
Method Article
Improved UPLC-UV Method for the Quantification of Vitamin C in Lettuce Varieties (Lactuca sativa L.) and Crop Wild Relatives (Lactuca spp.)
* These authors contributed equally
In This Article
Summary
We present a fast and reliable method to quantify vitamin C in Lactuca spp. using UPLC-UV, potentially transferable to other plants. The key steps are the sample preparation and vitamin C extraction under stable conditions, the reduction of dehydroascorbic acid to ascorbic acid and the optimization of the chromatographic procedure.
Abstract
Vitamins, especially vitamin C, are important micronutrients found in fruits and vegetables. Vitamin C is also a major contributor to their antioxidant capacity. Lettuce is one of the most popular vegetables among consumers worldwide. An accurate protocol to measure vitamin C content in lettuce and other related species is crucial. We describe here a method using the ultra-high-performance liquid chromatography-ultraviolet (UPLC-UV) technique, in which sample preparation, vitamin extraction and chromatography conditions were optimized.
Samples were collected to represent the entire plant, frozen at -80 °C and lyophilized to prevent undesirable oxidation and make their manipulation easier. The extraction of vitamin C was carried out in acidic media, which also contributed to its stability. As vitamin C can be present in two different interconvertible forms, ascorbic acid (AA) and dehydroascorbic acid (DHAA), both compounds should be measured for accurate quantification. The DHAA was quantified indirectly after its reduction to AA because AA shows a higher absorptivity than DHAA in the UV range of the spectrum. From the same extract, two measurements were carried out, one before and one after that reduction reaction. In the first case, we were quantifying the AA content, and in the second one, we quantified the sum of AA and DHAA (TAA: total ascorbic acid) in the form of AA. Then, DHAA quantity was indirectly obtained by subtracting AA coming from the first measurement from TAA. They were determined by UPLC-UV, using a commercial AA standard to build a calibration curve and optimizing the chromatographic procedure, to obtain AA peaks that were completely resolved in a short time. This protocol could be easily extrapolated to any other plant material with slight or no changes. Its accuracy revealed statistically significant differences otherwise unperceived. Other strengths and limitations are discussed more in depth in the manuscript.
Introduction
Cultivated lettuce (Lactuca sativa L.) is one of the most produced and consumed leafy vegetables worldwide, with a total production of about 27.3 million tons in 20181. Lettuce is perceived as healthy by consumers. The nutritional properties are mainly attributed to the source of antioxidant compounds in the crop, such as vitamin C, among others like polyphenols and vitamin E2. Vitamin C is an essential micronutrient for humans unlike many other vertebrates, as we are unable to produce it due to mutations present in the gene coding for the last step enzyme in the biosynthetic pathway3. It is required for a normal cell metabolism and it also plays an important role in immune responses mainly due to its antioxidant activity3,4.
Total vitamin C is made up of ascorbic acid (AA) and dehydroascorbic acid (DHAA). AA is the most biologically active form of the vitamin, but DHAA (its oxidation product) also shows biological activity and it can be easily converted into AA in the human body5. Therefore, quantifying both forms is important to determine the total vitamin C content of any horticultural crop, lettuce included.
A wide variety of approaches based on different analytical techniques have been used to measure vitamin C in vegetables, such as enzymatic, spectrophotometric, and titrimetric methods6,7,8. Although these methods are simple, they are not chemically specific for AA9. Consequently, chromatographic methods are preferred, especially the high-performance liquid chromatography-ultraviolet (HPLC-UV) technique, because of their higher accuracy10. HPLC-UV has been used to determine vitamin C in a great diversity of crops, like broccoli, spinach and lettuce11,12,13. However, the simultaneous quantification of AA and DHAA is complicated due to the low absorptivity of DHAA in the UV range of the spectrum. Alternatively, DHAA can be determined indirectly by using a reducing agent that converts DHAA to AA, measuring total ascorbic acid (TAA), and then calculating the difference between TAA and AA. Due to the necessity of a reduction reaction, in some studies, only AA has been quantified14, which could actually represent an underestimation of vitamin C activity. That additional reduction reaction is also needed to determine DHAA indirectly even when the last advance in liquid chromatography techniques, ultra-high performance liquid chromatography (UPLC), is used. That step also benefits from the advantages that UPLC exhibits when compared to HPLC: higher efficiency and resolution, increased sensitivity, shorter time analysis and lower solvent consumption15. In consequence, UPLC-UV technique has been utilized to quantify vitamin C in different crops16.
In addition, AA is a very labile molecule; thus, it is important to develop a protocol that prevents its degradation during lettuce storage and vitamin C analysis9. In this context, the following protocol offers a rapid and improved quantification of vitamin C content in lettuce by UPLC-UV, as well as an efficient extraction procedure. Not only elite cultivars have been included in the present study, but also traditional landraces and some wild relatives due to their potential interest in crop breeding, specifically in the improvement of the nutritional value of lettuce.
Protocol
1. Plant material preparation
- Sample at least two leaves per plant in 50 mL polypropylene tubes, an outer (older) and an inner (younger) one in order to represent more accurately the whole plant. Collect at least three biological replicates for each sample.
- Freeze them immediately using liquid nitrogen and store them at -80 °C until use. Make sure the liquid nitrogen does not get into the tubes; otherwise they could explode when removed due to the gas expansion during vaporization.
CAUTION: Gloves and a face shield are required due to the potential hazards associated with using liquid nitrogen. - Remove the caps from the tubes and place them on the trays within the freeze dryer chamber of the lyophilizer (Table of Materials) programmed as follows: -25 °C for 72 h, -10 °C for 10 h, 0 °C for 10 h, and 20 °C for at least 4 h. Maintain the condenser temperature and the vacuum constant during the freeze-drying process at -80.2 °C and 112 mTorr, respectively.
- When the material is completely dry (between 4 and 7 days depending on the plant and the degree of compaction into the tube), preserve at 4 °C, -20 °C or -80 °C for short (days to weeks), medium (months) or long (years) storage, respectively. The inclusion of bags containing silica gel beads in the sample-containing tubes is recommended.
- Place the lyophilized samples into 20 mL polypropylene tubes together with 10 mm diameter stainless steel balls and grind them with a multitube vortexer using the intensity and time needed to obtain a fine dust.
NOTE: During the entire process, protect the samples from exposure to direct light.
2. Reagent and solution preparation
- Prepare the solvent extraction solution: 8% acetic acid (v/v), 1% MPA (meta-phosphoric acid) (w/v), 1 mM EDTA (ethylenediaminetetraacetic acid).
- Calculate the total volume of solvent needed to process the whole set of samples taking into account that 5 mL will be added to each. To prepare 1 L of the solution, add to a flask: 30 g of MPA, 0.372 g of EDTA dehydrate, 80 mL of acetic acid and 500 mL of ultrapure water (scale volumes and quantities accordingly). Seal the flask mouth with plastic film.
- Once dissolved with the help of a magnetic stirrer, use a volumetric flask to accurately measure 1 L, adding the necessary ultrapure water.
- Prepare the reduction reaction buffer (0.5 M Tris (2-amino-2-(hydroxymethyl)-1,3-propanediol) pH 9.0) and reducing solution (40 mM DTT (1,4-Dithiothreitol) with 0.5 M Tris pH 9.0).
- Calculate the total volume of reducing solution needed to process the whole set of samples taking into account that 200 µL will be added to each of them. To prepare 100 mL of the buffer, add to a beaker: 6.055 g of Tris and 90 mL of ultrapure water (scale volumes and quantities accordingly). Seal the beaker mouth with plastic film.
- Once dissolved with the help of a magnetic stirrer, adjust the solution to pH 9.0 by adding 2 M HCl and use a volumetric flask to accurately measure 100 mL, adding the necessary ultrapure water.
- To prepare 100 mL of the reducing solution, add to a beaker: 0.629 g of DTT (purity: 98%) and 90 mL of the buffer (0.5 M Tris pH 9.0) previously prepared (2.2.1 to 2.2.2). Scale volumes and quantities accordingly. Seal the beaker mouth with plastic film.
- Once dissolved with the help of a magnetic stirrer, use a volumetric flask to accurately measure 100 mL, adding the necessary volume of buffer 0.5 M Tris pH 9.0.
NOTE: The reducing solution is very unstable. That is why a freshly made solution is strongly recommended.
- Sulphuric acid (0.4 M H2SO4)
- Calculate the total volume of 0.4 M sulphuric acid needed to process the whole set of samples taking into account that 200 µL will be added to each. To prepare 100 mL of the solution, add to a beaker: 80 mL of ultrapure water and then 2.22 mL of H2SO4 (purity: 96%, density: 1.84 g mL-1). Use a volumetric flask to accurately measure 100 mL, adding the necessary ultrapure water.
CAUTION: Sulphuric acid is very corrosive, so it must be handled using protective equipment and under hood. In addition, the acid should be added to ultrapure water, and not water to acid, to reduce fumes and avoid accidents.
- Calculate the total volume of 0.4 M sulphuric acid needed to process the whole set of samples taking into account that 200 µL will be added to each. To prepare 100 mL of the solution, add to a beaker: 80 mL of ultrapure water and then 2.22 mL of H2SO4 (purity: 96%, density: 1.84 g mL-1). Use a volumetric flask to accurately measure 100 mL, adding the necessary ultrapure water.
- Hydrochloric acid (2 M HCl).
- To prepare 100 mL of 2 M hydrochloric acid, add to a beaker: 80 mL of ultrapure water and then 6.13 mL of HCl (purity: 37%, density: 1.19 g mL-1). Seal the beaker mouth with plastic film. Use a volumetric flask to accurately measure 100 mL, adding the necessary ultrapure water. Scale volumes accordingly.
CAUTION: Hydrochloric acid is very corrosive, so it has to be handled using protective equipment and under hood. In addition, the acid should be added to ultrapure water, and not water to acid, to reduce fumes and avoid accidents.
- To prepare 100 mL of 2 M hydrochloric acid, add to a beaker: 80 mL of ultrapure water and then 6.13 mL of HCl (purity: 37%, density: 1.19 g mL-1). Seal the beaker mouth with plastic film. Use a volumetric flask to accurately measure 100 mL, adding the necessary ultrapure water. Scale volumes accordingly.
- AA standard (stock and dilutions)
- Weigh exactly 10 mg of AA standard (purity: 99%) using a precision balance and add 90 mL of mobile phase (ultrapure water pH 2.0 with formic acid).
- Once dissolved with the help of a magnetic stirrer, use a volumetric flask to accurately measure 100 mL, adding the necessary volume of ultrapure water pH 2.0 with formic acid.
NOTE: Protect this stock solution from the exposure to light. - Prepare five dilutions from the stock of the AA standard to obtain a calibration curve following the instructions in Table 1 and proceed with step 5.2.
| Standard | [AA] (µg mL-1) | AA (100 µg mL-1) solution (µL) | Mobile phase (µL)a |
| 1 | 0.5 | 5 | 995 |
| 2 | 2.5 | 25 | 975 |
| 3 | 5 | 50 | 950 |
| 4 | 10 | 100 | 900 |
| 5 | 25 | 250 | 750 |
| aUltrapure water pH 2.0 acidified by formic acid. | |||
Table 1: Protocol to prepare five standards of AA (ascorbic acid). Volumes of solute and solvent to prepare each of the different concentrations of the standards are indicated.
3. Extraction of AA and DHAA
NOTE: It is recommended to work under conditions of low light intensity during the extraction steps.
- To a 15 mL polypropylene centrifuge tube, add 50 mg of lyophilized ground sample and 5 mL of the extraction solvent (step 2.1).
- Shake the mixture using a vortex for 5 s and then an orbital shaker for 10 min at 2000 rpm.
- Introduce the tube in an ultrasonic bath for 10 min at room temperature with ultrasound activated.
- Centrifuge at 4,000 x g for 10 min at 4 °C.
- Take the supernatant, pass it through a 0.22 µm regenerated cellulose filter and store it in a 5 mL amber vial. This is Extract 1, which contains AA and DHAA.
NOTE: The protocol can be paused here by freezing the extracts at -80 °C and protecting them from exposure to light as AA and DHAA are very unstable and degrade easily in the presence of light, at high temperatures or under oxidizing atmospheres (Supplemental File 1).
4. DHAA reduction to AA to extract TAA
- Transfer 200 µL of Extract 1 to a 2 mL amber vial for liquid chromatography and add 200 µL of the reducing solution (step 2.2). Close the vial with a PTFE-silicone plug with pre-opening and shake it with a vortex for 5 s.
- Allow the solution to stand for 30 min at room temperature and protect from light.
- Add 200 µL of 0.4 M H2SO4 to stop the reaction and stabilize AA in acidic pH. The resulting solution is Extract 2, which contains only AA and is actually TAA.
5. Determination
- UPLC-UV preparation
- Prepare the working solutions described in Table 2, suitably filtered through 0.22 µm filters, sonicated for at least 10 min and place them in the UPLC system.
- Switch on the three UPLC modules and wait for the internal calibration process to finish.
- Open the software (e.g., Empower 3) and load the instrumental program described in Table 2: Empower 3 | Run Samples | Vitamin C method | UPLC_PDA | Use QuickStart.
- Once the software is loaded with the correct program, access the UPLC management console: Quaternary Solvent Manager | Click right mouse button | Launch Console.
- Proceed to the preparation and stabilization of the UPLC instrument: System | Control | Startup.
- Purge all UPLC lines for at least 5 min: Prime Solvents | QSM | Check A, B, C, D and Seal Wash | Duration of prime > 5 min.
- Purge and clean the injector: Prime Solvents | SM | Check Wash solvent (> 45 s) and Check Purge solvent (> 35 cycles).
- Equilibrate UPLC to method conditions: Equilibrate to Method | QSM | Flow (0.3 mL min-1) | Solvent A (2%) | Solvent B (0%) | Solvent C (98%) | Solvent D (0%); Equilibrate to Method | SM | Sample (5 °C) | Column (30 °C) and Equilibrate to Method | Other | Check Lamp On | Press Start.
- Wait for at least 1 h (even more time is recommended) for the equipment to stabilize. Stability can be verified checking the pressure in the column in the Launch Console: System | Quaternary Solvent Manager | QSM System Pressure. Ensure that there are no identifiable trends in pressure changes (either increases or decreases) and the delta value is less than 10 psi.
- In the QuickStart screen, fill the matrix with the names of the standards and samples to be analyzed.
- AA determination in the standards
- Transfer 1 mL of each of the five AA standards previously prepared (step 2.5.3) to 2 mL amber vials for liquid chromatography. Close the vial with a PTFE-silicone plug with pre-opening and inject 5 µL in the UPLC instrument.
- Carry out the chromatography following the procedure described in Table 2 starting from most diluted to most concentrated.
- AA determination in the samples
- Pipette 200 µL of Extract 1 in a 2 mL amber vial for liquid chromatography and add 800 µL of ultrapure water. Close the vial with a PTFE-silicone plug with pre-opening and inject 5 µL in the UPLC instrument.
- Carry out the chromatography following the procedure described in Table 2.
- TAA determination in the samples
- Add 400 µL of ultrapure water to Extract 2. Close the vial with a PTFE-silicone plug with pre-opening and inject 5 µL in the UPLC instrument.
- Carry out the chromatography following the procedure described in Table 2.
| Components and parameters | Description |
| Instrument | Acquity UPLC H-Class |
| Detector | PDA eλ Detector λabs for AA=245 nm |
| Software | Empower 3 |
| Column | Acquity UPLC HSS T3 (150 mm x 2.1 mm x 1.8 µm) |
| Channel A | CH3OH |
| Channel B/Wash | H2O:CH3OH (50:50 v:v) |
| Channel C | Ultrapure water pH 2.0 acidified by formic acida |
| Channel D/Seal Wash | Ultrapure water:acetonitrile (90:10 v:v) |
| Mobile phase | 0.3 mL min-1 of 2%A + 98%C (isocratic mode) |
| Column temperature | 30 °C |
| Autosampler temperature | 5 °C |
| Injection volume | 5 µL |
| AA retention time | 1.874 min |
| Running time | 3 min |
| aUndetermined volume of formic acid used until pH adjustment | |
Table 2: Chromatographic procedure optimized to determine AA (ascorbic acid) in extracts from lettuce and wild relatives. Description of the components, conditions and solutions employed.
6. Quantification of AA and DHAA
- Statistical analysis
- Determine the analytical parameters of the chromatographic method as described by Bertolín et al.18 (Table 3).
NOTE: The values of the parameters presented in Table 3 will need to be defined under specific experimental conditions.
- Determine the analytical parameters of the chromatographic method as described by Bertolín et al.18 (Table 3).
| Analytical parameters of the method | Values |
| Linear range (μg mL-1) | 0.5-25 |
| Linear equation | y=53,143.03x |
| R2 | 0.99998 |
| Limit of detection (mg AA g-1 of dry matter) | 0.013 |
| Limit of quantification (mg AA g-1 of dry matter) | 0.045 |
| Repeatability (CV, %)a | 1.75 |
| Intermediate precision (CV, %)a | 4.22 |
| Recovery (Rec, %)b | 95.6±2.4 |
| aCV: coefficient of variation | |
| bThe recovery assay was performed with 10 aliquots containing 50 mg of the same sample, 5 spiked with 2 mg of AA g-1 of dry matter, and 5 non-spiked. %Rec=([AA]spiked sample-[AA]sample)/([AA]spiked)x100. | |
Table 3: Optimized analytical parameters for the detection and quantification of AA (ascorbic acid) and TAA (total ascorbic acid). The linear range, the equation and the coefficient of determination of the calibration (R2) curve, as well as the limits of detection and quantification of AA (the same for TAA), and the repeatability, intermediate precision and recovery were obtained with a sample injection volume of 5 μL.
- Calculate the AA and TAA concentration.
- Open the standard and sample chromatograms: QuickStart | Browse Project | Channels | “name of standard or sample” | PDA Ch1 245 nm@1.2 nm.
- Integrate the corresponding peak (AA or TAA) in the standards and samples by clicking on its starting point (approximately 1.790 min) and dragging it with the mouse to its end point (approximately 1.910 min).
- Build a calibration curve representing the absorbance values determined chromatographically (step 5.2.) against the concentration of the five AA standards prepared above (Table 1).
- Interpolate the absorbance values of the samples determined in steps 5.3 and 5.4 and obtain the AA and TAA concentration, respectively, with the following formula:

where y is the integrated peak area, x is the AA or TAA concentration in ppm and m and n are the slope and the y-intercept of the obtained regression line, respectively. - For calculating the concentration of DHAA, apply the following formula:

NOTE: To obtain the total concentrations of the DHAA, AA and TAA in mg g-1 of dry weight, the values obtained directly interpolating in the calibration curve will have to be multiplied by the total extract volume and the dilution factor applied, and then divided by the weight of the sample used to carry out the extraction.
Results
Vitamin C quantification in Lactuca matrixes requires the development of a chromatographic approach that can ensure reliable results. Figure 1A shows a chromatogram resulting from a non-optimized protocol (Supplemental File 2), which presents an AA peak together with an unidentified minor “shoulder”. Nevertheless, after improving the extraction and chromatographic conditions, a resolved AA peak without interferences of unknown compounds was achieved (Figure 1B). In addition, the use of UPLC-UV equipment instead of HPLC-UV allowed us to reduce the retention time (RT) for AA: 1.874 min in the optimized chromatograms versus 2.980 min in the non-optimized ones (Figure 1), as well as the running times, 3 and 7 minutes for the optimized and non-optimized protocols, respectively.

Figure 1: Chromatograms of AA in the same lettuce sample (commercial cultivar ‘Begoña’). (A) HPLC-UV chromatogram resulting from a non-optimized protocol (conditions described in Supplemental File 2). (B) UPLC-UV chromatogram obtained with the optimized protocol (conditions described in Table 2). Please click here to view a larger version of this figure.
{kind=link}
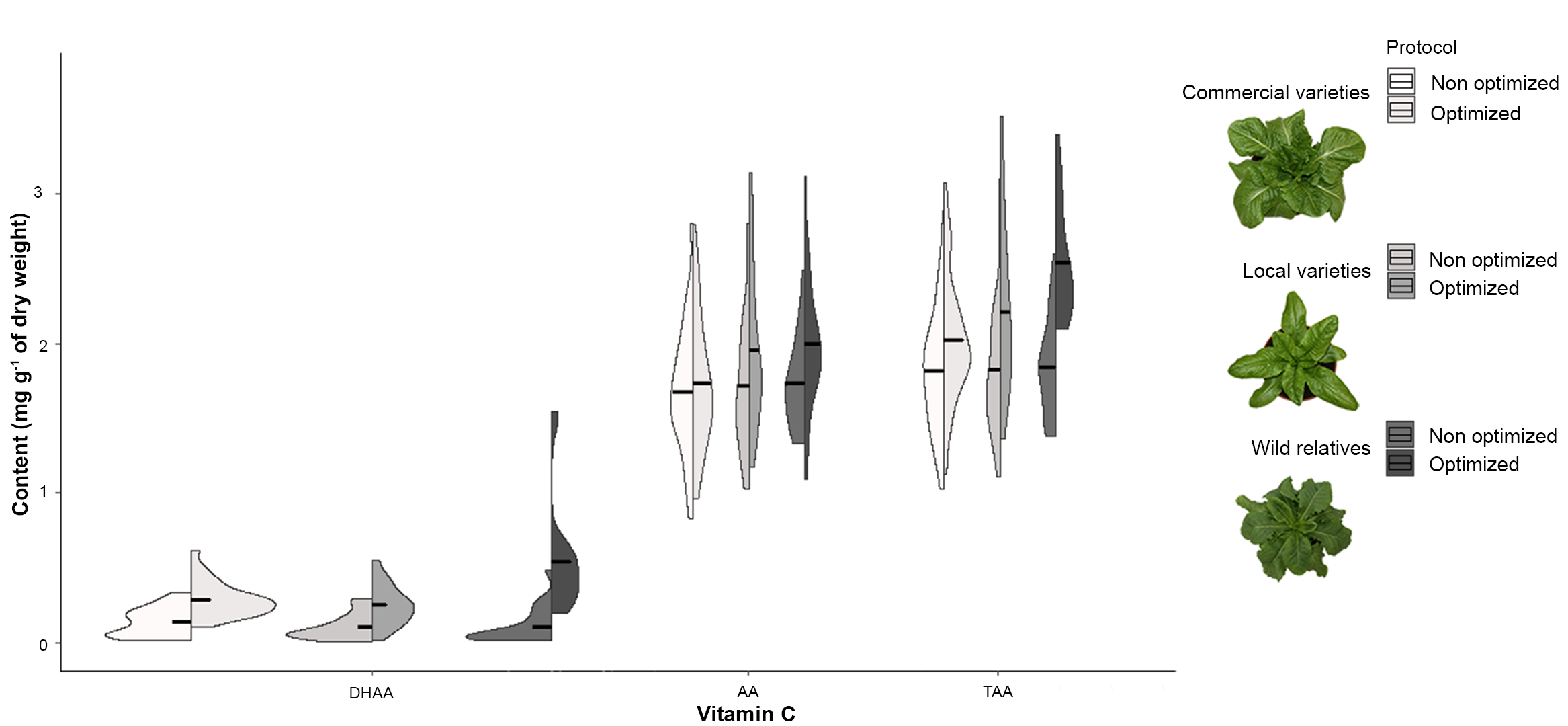
Interferences in AA peaks, like those observed in Figure 1A, consistently resulted in underestimation of vitamin C (AA, DHAA and TAA) content (Figure 2) due to an insufficient separation during the chromatographic process as the overlapping peak areas were integrated by a vertical drop at the deepest point between them. This bias is especially noticeable in the case of the crop wild relatives, particularly in DHAA and TAA content (Figure 2).

Figure 2: Distribution of the content of vitamin C. Split violin plots of DHAA, AA and TAA content (mg g-1 of dry weight) in commercial and traditional lettuce varieties and some wild relatives using non-optimized and optimized protocols. Black lines show the mean values. Please click here to view a larger version of this figure.
{kind=link}
Furthermore, the use of a non-optimized protocol prevented us from extracting any useful conclusion from the results as they showed all samples, both types of lettuces and the wild relatives, having a similar vitamin C content. In contrast, the optimized protocol allowed us to detect statistically significant differences among them for DHAA and TAA content (Table 4), the richest ones being the wild species (Figure 2).
| Non optimized | Optimized | |||
| F-ratio | p-value | F-ratio | p-value | |
| DHAA | 0.460 | 0.637ns | 5.613 | 0.009** |
| AA | 0.070 | 0.932ns | 1.020 | 0.374ns |
| TAA | 0.015 | 0.985ns | 4.438 | 0.022* |
| ns, * and ** indicate non significant and significant at p<0.05 and 0.01, respectively. | ||||
Table 4: Variation in the content of vitamin C. F-ratios (quotients of two variances, the between-group variance and the within-group variance) and significance values from the one-way ANOVA considering the type of Lactuca (commercial lettuce varieties, traditional lettuce varieties, and crop wild relatives) for DHAA, AA and TAA content in non-optimized and optimized protocols.
Supplemental File 1: AA and TAA stability at 5 °C over 24 h. (A) AA and TAA peak areas throughout 24 h. (B) AA and TAA content (mg g-1 of dry weight) throughout 24 h. Bars represent the standard deviations of two technical replicates (n=2) kept in the autosampler at 5 °C and protected from exposure to light. Please click here to download this file.
Supplemental File 2: Main differences between the optimized and the non-optimized protocol for TAA, AA and DHAA extraction and quantification. The samples used were the same in both cases. Please click here to download this file.
Discussion
Vitamin C is a very important nutrient, but it is a very labile compound too, so its UPLC-UV quantification is dependent on multiple factors, such as sample storage and preparation, extraction method and chromatographic conditions. Therefore, a fast and simple procedure to prevent AA (with antioxidant power) oxidation to DHAA (without antioxidant properties) was needed. It was also crucial to avoid high pH and temperature conditions, as well as intense light and an oxidizing atmosphere during sample treatment to promote the stability of the compound.
To minimize AA oxidation, the following measures were taken. First of all, samples were lyophilized as a starting material for both protocols to ensure accurate quantification of vitamin C content and to easily manipulate samples. This option was preferred over fine grinding, commonly found throughout the literature19, as the dust thaws very quickly so the water becomes available again. During the extraction procedure, a higher volume of a more acidic solution (8% acetic acid and 1% MPA) was used as extractant in the optimized protocol (Supplemental File 2), which also acted as a stabilizer by preventing AA degradation. This solution also contained EDTA as a chelating agent to increase stabilization16, unlike the extractant in the non-optimized protocol (Supplemental File 2). Moreover, we tested if the extraction procedure could be enhanced by using two consecutive extractions with 2.5 mL of extractant instead of a single one with 5 mL and under a N2 atmosphere instead of the standard atmospheric conditions. The best results were reached using only one extraction under an unmodified atmosphere, which simplified the protocol by making unnecessary additional steps (data not shown). Other minor changes were also introduced in the protocol in order to enhance the extraction (i.e., sonication), obtain a clearer extract (finer filtration) and reduce the protocol duration (Supplemental File 2). Regarding the chromatographic conditions, the validation of the method was carried out as reported before18, guaranteeing good analytical parameters (Table 3). Besides, the use of ultrapure water with formic acid (pH 2.0) and methanol (98:2 v:v) with a 0.3 mL min-1 flow, instead of monopotassium phosphate 30 mM (pH 3.0) at 1 mL min-1 as the mobile phase (Supplemental File 2), resulted in an improved method. The most important advancement was likely using a UPLC system instead of an HPLC, which allowed us greater control of impacting conditions (like the temperature) and resulting in resolved AA peaks without interferences by unknown compounds, in a shorter time and consuming less volume of extract (Supplemental File 2).
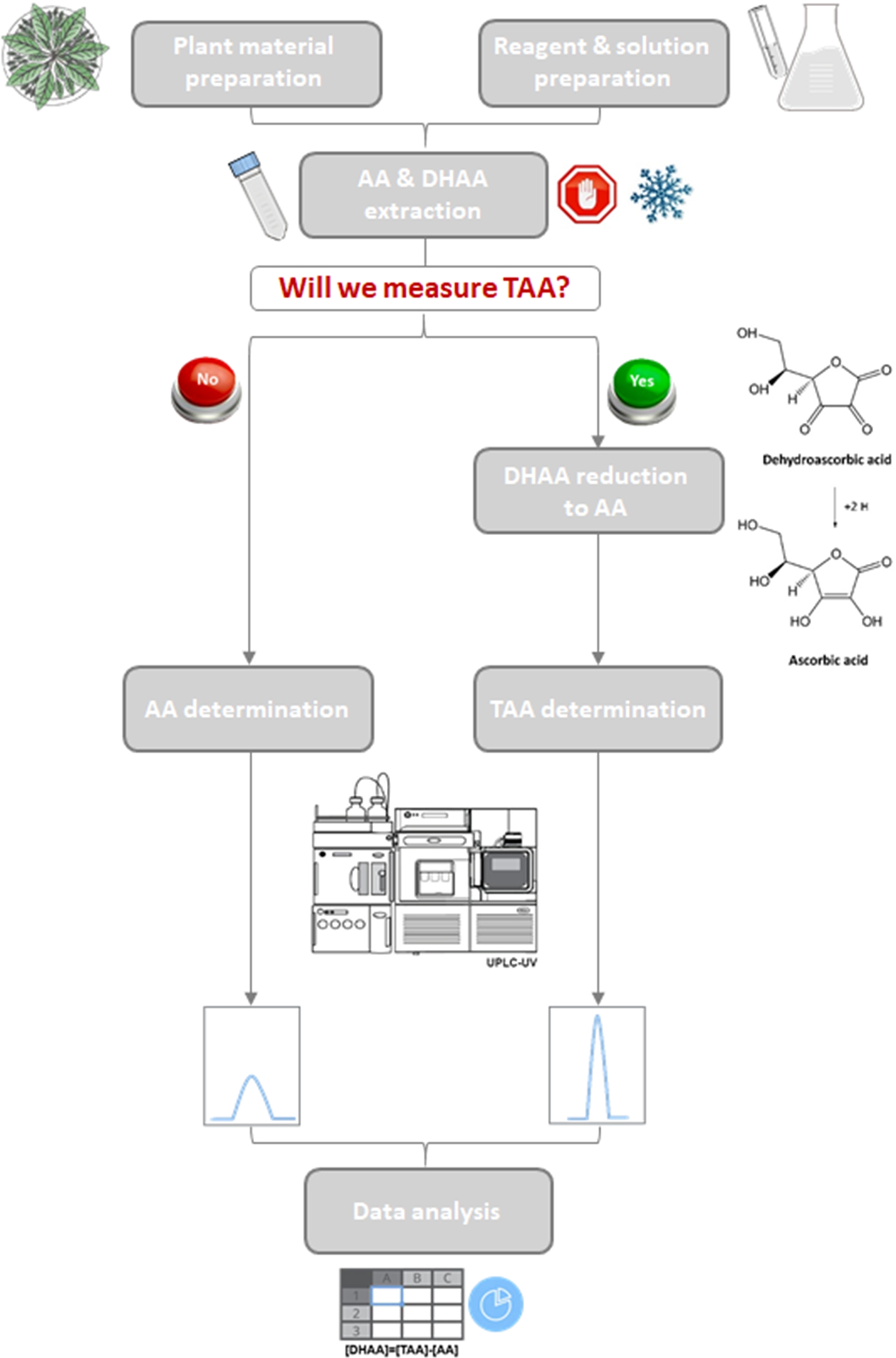
Nevertheless, there are two main limitations of this method. The first one is that DHAA cannot be measured directly using an UV detector due to its low absorptivity in the UV range of the spectrum. It is important to quantify the DHAA content because it presents certain biological activity and is easily convertible to AA in the human body5. For that, an additional reaction to reduce DHAA to AA is needed, together with a second chromatographic run in order to measure TAA and then determine DHAA indirectly by subtracting AA content from TAA (Figure 3). In this sense, the reduction step has been optimized by using a higher concentration of the reducing agent (DTT), increasing the reaction time from 5 to 30 min, and stopping the reaction with sulfuric acid (Supplemental File 2). The low stability of AA constitutes the second limitation of the method. As AA starts to degrade 4 h after extraction (Supplemental File 1), it is necessary to quantify it in this time interval. So, the number of samples to extract is conditioned by the chromatographic procedure. That is why we propose to freeze them at that step in this protocol, though in that case, not all of them could be placed in the UPLC autosampler to be measured automatically. Fortunately, the reduced RT for AA allowed us to obtain 3 min chromatograms, much shorter than the 7 min chromatograms obtained using HPLC (Supplemental File 2). Hence, vitamin C content could be determined in a high number of samples in a 4 h window.

Figure 3: Workflow of the quantification of vitamin C in lettuce and some wild relatives.
Schematic diagram of the optimized protocol showing two branches for the determination of only AA or AA + DHAA (TAA). Please click here to view a larger version of this figure.
{kind=link}
As vitamin C is an essential nutrient for humans and due to its important health benefits, it has become the object of many studies. Therefore, it has been quantified in a great variety of crops, including lettuce, one of the most consumed vegetables worldwide. Simple classical methods have been gradually replaced by liquid chromatography techniques because they are more specific and accurate10. However, due to the need of an additional reaction to quantify both, AA and DHAA via HPLC, in some studies on lettuce, only AA14 or only TAA11 (without quantifying AA before the reduction of DHAA into AA) have been measured. Furthermore, only a few authors have quantified AA and DHAA, despite the contribution of both molecules to vitamin C antioxidant activity2. Nevertheless, UPLC technique has become more important in recent times because of its higher performance when measuring vitamin C in several crops16. Comparing the results obtained in this study with the two methodologies, UPLC and HPLC, these advantages have been confirmed: well-defined AA peaks thanks to a higher sensitivity, and in very short times, have been achieved, which also implies fewer resources consumed. Despite of UPLC efficiency, only Chen et al.20 have applied this technique to measure the vitamin C content in lettuce, which still led to an underestimation as only the AA form was quantified.
In summary, this work represents the first successful attempt to determine the total vitamin C content not only in different lettuce varieties but also in some of their wild relatives. Vitamin C quantification is also essential to select lettuces with higher antioxidant activity within breeding programs. In this sense, the increased total vitamin C content in lettuce wild relatives found here and the increased AA content reported in previous studies14, as well as other antioxidant compounds21, broadens the suitable candidates to improve the nutritional value of lettuces.
In conclusion, even with some limitations inherent to vitamin C’s nature, like its gradual degradation few hours after being extracted or the need of a reduction reaction due to the low DHAA UV-absorptivity, it offers a less labor-intense and a less time-consuming method to measure vitamin C content. Additionally, it is also very robust and shows a high sensitivity and power of resolution. Moreover, it is easily transferable not only to other plant materials with slight or no changes, but also to processed products that supply the dietary intake of vitamin C to humans, which gives rise to a wide range of future applications in the emerging field of testing for reliable food quality.
Disclosures
The authors have nothing to disclose.
Acknowledgements
We gratefully acknowledge the Vegetable Germplasm Bank of Zaragoza (BGHZ-CITA, Spain) and the Centre for Genetic Resources (CNG, Wageningen, Netherlands) for supplying the seeds needed for this work. We thank J. A. Aranjuelo, A. Castellanos and “laboratorio de valoración nutritiva” from CITA for technical support and D. L. Goodchild for reviewing the English language. This work was funded by the projects RTA2017-00093-00-00 from the National Institute for Agricultural and Food Research and Technology (INIA) and LMP164_18 from the Government of Aragón; and by the Operational Programme FEDER Aragón 2014-2020 and the European Social Fund from the European Union [Grupos Consolidados A12-17R: “Grupo de investigación en fruticultura: caracterización, adaptación y mejora genéica” and A14-17R: “Sistemas agroganaderos alimentarios sostenibles” (SAGAS)]. I. M.L. was supported by a predoctoral contract for training doctors from the Spanish Ministry of Science, Innovation and Universities (MCIU) and the Spanish State Research Agency (AEI).
Materials
| Name | Company | Catalog Number | Comments |
| 1,4-Dithiothreitol (DTT) ≥98% (Ellman′s reagent) | Roche | 10197777001 | |
| 10 mm Φ stainless steel ball | Euro Aznar Supplies S.L. | 20112100 | |
| 15 mL polypropylene tube for centrifuge | Deltalab | 429946 | |
| 2 mL HPLC amber vial | Agilent | 5190-9063 | |
| 2 mL syringe with needle | Deltalab | JS2 | |
| 20 mL polypropylene tubes | Deltalab | 202840 | |
| 2-Amino-2-(hydroxymethyl)-1,3-propanediol (TRIS) ≥99.9% (titration) | Roche | 10708976001 | |
| 50 mL polypropylene tube | Deltalab | 429951 | |
| Acetic acid (CH3COOH) ≥99% purity glacial, ReagentPlus | Sigma-Aldrich | A6283 | |
| Acetonitrile, HPLC super gradient grade (99.9+% CH3CN-0.2 µm filtrated) | Chem-lab | CL00.0189 | |
| Acquity UPLC HSS T3 column (150 mm x 2.1 mm x 1.8 µm) | Waters | 186003540 | |
| Beaker 1 L, 200 mL | Deltalab | 191725, 191722 | |
| Dipotassium phosphate (KH2PO4) | Panreac | 766384 | Only in the non-optimized protocol (Supplemental File 2) |
| Ethylenediaminetetraacetic Acid Disodium salt (EDTA x 2H2O) 99-101% assay, ACS reagent | Panreac | 131669 | |
| Fisherbrand Analog MultiTube Vortexer | Thermo Fisher Scientific | 15549004 | |
| Formic acid 98-100% for HPLC LiChropur | Supelco | 5438040100 | |
| Freeze dryer VirTis genesis 25EL | VirTis | na | |
| Heidolph Multi Reax mixer | Heidolph | na | |
| Heidolph Reax top mixer | Heidolph | na | |
| Hewlett Packard HPLC 1050 equipped with an eλ Detector | Hewlett Packard | na | Only in the non-optimized protocol (Supplemental File 2) |
| Hydrochloric acid (HCl) 37% purity, ACS reagent | Sigma-Aldrich | 320331 | |
| L-Ascorbic acid ≥99%, ACS reagent | Sigma-Aldrich | 255564 | |
| Meta-phosphoric acid (MPA) ACS reagent, chips, 33.5-36.5% | Sigma-Aldrich | 239275 | |
| Methanol ≥99.9% (HPLC supergradient grade) | ChemLab | CL00.0189.2500 | |
| Micropipettes 10-1000 μL | Socorex | na | |
| Nucleosil 120 C18 Tracer column (250 mm x 4 mm x 5 µm) | Merck (Sigma-Aldrich) | 54919 | Only in the non-optimized protocol (Supplemental File 2) |
| PTFE-silicone cap with preaperture | Agilent | 5190-9067 | |
| Refrigerated centrifuge Gyrozen 1248R | Gyrozen | na | |
| Regenerated cellulose filters 0.22 µm (13 mm) | Agilent | 1015190-5108 | |
| Regenerated cellulose filters 0.45-µm (13 mm) | Labbox | SFCA-145-100 | Only in the non-optimized protocol (Supplemental File 2) |
| Spectrophotometer Heλios β | Thermo Scientific Corporation | na | |
| Sulfuric acid (H2SO4) 95.0-98.0% purity, ACS reagent | Sigma-Aldrich | 258105 | |
| Ultrapure water | WasserLab | na | |
| Ultrasons H-D | Selecta | na | |
| Volumetric flasks 1 L, 100 mL | DeltaLab | 191489, 191486 | |
| Waters Acquity UPLC H-Class equipped with a PDA eλ Detector | Waters | na |
References
- FAOSTAT. Statistics of the Food and Agriculture Organization of the United Nations. FAOSTAT. , Available from: http://www.fao.org/faostat/en/#data/QC (2018).
- Llorach, R., Martínez-Sánchez, A., Tomás-Barberán, F. A., Gil, M. I., Ferreres, F. Characterisation of polyphenols and antioxidant properties of five lettuce varieties and escarole. Food Chemistry. 108 (3), 1028-1038 (2008).
- Carr, A. C., Frei, B. Toward a new recommended dietary allowance for vitamin C based on antioxidant and health effects in humans. American Journal of Clinical Nutrition. 69 (6), 1086-1107 (1999).
- Carr, A. C., Vissers, M. C. M. Synthetic or food-derived vitamin C-Are they equally bioavailable. Nutrients. 5 (11), 4284-4304 (2013).
- Lee, S. K., Kader, A. A. Preharvest and postharvest factors influencing vitamin C content of horticultural crops. Postharvest Biology and Technology. 20 (3), 207-220 (2000).
- Shekhovtsova, T. N., Muginova, S. V., Luchinina, J. A., Galimova, A. Z. Enzymatic methods in food analysis: determination of ascorbic acid. Analytica Chimica Acta. 573-574, 125-132 (2006).
- Zhan, L., et al. Light exposure during storage preserving soluble sugar and L-ascorbic acid content of minimally processed romaine lettuce (Lactuca sativa L.var. longifolia). Food Chemistry. 136 (1), 273-278 (2013).
- Malejane, D. N., Tinyani, P., Soundy, P., Sultanbawa, Y., Sivakumar, D. Deficit irrigation improves phenolic content and antioxidant activity in leafy lettuce varieties. Food Science and Nutrition. 6 (2), 334-341 (2017).
- Tarrago-Trani, M. T., Phillips, K. M., Cotty, M. Matrix-specific method validation for quantitative analysis of vitamin C in diverse foods. Journal of Food Composition and Analysis. 26 (1-2), 12-25 (2012).
- Klimczak, I., Gliszczyńska-Świgło, A. Comparison of UPLC and HPLC methods for determination of vitamin C. Food Chemistry. 175, 100-105 (2015).
- Złotek, U., Świeca, M., Jakubczyk, A. Effect of abiotic elicitation on main health-promoting compounds, antioxidant activity and commercial quality of butter lettuce (Lactuca sativa L.). Food Chemistry. 148, 253-260 (2014).
- Koh, E., Charoenprasert, S., Mitchell, A. E. Effect of organic and conventional cropping systems on ascorbic acid, vitamin C, flavonoids, nitrate, and oxalate in 27 varieties of spinach (Spinacia oleracea L). Journal of Agricultural and Food Chemistry. 60 (12), 3144-3150 (2012).
- Kałuzewicz, A., et al. The influence of short-term storage on the content of flavonoids and vitamin C in Broccoli. European Journal of Horticultural Science. 77 (3), 137-143 (2012).
- van Treuren, R., van Eekelen, H. D. L. M., Wehrens, R., de Vos, R. C. H. Metabolite variation in the lettuce gene pool: towards healthier crop varieties and food. Metabolomics. 14 (11), 1-14 (2018).
- Swartz, M. E. UPLC: An introduction and review. Journal of Liquid Chromatography & Related Technologies. 28 (7), 1253-1263 (2005).
- Spínola, V., Mendes, B., Câmara, J. S., Castilho, P. C. An improved and fast UHPLC-PDA methodology for determination of L-ascorbic and dehydroascorbic acids in fruits and vegetables. Evaluation of degradation rate during storage. Analytical and Bioanalytical Chemistry. 403 (4), 1049-1058 (2012).
- Ball, G. F. M. Water-soluble vitamin assays in human nutrition. , Springer. Boston, MA. (1994).
- Bertolín, J. R., et al. Simultaneous determination of carotenoids, tocopherols, retinol and cholesterol in ovine lyophilised samples of milk, meat, and liver and in unprocessed/raw samples of fat. Food Chemistry. 257, 182-188 (2018).
- Spínola, V., Llorent-Martínez, E. J., Castilho, P. C. Determination of vitamin C in foods: Current state of method validation. Journal of Chromatography A. 1369, 2-17 (2014).
- Chen, Y., et al. radiation is beneficial for yield and quality of indoor cultivated lettuce. Frontiers in Plant Science. 10, 1-10 (2019).
- Damerum, A., et al. Elucidating the genetic basis of antioxidant status in lettuce (Lactuca sativa). Horticulture Research. 2 (15055), 1-13 (2015).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved