., the proportion of mutant cells) is an inadequate indicator of the mutation rate.
The overall goal of the fluctuation assay is to estimate the spontaneous mutation rate of a particular genotype of bacteria or other single-celled organism growing in a particular liquid environment. The fluctuation assay remains the most appropriate tool for studying environmental dependency of microbial mutation rates and allows rapid and inexpensive mutation rate estimation. Alternative approaches to mutation rate estimation, such as maximum-depth sequencing6, population sequencing7, mutation accumulation experiments8, or comparing genome sequences of an offspring to those of the parents9 are much more laborious, and thus poorly suited to potentially detecting environmental dependencies. However, dynamic aspects of a mutation's generation and repair are largely inaccessible to a fluctuation assay or to any of the methods of assaying a mutation rate listed above. To study how the number of mutations changes in time, space, or among individual cells within a population, single cell approaches11,12 are necessary, which, in addition to being more laborious than fluctuation assays, require highly specialized skills and equipment.
In practice, a fluctuation assay is counting cells gaining a phenotypic marker due to a mutation that occurs in an environment lacking selection for that marker. The meta-analysis of hundreds of published assays10 shows that at least 39 different phenotypic markers have been used since the assay's inception in 1943. The fluctuation assay can be used to compare averages and environmental dependency of mutation rates among laboratory, clinical, nonmutator, and mutator strains growing in permissive environments. The assay allows for the mutation rate estimation in cells with different genetic backgrounds growing in either minimal or rich environments. The assay is suitable not only for populations growing as a monoculture but can also be used for studying the effects of cell-cell interactions on mutation rates11. When the strain of interest is cocultured with a second strain, and a neutral marker is used to distinguish the strains, mutation rates can be assayed for two strains in the same tube at the same time.
Fluctuation assays have revealed that the spontaneous mutation rate depends upon both a cell's genotype and its environment12 and is a trait that itself evolves13. Whenever the mutation rate of one particular genotype changes with the environment, it is described as mutation-rate plasticity11. Plastic mutation rates have been most thoroughly addressed for stress-induced mutagenesis (SIM)14. In addition, using fluctuation assays, it has been recently shown that the density to which a population of cells grows (typically a batch culture at carrying capacity) is closely associated with mutation rates across bacteria and unicellular eukaryotes. The mutation rate per genome per generation decreases in dense populations by as much as 23-fold10,11. This density-associated mutation rate plasticity (DAMP) can depend on a quorum-sensing system15 and act independently of SIM16.
Here, a detailed protocol is presented for the fluctuation assay used to study Escherichia coli strain K-12 gaining resistance to the antibiotic rifampicin in a glucose minimal media environment. However, this protocol should be viewed as a basic template that can be utilized to study a wide variety of microbes by simply modifying the culture conditions and phenotypic markers of mutation. The protocol has evolved from its inception5,17,18,19,20,21,22,23,24,25,26,27,28,29 through its use on a wide range of microbes and even cancer cells30 and has been modified to increase the throughput, which was essential for properly testing environmental dependencies of microbial mutation rates10,11,16. The protocol described here does not cover all the methodological and analytical issues of the fluctuation assay that have been already well discussed in the literature, particularly fitness effects of resistant mutations31, phenotypic delay32, cell death33, and the suitability of various algorithms available to estimate mutation rates26,34. This can be important, for instance, when the environmental dependence of fitness effects can give rise to erroneous variation in the mutation rate estimates35. However, we note that the analytical tools that we use here can deal with the variation in mutant fitness and cell death. As addressed in the notes and discussion, it is also recommended that multiple phenotypic markers that are unlikely to have the same environmentally dependent fitness effects be considered. This protocol will enable people to routinely assay environmental dependencies of mutation rates in the diversity of microbial strains and environments. Assaying mutations in different environments has not yet been thoroughly tested and once population density is considered, fluctuation assays can give a more precise estimate of mutation rate10. This protocol will enable more fluctuation assays to be performed, as is necessary for understanding the mechanisms underpinning mutation rates, which in turn is vital for understanding evolution, carcinogenesis, aging, and antimicrobial resistance.
Protocol
1. Day 1: Inoculation and Acclimation of Cultures
- Inoculate 3 mL of liquid lysogeny broth (LB, see Supplementary Table 1) with a scrape of ice from the E. coli MG1655 glycerol stock (18% glycerol, -80 °C). Shake the LB culture at 120 rpm for ~7 h at 37 °C.
NOTE: In this experiment, E. coli K12 MG1655 growing in LB is used, but this assay can be performed with any E. coli strain or any other culturable microbial species. Incubation temperature, incubation times, and nutrient level of the growth media may all be subject to variation by species or strain. - Dilute the culture 2,000-fold using the saline solution. Add 100 µL of the diluted solution to three 50 mL screw cap conical bottom polymer tubes (50 mL tubes) with 10 mL of liquid Davis minimal medium (DM, see the Supplementary Table 1), respectively containing 80 mg/L, 125 mg/L, or 250 mg/L of glucose. This is the same medium (i.e., environment) in which the mutation rate will be estimated. Shake the cultures at 120 rpm overnight at 37 °C.
NOTE: The choice of media is subject to variation by species, strain, or research question.
2. Day 2: Generation of Mutants in Parallel Cultures
- First, prepare the environments in which the bacteria will be cultured. Always prepare 10% more than required (i.e., twenty 1 ml cultures require 22 mL). Prepare 22 mL of 1) DM with 80 mg/L of glucose, 2) DM with 125 mg/L of glucose, 3) DM with 250 mg/L of glucose, 4) DM with 80 mg/L of glucose, and 5) DM with 250 mg/L of glucose in five 50 mL tubes. Label them as GLC-80A, GLC-125, GLC-250A, GLC-80B, and GLC-250B, respectively.
- Prepare inocula for the environments. Ensure that the inoculum for 1 mL of the environment contains 1,000−5,000 cells. Do this by following the steps below.
- Measure the optical density (OD) of overnight cultures (from step 1.2) at 600 nm.
- Dilute each overnight culture in order to reach a final density of 1,000-5,000 cells per mL of DM with glucose. In our hands, if the OD measured in 2.2.1 is 0.3, this means making a 100-fold dilution (in saline solution) of the overnight culture, then adding 11µl of this solution to the 22 mL environment.
- Prepare parallel cultures.
NOTE: This protocol is written for one 96 deep well plate, performing five fluctuation assays (the maximum reasonable number on one 96 well plate), using three environments. With experience, multiple deep-well plates may be run in parallel.- Create a random layout of parallel cultures for one 96 deep well plate. The Supplementary R script LayoutGenerator.R (see the layout in Figure 1B) can be used for this. Position each parallel culture on the 96 deep well plate according to the layout.
NOTE: Running the LayoutGenerator.R will ensure that the first assay has 20 parallel cultures, and that the second, third, fourth, and fifth assays have 19 parallel cultures each. - Transfer 1 mL of inoculated media into each well of a 96 deep well plate according to the randomized layout.
- Fix the lid of the deep well plate with the tape. Do not fix the lid tightly, because the culture growth is sensitive to the amount of aeration.
- Weigh the entire plate with the lid and the tape and shake the plate at 250 rpm for 24 h at 37 °C. Place 2 L of distilled water in the incubator to stabilize the amount of evaporation among experimental sets.
- Create a random layout of parallel cultures for one 96 deep well plate. The Supplementary R script LayoutGenerator.R (see the layout in Figure 1B) can be used for this. Position each parallel culture on the 96 deep well plate according to the layout.
- Determine the inoculum size by plating 10 µL of each of the inoculated media on the non-selective Tetrazolium (TA) agar plate (see Supplementary Table 1). Use a sterile L-shaped spreader until the agar surface is dry. Incubate TA agar plates lid down overnight at 37 °C.
NOTE: TA agar is a rich agar that contains the sugar L-arabinose and the water-soluble dye 2,3,5-triphenyltetrazolium chloride, which is colorless in its oxidized form. When bacteria reduce the dye, it turns red due to the formation of formazan. Colonies on TA agar that cannot utilize L-arabinose are dark red. Other strains, such as MG1655, are pinkish. Use of TA agar rather than standard LB agar is recommended because colored bacterial colonies are easier to spot, which makes colony counting more reliable and quicker. - Prepare selective TA agar containing rifampicin in 6 well plates. Pipette 5 mL of the selective TA agar into each well of the 6 well plates. Prepare the antibiotic rifampicin just before it is added to the TA agar.
NOTE: When cycloserine is used as a marker, use Davis minimal medium with 250 mg/L of glucose supplemented with agar and L-arabinose and 2,3,5-triphenyltetrazolium chloride as a selective agar. Namely, TA agar does not select only cells that are resistant to cycloserine, because tryptone and yeast extract (both the essential components of TA agar) contain the amino acid D-alanine. This amino acid antagonizes the antimicrobial effect of the cycloserine and allows all cells to make a colony.- Leave the selective and remaining non-selective plates in the dark (e.g., in a box) at room temperature.
3. Day 3: Plating Cultures on Selective and Non-selective Agar Plates
- Count colony forming units (CFU) on the non-selective agar plates and determine the size of the inocula by multiplying CFU by 100. This is a ratio between a volume of the parallel culture (1 mL = 1,000 µL) and the plated volume (10 µL).
NOTE: If non-selective plates are stored refrigerated the CFU can be counted later. - After 24 h of incubation, weigh the entire deep well plate to determine the amount of evaporation. This is likely to be around 10%.
- Calculate the average volume of a parallel culture after 24 h of incubation (V[24h]) in microliters using a starting volume V[0h] = 1,000 µl and the plate's weight converted into microliters, where the density of the growth medium is measured in mg/µl. This study uses a density of 1 mg/µl:
![Volume change formula, V[24h]=V[0h]-(1/density)×(weight[0h]-weight[24h])/96; mathematical equation.](/files/ftp_upload/60406/60406eq1.jpg "figure-protocol-6402")
- Transfer the three randomly chosen cultures per assay (15 in total, highlighted with a black circle in the layout in Figure 1B) into labeled microcentrifuge tubes. Leave them on the bench to be used later (see step 3.6) for determining the final population size (or Nt).
- Plate the remaining 81 parallel cultures from the deep well plate onto the selective TA agar containing rifampicin. Pipette one entire parallel culture from the 96 deep well plate into one well of a 6-well plate. Ensure that any 6 well plate contains parallel cultures from more than one fluctuation assay.
- Remove the lids from the selective agar plates, and leave uncovered in sterile conditions. Dry out all the liquid on the surface of the selective TA agar.
NOTE: The agar being completely dry is critical. However, do not overdry the selective TA agar, because it must not be allowed to crack.
- Remove the lids from the selective agar plates, and leave uncovered in sterile conditions. Dry out all the liquid on the surface of the selective TA agar.
- While the selective TA agar plates are drying, which can take up to several hours, determine Nt of the 15 cultures prepared in step 3.4 using colony forming units (CFU).
- Determine CFU by diluting the cultures from the microcentrifuge tubes. Use five 10-fold dilution steps, mixing and vortexing 900 µL of saline solution with 100 µL of the culture on each step. Plate 40 µL of the last dilution (with a dilution factor of 105) on the non-selective TA agar and incubate plates lid down overnight at 37 °C.
NOTE: The dilution series in 96 well plates can be performed with a multichannel pipette, which can increase the speed of this step.
- Determine CFU by diluting the cultures from the microcentrifuge tubes. Use five 10-fold dilution steps, mixing and vortexing 900 µL of saline solution with 100 µL of the culture on each step. Plate 40 µL of the last dilution (with a dilution factor of 105) on the non-selective TA agar and incubate plates lid down overnight at 37 °C.
- Once all the wells on a 6 well plate are free of the culture liquid, put the lid back on and place the 6 well plate with the lid down on the bench until all 6 well plates are dry. Once all are dry, incubate the plates lid down at 37 °C for 44-48 h.
NOTE: For other markers (nalidixic acid, cycloserine, hygromycin B, or 5-FOA) incubate the plates for 68-72 hours. Make sure that the humidity in the incubator is high. It is critical that the selective agar plates not dry out during the incubation period.
4. Day 4: Determination of the Number of the Cells in the Cultures
- Count the CFU on the non-selective agar plates. Estimate the number of viable cells in the culture by multiplying the CFU with the dilution factor (105) and ratio between the calculated average volume V[24h] in microliters (see step 3.3) and plated volume (40 µl):
![Nt calculation formula, CFU, dilution factor, V[24h]/40, microbial growth analysis equation.](/files/ftp_upload/60406/60406eq2.jpg "figure-protocol-9310")
- Nt of a particular genotype growing in a particular environment is the mean of these values from the three cultures.
5. Day 5: Estimation of the Mutation Rate
- Count the number of colonies resistant to the antibiotic on the selective TA agar plates (i.e., the numbers of resistant cells that arose through spontaneous mutation in the deep-well plate on days 2−3). Record the distribution among parallel cultures of the observed number of mutants for a particular assay (e.g., 16 or 17 values, including zero counts).
NOTE: If selective plates are stored refrigerated, colonies resistant to the antibiotic can be counted later.- Use each distribution to estimate the number of mutational events, m, using the R-package flan36.
NOTE: There is also the R-package rSalvador with similar functionality29. - Save the distribution of the observed mutants for one assay in a text file as a single column.
- Use each distribution to estimate the number of mutational events, m, using the R-package flan36.
- Use Shinyflan software (http://shinyflan.its.manchester.ac.uk/) to estimate m as detailed below.
- In the tab Hypothesis Testing leave values as defaults (i.e., Unknown Fitness ticked, Estimation Method = Maximum Likelihood (ML), Distribution of Mutant Lifetime = Exponential (LD model), Winsor Parameter = 1,024, Mutation Number and Fitness = 1, Confidence Level = 0.95, Number of Class and Maximal Value = 100).
- Click Browse and select the text file with the distribution of observed mutants. Try first with the Supplementary Data file.
- After uploading the file, click on Perform Test. On the right side under the Result of the Test, under the One Sample ML-Test (LD model), find the Mutation Number. This is m, the expected number of mutational events.
- Under the 95 Percent Confidence Interval for the Mutation Number find the upper and lower bound of m.
- Once m and Nt (determined by CFU) are available, estimate the mutation rate of a particular genotype in a particular environment as
 . Divide the upper and lower bounds on m by Nt in the same way to generate confidence intervals on the mutation rate (NB this does not account for the uncertainty in Nt).
. Divide the upper and lower bounds on m by Nt in the same way to generate confidence intervals on the mutation rate (NB this does not account for the uncertainty in Nt).
NOTE: Results are presented as the mutation rate per rpoB locus per generation. Mutation rate per base pair is generated by dividing the mutation rate per rpoB locus by 79, which is our current knowledge of how many point mutations within an rpoB gene confer resistance to rifampicin37. Multiplying the mutation rate per nucleotide by the size of the chromosome (E. coli K-12 MG1655 = 4,639,675 bp), gives the mutation rate per genome. - Repeat steps 5.1−5.3 with the remaining four fluctuation assays.
Results
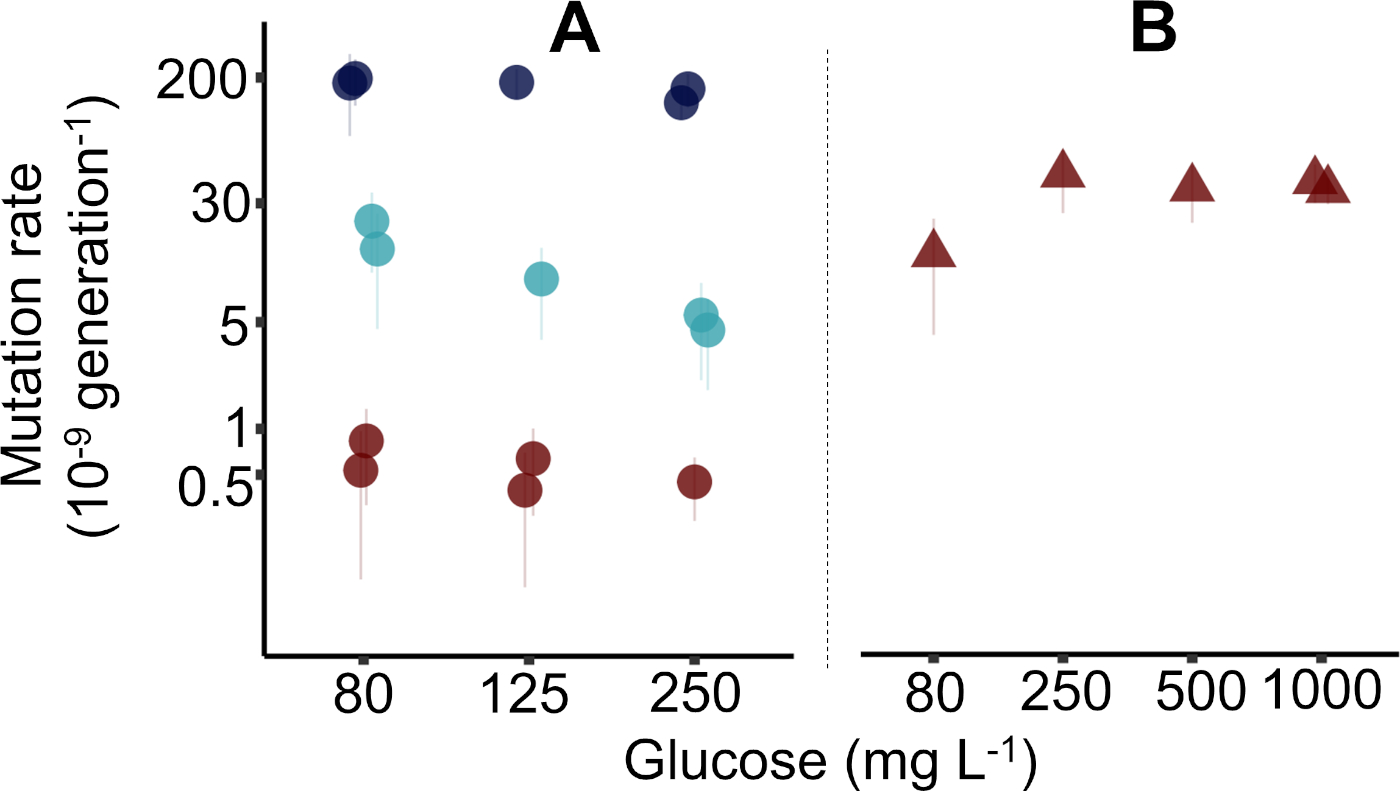
Results were gathered with the reported protocol in four different weeks by three different individual researchers, where each week a 96 deep well plate was used to generate five mutation rate estimates. Altogether three 96 deep well plates generated 15 mutation rate estimates (± 95% confidence interval, CI) in E. coli K-12 MG1655 (Figure 2A, as used in the protocol), while one 96 deep well plate was used for five estimates (± 95% CI) of E. coli K-12 BW25113 ΔmutT mutant (Figure 2B). In Figure 2 the median precision with interquartile range of estimating m is 17.5% (1.00%−28.9%, n = 20). Precision is a coefficient of the variation of the expected number of mutational events (m) calculated as σm / m x 100% (where σ is calculated as shown previously38). Raw data for Figure 2 is available in the Supplementary Data File "Krasovec_etal_JoVE_data.csv". The Supplementary Data File is accompanied by an R script to generate Figure 2. See also Supplementary Table 2 for more details on bacterial strains, and Supplementary Table 3, where columns in the data file are explained. Data points acquired with rifampicin and nalidixic acid were previously published in Krašovec et al.10 and have the same IDs here as when first published.
In Figure 2A the MG1655 mutation rates of three different phenotypic markers, cycloserine, rifampicin, and nalidixic acid are presented. Here, mutation rates were assessed in Davis minimal medium with 80, 125, and 250 mg/L of glucose, as explained in the protocol. In one case 1,000 mg/L of glucose was used (Figure 1B). In practice, any initial glucose concentration can be used. As expected, mutation rates were higher for cycloserine resistance, lowest for nalidixic acid resistance, and the rate of rifampicin resistance was in the middle. This is in accordance with the known target sizes for these three resistances, where the largest is for the cycloserine and the smallest for the nalidixic acid. The discussion and Figure 3 give details on how to exploit the target sizes, volumes of parallel cultures, and level of nutrients in the environment to optimize the measuring of microbial mutation rates with the fluctuation assay.
The fluctuation assay clearly shows which strain is a constitutive mutator and which has normal mutation rates. The ΔmutT strain had an approximately 50x higher mutation rate to nalidixic acid resistance (Figure 2B) as MG1655 (Figure 2A). However, to determine the mutation rate of a particular genotype in different environments, doing at least five replicates per genotype per environment using only one phenotypic marker is recommended. For a detailed outline of the statistical methods previously used to analyze this type of experiment, see the supplementary information in Krašovec et al.10.

Figure 2: Representative figure of mutation rates estimated by the fluctuation assay in Escherichia coli populations. (A) The mutation rate as determined by the reported protocol in wild type MG1655 (circles). The figure shows the mutation rates in the presence of rifampicin (light blue circles), nalidixic acid (red circles), and cycloserine (dark blue circles) resistance. For nalidixic acid, larger culture volumes of 10 mL were used (see Supplementary Data File). Data for rifampicin and nalidixic acid is replotted from Figure 2a in Krašovec et al.10. (B) The mutation rate to nalidixic acid resistance in the Keio39 ΔmutT mutant (red triangles). Note the logarithmic scale on the mutation rate axis. Error bars = 95% confidence intervals calculated as explained in the protocol. Data is replotted from Figure 4a in Krašovec et al.10. Please click here to view a larger version of this figure.
{kind=link}

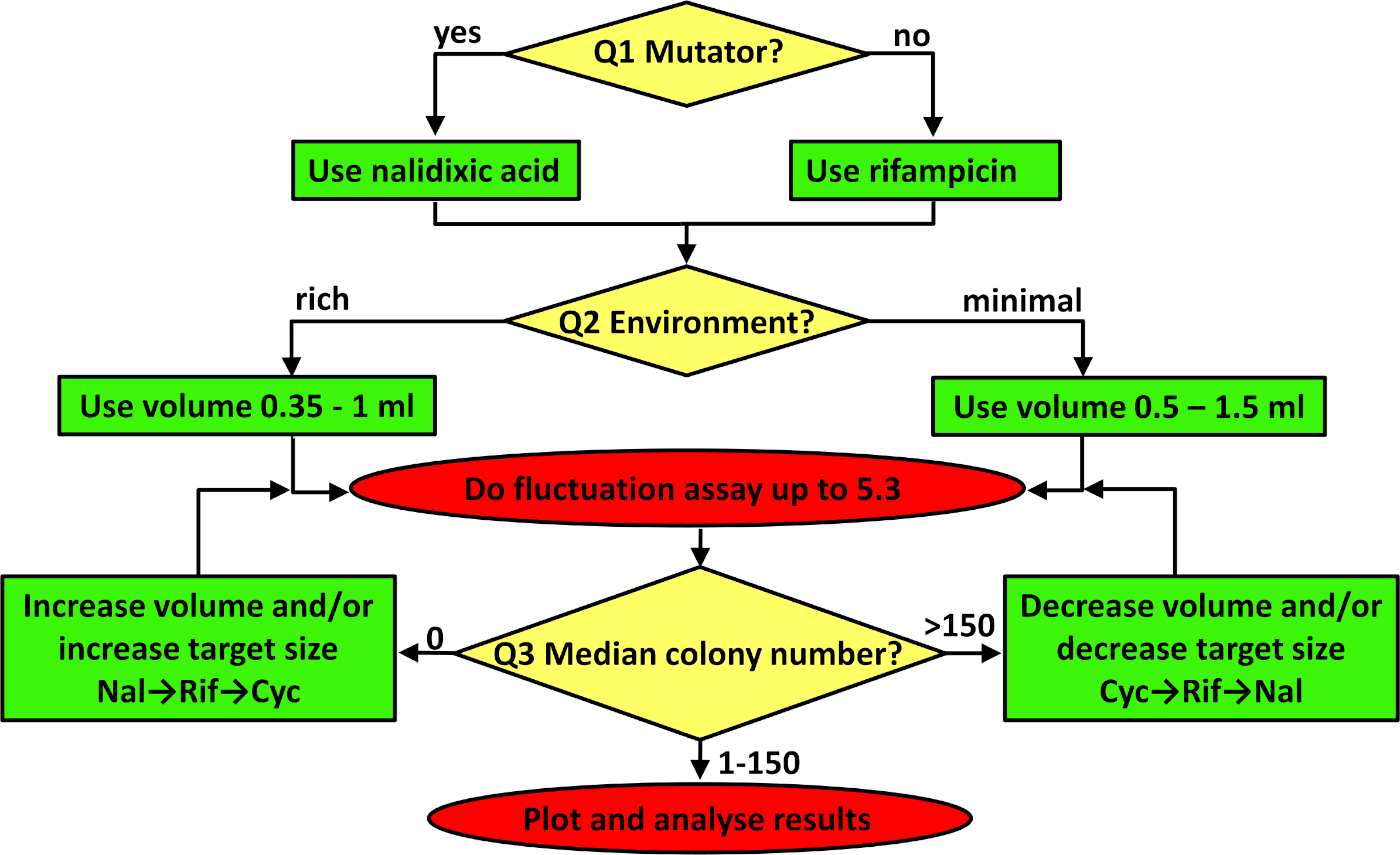
Figure 3: A flowchart for troubleshooting the fluctuation assay. The flowchart is to be followed from the top, addressing the three questions in the yellow diamonds in turn, adjusting the protocol according to the contents of the resulting green boxes, and implementing the protocol as indicated in the red ovals. See the first three paragraphs of the discussion for more details on how to troubleshoot. Please click here to view a larger version of this figure.
{kind=link}
Supplementary Table 1: Formulas for the media used in the protocol. Please click here to view this table (Right click to download).
Supplementary Table 2: Bacterial strains. Please click here to view this table (Right click to download).
Supplementary Table 3: Detailed description of the columns in the raw data file Krasovec_etal_JoVE_data.csv. Please click here to view this table (Right click to download).
Supplementary Data Files. Please click here to download these files.
Discussion
Any estimate of a mutation rate needs to maximize the precision achieved in order to ensure repeatability and reproducibility within and between studies34. For a fluctuation assay, there are three critical considerations. The first two have been set in the protocol given but will need troubleshooting (see Figure 3) if the protocol is adapted to work with different strains or environments. First is to choose the appropriate phenotypic marker. For bacteria, it is recommended to estimate rates at one of two marker loci, rpoB or gyrA, conferring resistance to antibiotics rifampicin and nalidixic acid, respectively. The target size for mutations to antibiotic resistance at these two loci is different. There are 79 and 20 unique mutations conferring resistance to rifampicin37 and nalidixic acid40 respectively. In practice, this means that on average, rifampicin resistant mutants are more frequently observed. So, the first question (Q1 in Figure 3) that needs to be answered is whether or not the strain is a mutator. When constitutive mutators are studied, where many observed mutant colonies are expected, it is better to use a marker with a smaller target size (e.g., nalidixic acid). See Figure 2B, where mutation rates of the constitutive mutator E. coli K-12 BW25113 ΔmutT were estimated using nalidixic acid as a marker. When working with nonmutator bacterial strains that have a wild type (i.e., normal) mutation rate, rifampicin is a better choice (see Figure 2A). If for some reason more observed mutants are needed, a relevant marker is resistance to a cycloserine. For this marker, resistance mutations can emerge in more than ten genes41, meaning that the target size is even larger than for rifampicin. When studying yeast and archaea it is recommended to estimate mutation rates in 25S ribosomal proteins and URA3, conferring resistance to hygromycin B and 5-fluoro-orotic acid (5-FOA), respectively.
The second critical consideration is the volume of parallel cultures. Which volume to use depends on the actual number of observed mutants. The expected number of observed mutants is affected by the target size for the chosen phenotypic marker, the strain's capacity to repair and avoid mutations (the average mutation rate), and the carrying capacity of the environment, which is affected both by the media used and the culture volume. If no parallel cultures contain mutant colonies resistant to rifampicin, then cycloserine should be used or the number of plated cells should be increased. This can be achieved by adding more glucose to a minimal medium or by growing cells in a richer (or complete) environment. However, in many cases, such an increase in population density is associated with a reduction in mutation rate, resulting in limited, if any, increase in the number of mutant colonies observed10. If increasing the nutrients is not a solution, then increasing the number of cells by increasing the volume of each parallel culture is an option. When the environment includes minimal salts with sugar as the only source of carbon and energy (i.e., the answer to Q2 in Figure 3 is "minimal medium"), then volumes between 0.5 and 1.5 mL should be used. If the environment is rich, then volumes of parallel cultures should be between 0.35−1 mL. The last question concerns the median number of resistant colonies. If too few mutant colonies are observed (i.e., the answer to Q3 in Figure 3 is 0) and the environment is not to be modified, then the volume of parallel cultures should be increased or the antibiotic with a larger target size (e.g., cycloserine) should be used. On the other hand, if a lot of mutant colonies are observed on all selective plates (more than ~150 per plate, see Figure 3), then the number of plated cells should be decreased, which usually means using a lower volume or switching to an antibiotic with a smaller target size (e.g., nalidixic acid).
Once the volume is chosen, it is best that all parallel cultures on one 96 deep well plate have the same volume. That allows more precise determination of the actual volume of parallel cultures from the weight of the plate. When mutation rates of a particular genotype are compared between different environments, it is again best to use the same volume of parallel cultures in all environments. If nalidixic acid is used for estimating wild type (i.e., normal) mutation rates or some other phenotypic marker is used that has an even smaller target size than nalidixic acid, the volume must be increased even more. One option is to make parallel cultures in 50 mL tubes with volumes of up to 15 mL. For instance, 10 mL parallel cultures were prepared in 50 mL tubes when estimating the E. coli K-12 MG1655 mutation rate to nalidixic acid (see Figure 2A). The parallel 10 mL cultures were then plated on the selective TA agar and poured into large 150 mm plates instead of standard 90 mm Petri dishes. The downside of preparing parallel cultures in 50 mL tubes is that the throughput is considerably lower compared to assaying mutation rates in a 96 deep well plate. One solution is to decrease the number of parallel cultures. However, this will affect the precision for the estimate of m, which depends on the expected number of mutational events and the number of parallel cultures26. Obtaining a distribution of observed mutants with 14−17 parallel cultures (as was done in Figure 2), is a good balance between a solid throughput and an acceptable precision level26 of 20%. A median precision level of 17.5% is similar to the median precision with an interquartile range of 16.4% (5.7%−38.9%, n = 580) calculated from a much larger data set10. Thus, it is recommended that when preparing parallel cultures in 96 deep well plates or 50 mL tubes the distribution of observed mutants is obtained with at least 14 parallel cultures. When mutation rates are estimated in different environments it is recommended to test precision levels by doing a multiplate experiment, where all 96 parallel cultures on one plate are grown in the same environment. In addition, when preparing parallel cultures, it is critical that inocula contain a low number of cells, because it reduces the chances of any resistant cells being present in the inoculum. Preexisting resistant mutants are not wanted in the inoculum, because they will increase in numbers and create a lawn on selective plates and estimation of the mutation rate will not be possible. For instance, in most nonmutator E. coli populations, mutation rate to rifampicin resistance is in the order of ~10-8. Thus, to avoid inoculating the culture with a preexisting resistant mutant, one must inoculate with fewer than 108 cells (e.g., 103−104 cells). The final critical step is to ensure that before selective agar plates are incubated, the surface on the selective agar is completely dry. Spreaders cannot be used if 6-well plates are used and the initial volume of a parallel culture is 1 mL, for example. Plates must be left uncovered in sterile conditions to let the surface liquid dry out. The time this takes can be highly variable, dependent on ambient conditions and the condition of the plates. This time should be minimized but can be up to several hours.
The fluctuation assay has inherent constraints. It assays phenotypic markers of mutation only in a small subset of the genome. The assay thus requires large populations that go through a sufficient number of generations to observe enough mutations to estimate a rate at all. This means that fluctuation assays can only be used on organisms that are capable of going through a large number of generations rapidly, like bacteria, baker's yeast42, or liquid-culture mammalian cells30. Also, mutations are rare events occurring in the specific biochemical circumstances of a particular cell. The fact that fluctuation assays look across large populations of cells over time means that those circumstances can differ substantially. Using this assay, it is thus difficult to study the progression of mutation rates of a particular population from the lag phase to early and late exponential phase and finally to a stationary phase. Any differentiation of mutation rates among single cells within the population are completely hidden from the fluctuation assay. Single-cell mutation dynamics can be studied with a single-molecule tracking of DNA repair protein MutS43 or by counting foci of accumulated MutL proteins44. Recent advances in high-throughput sequencing have also made it possible to directly estimate mutation rates from parent-offspring trios9,45 and multigeneration pedigrees46. Such methodological advances are beginning to permit the direct counting of mutations occurring within a single generation. However, this direct approach needs expensive and state-of-the-art technologies like fluorescence microscopy, microfluidics, or whole-genome sequencing. On the other hand, the fluctuation assay is relatively inexpensive and only standard laboratory equipment is needed. Doing more fluctuation assays will also facilitate the generation of novel hypotheses that may be tested with more direct single-cell approaches.
There is a long-standing interest in the study of mutations, so the fluctuation assay will likely remain a widely used method. The number of citations of the seminal paper by Luria and Delbrück5 in the last 4 years (2015-2018) have all been among the top five for citations of this paper. However, due to a large amount of precise manual work needed for properly carrying out a fluctuation assay, most studies only conduct a handful of fluctuation assays. This, however, is insufficient to reveal the environmental dependencies of the mutation rate. By streamlining fluctuation assays using multiwell plates, as explained in this paper, the current maximum throughput possible is 11 deep well plates (55 fluctuation assays) in parallel, as described here. Running two sets of fluctuation assays staggered by a day in parallel, allows carrying out up to 110 assays per week. Another step change in throughput may yet be possible by automating various steps of the fluctuation assays from the purely manual protocol given. Also, for studying environmental dependencies of the mutation rate, population density needs to be taken into account. Previous results10 show that when known factors that affect mutation rate are accounted for, controlling for population density can reduce variation in mutation-rate estimates by more than 90%. To control the density, we recommend that Nt (used for estimating mutation rate) be determined independently from the method used to determine population density. In bacteria, Nt can be determined by CFU and density, for instance, with an ATP-based luminescence assay10.
High throughput and controlling density are both essential when studying how an organism's ecological context affects spontaneous mutation rate. Knowing the existence of mutation rate plasticity is important, but understanding its causes and effects are key challenges that need to be met if mutation rate plasticity is to be incorporated into a wider biological context. The fluctuation assay is a great tool that can be used to test many hypotheses, because results are obtained rapidly, and assays are inexpensive relative to other methods. The stage is set, for instance, to study environmental dependencies of mutation rate in bacterial communities and microbiomes. Adapting the fluctuation assay to cocultures can test the hypothesis that strains influence each other's mutation rates via small molecules. Doing thousands of fluctuation assays with cocultures can determine if strains vary both in their ability to modify each other's mutation rates and in their susceptibility to have their mutation rate modified by others. Perhaps variation among strains in susceptibility to mutation rate manipulation is attributable to specific genetic variation. This may transform our views on how evolution works in complex communities, not least in examples of wide importance such as how antimicrobial resistance emerges.
Disclosures
The authors have nothing to disclose.
Acknowledgements
RK was supported by BB/M020975/1 and University of Manchester School of Biological Sciences. HR was supported by BB/J014478/1. GG was supported by BBSRC Doctoral Training Partnership BB/M011208/1. DRG was supported by UKRI award number MR/R024936/1.
Materials
| Name | Company | Catalog Number | Comments |
| 1.5 mL Microcentrifuge tubes | Starlab International GmbH | S1615-5550 | |
| 2,3,5-Triphenyltetrazolium chloride | Sigma-Aldrich | T8877-10g | |
| 6-well plates | Greiner Bio-One | REF 657102 | |
| 90 mm Petri Dishes Triple Vented | ThermoFisher Scientific | REF 120189 | |
| 96 deep-well plate (Masterblock 2 mL) | Greiner Bio-One | REF 780270 | |
| Ammonium sulfate | Fisher Chemical | A/6440/53 | |
| Bacto Agar | Becton, Dickinson and Company | REF 214010 | |
| Bacto yeast extract | Becton, Dickinson and Company | REF 212750 | |
| Cycloserine | Sigma-Aldrich | 1158005-250MG | Only for assaying an alternative phenotypic marker |
| D-Glucose anhydrous | Fisher Chemical | G/0500/61 | |
| 50 mL Centrifuge Tube | Corning | REF 430828 | |
| L-(+)-Arabinose | Sigma-Aldrich | A3256-500g | |
| Magnesium sulfate heptahydrate | Fisher Chemical | M/1050/53 | |
| Nalidixic acid | Sigma-Aldrich | N8878-5G | Only for assaying an alternative phenotypic marker |
| Potassium phosphate dibasic trihydrate | Sigma-Aldrich | P5504-500g | |
| Potassium phosphate monobasic | Sigma-Aldrich | P0662-500g | |
| Rifampicin | EMD Millipore Corp, USA | 557303-1GM | |
| Sodium chloride | Fisher Chemical | S/3160/60 | |
| Spectophotometer | Jenway | 6320D | |
| Thiamine hydrochloride | Sigma-Aldrich | T4625-25g | |
| Trisodium citrate dihydrate | Sigma-Aldrich | S1804-500g | |
| Tryptone | Fisher Chemical | 1278-7099 |
References
- de Vries, H. Die mutationstheorie. Versuche und beobachtungen über die entstehung von arten im pflanzenreich. 1, (1901).
- Muller, H. J. Artificial transmutation of the gene. Science. 66 (1699), (1927).
- Muller, H. J. The measurement of gene mutation rate in Drosophila, its high variability, and its dependence upon temperature. Genetics. 13 (4), 279-357 (1928).
- Sturtevant, A. H. Essays on evolution. I. On the effects of selection on mutation rate. The Quarterly Review of Biology. 12 (4), 464-467 (1937).
- Luria, S. E., Delbrück, M. Mutations of bacteria from virus sensitivity to virus resistance. Genetics. 28 (6), 491-511 (1943).
- Jee, J., et al. Rates and mechanisms of bacterial mutagenesis from maximum-depth sequencing. Nature. 534 (7609), 693-696 (2016).
- Fusco, D., Gralka, M., Kayser, J., Anderson, A., Hallatschek, O. Excess of mutational jackpot events in expanding populations revealed by spatial Luria-Delbrück experiments. Nature Communications. 7, (2016).
- Halligan, D. L., Keightley, P. D. Spontaneous mutation accumulation studies in evolutionary genetics. Annual Review of Ecology Evolution and Systematics. 40 (1), 151-172 (2009).
- Jónsson, H., et al. Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature. 549, (2017).
- Krašovec, R., et al. Spontaneous mutation rate is a plastic trait associated with population density across domains of life. PLoS Biology. 15 (8), (2017).
- Krašovec, R., et al. Mutation rate plasticity in rifampicin resistance depends on Escherichia coli cell-cell interactions. Nature Communications. 5, 3742 (2014).
- Massey, R. C., Buckling, A. Environmental regulation of mutation rates at specific sites. Trends in Microbiology. 10 (12), 580-584 (2002).
- Lynch, M., et al. Genetic drift, selection and the evolution of the mutation rate. Nature Review Genetics. 17 (11), 704-714 (2016).
- Foster, P. L. Stress-induced mutagenesis in bacteria. Critical Reviews in Biochemistry and Molecular Biology. 42 (5), 373-397 (2007).
- Krašovec, R., et al. Where antibiotic resistance mutations meet quorum-sensing. Microbial Cell. 1 (7), 250-252 (2014).
- Krašovec, R., et al. Opposing effects of final population density and stress on Escherichia coli mutation rate. The ISME Journal-Multidisciplinary Journal of Microbial Ecology. 12, 2981-2987 (2018).
- Lea, D., Coulson, C. The distribution of the numbers of mutants in bacterial populations. Journal of Genetics. 49 (3), 264-285 (1949).
- Armitage, P. The statistical theory of bacterial populations subject to mutation. Journal of the Royal Statistical Society. Series B (Methodological. 14 (1), 1-40 (1952).
- Koch, A. L. Mutation and growth rates from Luria-Delbrück fluctuation tests). Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 95 (2-3), 129-143 (1982).
- Cairns, J., Overbaugh, J., Miller, S. The origin of mutants. Nature. 335 (6186), 142-145 (1988).
- Stewart, F. M., Gordon, D. M., Levin, B. R. Fluctuation analysis: the probability distribution of the number of mutants under different conditions. Genetics. 124 (1), 175-185 (1990).
- Drake, J. W. A constant rate of spontaneous mutation in DNA-based microbes. Proceedings of the National Academy of Sciences. 88 (16), 7160-7164 (1991).
- Ma, W. T., Sandri, G. V., Sarkar, S. Analysis of the Luria-Delbrück distribution using discrete convolution powers. Journal of Applied Probability. 29 (2), 255-267 (1992).
- Shapiro, J. A. Adaptive mutation - Who's really in the garden. Science. 268 (5209), 373-374 (1995).
- Matic, I., et al. Highly variable mutation rates in commensal and pathogenic Escherichia coli. Science. 277 (5333), 1833-1834 (1997).
- Rosche, W. A., Foster, P. L. Determining mutation rates in bacterial populations. Methods. 20 (1), 4-17 (2000).
- Rosenberg, S. M. Evolving responsively: Adaptive mutation. Nature Reviews Genetics. 2 (7), 504-515 (2001).
- Lynch, M. Evolution of the mutation rate. Trends in Genetics. 26 (8), 345-352 (2010).
- Zheng, Q. A new practical guide to the Luria-Delbrück protocol. Mutation Research. 781, 7-13 (2015).
- Boesen, J. J. B., Niericker, M. J., Dieteren, N., Simons, J. How variable is a spontaneous mutation-rate in cultured-mammalian-cells. Mutation Research. 307 (1), 121-129 (1994).
- Melnyk, A. H., Wong, A., Kassen, R. The fitness costs of antibiotic resistance mutations. Evolutionary Applications. 8 (3), 273-283 (2015).
- Sun, L., et al. Effective polyploidy causes phenotypic delay and influences bacterial evolvability. PLoS Biology. 16 (2), (2018).
- Frenoy, A., Bonhoeffer, S. Death and population dynamics affect mutation rate estimates and evolvability under stress in bacteria. PLoS Biology. 16 (5), (2018).
- Foster, P. L. Methods for determining spontaneous mutation rates. Methods in Enzymology. , 195-213 (2006).
- Wrande, M., Roth, J. R., Hughes, D. Accumulation of mutants in "aging" bacterial colonies is due to growth under selection, not stress-induced mutagenesis. Proceedings of the National Academy of Sciences of the United States of America. 105 (33), 11863-11868 (2008).
- Adrien, M., Rémy, D., Stéphane, D., Bernard, Y. flan: An R package for inference on mutation models. The R Journal. 9. 334, (2017).
- Garibyan, L., et al. Use of the rpoB gene to determine the specificity of base substitution mutations on the Escherichia coli chromosome. DNA Repair. 2 (5), 593-608 (2003).
- Stewart, F. M. Fluctuation tests: how reliable are the estimates of mutation rates?. Genetics. 137 (4), 1139-1146 (1994).
- Baba, T., et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Molecular Systems Biology. 2 (1), (2006).
- Yamagishi, J. -. I., Yoshida, H., Yamayoshi, M., Nakamura, S. Nalidixic acid-resistant mutations of the gyrB gene of Escherichia coli. Molecular and General Genetics MGG. 204 (3), 367-373 (1986).
- Chen, J., et al. Identification of novel mutations associated with cycloserine resistance in Mycobacterium tuberculosis. Journal of Antimicrobial Chemotherapy. 72 (12), 3272-3276 (2017).
- Lang, G. I., Murray, A. W. Estimating the per-base-pair mutation rate in the yeast Saccharomyces cerevisiae. Genetics. 178 (1), 67-82 (2008).
- Uphoff, S., et al. Stochastic activation of a DNA damage response causes cell-to-cell mutation rate variation. Science. 351 (6277), 1094-1097 (2016).
- Robert, L., et al. Mutation dynamics and fitness effects followed in single cells. Science. 359 (6381), 1283-1286 (2018).
- Kong, A., et al. Rate of de novo mutations and the importance of father/'s age to disease risk. Nature. 488 (7412), 471-475 (2012).
- Narasimhan, V. M., et al. Estimating the human mutation rate from autozygous segments reveals population differences in human mutational processes. Nature Communications. 8 (1), 303 (2017).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved