
Method Article
High-Throughput Cellular Profiling of Targeted Protein Degradation Compounds Using HiBiT CRISPR Cell Lines
In This Article
Summary
This protocol describes the quantitative luminescent detection of protein degradation kinetics in living cells that have been engineered using CRISPR/Cas9 to express antibody free endogenous protein detection tag fused to a target protein. Detailed instructions for calculating and obtaining quantitative degradation parameters, rate, Dmax, DC50, and Dmax50 are included.
Abstract
Targeted protein degradation compounds, including molecular glues or proteolysis targeting chimeras, are an exciting new therapeutic modality in small molecule drug discovery. This class of compounds induces protein degradation by bringing into proximity the target protein and the E3 ligase machinery proteins required to ubiquitinate and ultimately degrade the target protein through the ubiquitin-proteasomal pathway (UPP). Profiling of target protein degradation in a high-throughput fashion, however, remains highly challenging given the complexity of cellular pathways required to achieve degradation. Here we present a protocol and screening strategy based on the use of CRISPR/Cas9 endogenous tagging of target proteins with the 11 amino acid HiBiT tag which complements with high affinity to the LgBiT protein, to produce a luminescent protein. These CRISPR targeted cell lines with endogenous tags can be used to measure compound induced degradation in either real-time, kinetic live cell or endpoint lytic modes by monitoring luminescent signal using a luminescent plate-based reader. Here we outline the recommended screening protocols for the different formats, and also describe the calculation of key degradation parameters of rate, Dmax, DC50, Dmax50, as well as multiplexing with cell viability assays. These approaches enable rapid discovery and triaging of early stage compounds while maintaining endogenous expression and regulation of target proteins in relevant cellular backgrounds, allowing for efficient optimization of lead therapeutic compounds.
Introduction
Targeted protein degradation has emerged as one of the fastest growing areas in small molecule drug discovery, bolstered greatly by the therapeutic success of immunomodulatory molecular glue compounds (e.g., IMiD) for cancer treatment, and promising early clinical trial data of Proteolysis Targeting Chimera compounds1,2,3,4,5,6,7,8,9,10,11,12. Targeted protein degradation compounds function by bringing into proximity a target protein with E3 ligase machinery proteins1,2,3,4,5,6,7,8,9,10,11,12. This compound-induced recruitment of the target protein to the E3 ligase leads to the target protein ubiquitination and degradation via the ubiquitin proteasomal pathway (UPP)1,2,3,4,5,6,7,8,9,10,11,12. Historically, small molecule drug discovery screening programs have relied upon initial biochemical assays to assess activity and rank order compounds. This, however, has presented a significant challenge for targeted protein degraders whose ultimate activity, degradation via the proteasome, is dependent upon a cascade of cellular events1,2,4,5,6,11,12,13,14,15,16,17,18. The multiple pathways and complexity of protein complexes required for the successful target degradation necessitate cellular assay approaches for early screening and triaging of initial compounds. Currently, the availability of technologies to monitor target protein degradation in a high-throughput fashion in the context of the cellular environment is severely lacking14. Here we will present protocols for real-time kinetic live cell or endpoint lytic degradation activity assessment using CRISPR/Cas9 endogenously tagged HiBiT target cell lines18,19,20 to monitor the loss of the target protein via luminescent measurement after treatment with degrader compounds10,11,18,19.
To achieve successful degradation of therapeutic targets and to expand the druggable proteome, numerous approaches and types of degraders have emerged which can target a broad range of proteins for destruction, including those localized at or in the plasma membrane, lysosomes, mitochondrial membranes, cytoplasm, and the nucleus21–57. The two primary classes of compounds most extensively studied are molecular glues and protein targeting cimeras2,4,5,6,7,12,26. Molecular glues are monovalent, thus typically smaller in size, and facilitate a novel protein:protein interaction interface with a target protein upon binding to an E3 ligase component2,12,26. They are most commonly degraders that bind to the Cereblon (CRBN) E3 ligase component2,12,26,55,56,57. Recently though exciting new examples utilizing other E3 ligase machinery such as DCAF1558,59,60 and CDK/Cyclin recruitment to DDB145 show the potential for expansion of this class of compounds. In contrast, PROTACs are larger, bivalent molecules, consisting of a target binding ligand, most often an inhibitor, bridged via a chemical linker to a E3 ligase handle1,3,4,5,7,13. As such, these compounds are capable of direct binding to both the E3 ligase and the target protein1,3,4,5,7,13. Numerous proteins have been shown to be degraded via these bivalent molecules, and the most used E3 ligase handles recruit either CRBN or Von Hippel Lindau (VHL)1,3,4,5,7,13. However, the number of available handles for E3 ligase recruitment in chimeras targeting proteolysis design is rapidly growing, expanding the capabilities of this class of compounds with the potential to degrade diverse target classes as well as enhance cell- or tissue-type specificity24,48,61,62. Combined with the minimal requirement to engage a target protein, even with marginal affinity, degradation compounds hold promise for expanding the druggable proteome.
Characterizing the cellular dynamics of protein loss, as well as potential protein recovery post-treatment, is critical for understanding degradation compound function and efficacy. While it is possible to study endogenous protein level changes in relevant cellular systems with western blot antibody assays or mass spectrometry, these approaches are difficult to adapt to high-throughput screening formats, have limited quantification capability, or ability to measure kinetic changes at many timepoints14. To address these challenges, we have developed a plate-based cellular luminescent system for monitoring changes in endogenous protein levels, which utilizes genomic insertion via CRISPR/Cas9 of the 11 amino acid tag, HiBiT, to the loci of any key degradation targets18,19,20. This peptide complements with high affinity to its binding partner, LgBiT, to produce bright luminescence in the presence of its substrate18,19,20,63, thereby making these tagged endogenous proteins luminescent in cells or lysates18,19,20,63. The relative light units (RLUs) measured with a luminometer instrument are directly proportional to the tagged target protein levels18,19,20,63. With the development of stabilized luciferase substrates, real-time kinetic protein level measurements over 24-48 h time frames are possible18,53,64. This allows for the determination of a complete degradation profile for any given target at any given compound concentration, including quantitative analysis of initial degradation rate, degradation maximum (Dmax), and recovery after compound treatment18,53. If screening large libraries of degradation compounds, however, endpoint analysis can also readily be performed in 384-well format at various drug concentrations and designated times.
The protocols presented in this manuscript represent cellular screening strategies for targeted protein degradation compounds, applicable for all types of degraders. The use of HiBiT CRISPR cell lines along with these protocols, however, are not limited to protein degradation, rather they are general tools for monitoring any endogenous target protein level which could be modulated post-treatment to study impact of compounds or even resistance mechanisms20,65,66. A pre-requisite for these luminescent-based detection methods is a CRISPR endogenously tagged HiBiT target cell line, which is critical as it enables sensitive luminescent detection, while still maintaining endogenous target expression and native promoter regulation18,19,20. Significant advances have been made in utilizing CRISRP/Cas9 for insertion of genomic tags, particularly in scalability20 and with the high sensitivity of detection, in various formats including CRISPR pools or clones with either heterozygous or homozygous allelic insertions18,19,20. Use of exogenous expression of HiBiT or other reporter fusions in cells in lieu of endogenous tagging is possible, but significant caution should be taken using systems with protein overexpression14,18. These can lead to artefacts in understanding true compound potency and protein recovery dynamics14,18, including potential transcriptional feedback loops activated post target degradation. In addition, early stage compounds with low potency could be missed, and present themselves as false negatives in screening. As protein loss could result from compound-induced toxicity and cell death, the protocols described here contain highly recommended, but optional cell viability luminescent or fluorescent assays paired with the degradation protocol. There are two major sections to the protocol, lytic endpoint, and live cell kinetic screening. Within each of those sections, options are included for multiplexed cell viability measurements in endpoint or kinetic formats. Monitoring the changes of the tagged endogenous protein requires complementation with LgBiT in cells. Therefore, the kinetic screening section references important protocols for the introduction of this, which can be achieved via transient or stable expression and is essential for performing the live cell luminescent measurements. All approaches presented here allow rapid rank ordering and activity assessment of compounds, enabling early stage compound screening efforts and more rapid identification of lead degraders.
This protocol is designed for the study of degradation compounds in conjunction with a HiBiT CRISPR cell line. Protocols for generation of HiBiT CRISPR insertions for numerous targets have been outlined in several recent publications18,19,20.
Protocol
1. Endpoint degradation studies with HiBiT CRISPR target proteins in lytic format with optional cell viability fluorescence analysis
- Preparation and plating of mammalian adherent or suspension cell line
- Adjust the cell density to 2.22 x 105/mL by dilution in appropriate cell media used for passaging and cell growth.
- Dispense cells into plates with a minimum of 3 wells per experimental and control condition. Dispense 90 µL (20,000 cells) per well of cell suspension into 96-well white plates. For 384-well format, dispense 36 µL (8,000 cells) per well of cell suspension into 384-well white plates.
- Preparation and addition of compounds
- Prepare serially diluted PROTAC or degrader test compound plates at 1,000x final concentration in 100% DMSO. Then dilute it to 10x final concentration in the cell culture medium. Add an equal volume of DMSO to the medium, to be used as a no compound DMSO control.
- For 96-well format add 10 µL of 10x compound and control solutions to 90 µL of cells. For 384-well format add 4 µL of 10x compound and control solutions to 36 µL of cells.
- Incubate the plates in an incubator at 37 °C and 5% CO2 for the desired amount of time or in conditions that are optimal for their growth.
NOTE: As this is an endpoint assay, testing of multiple time points will require preparation of separate degradation plates for each time point, as described in above step 1.1.2. Incubation times to detect compound-mediated degradation are highly variable and are also likely dependent on compound concentration. Suggested initial time points would be 6 h and 24 h. - If measuring endpoint luminescent detection without the optional cell viability measurement, proceed directly to step 1.3 below. If performing multiplexing with cell viability measurement, proceed to the next section 1.4 below.
- Lytic measurement of cells
- Immediately prior to HiBiT lytic measurements, prepare 2x lytic detection reagent by adding 20 µL of lytic substrate and 10 µL of LgBiT protein per every 1 mL of the lytic buffer. Prepare enough 2x detection reagent for number of wells to be assayed, including extra volume to account for pipetting error (i.e., number of wells + 10%).
- Add prepared lytic detection reagent to cells. For 96-well format, add 100 µL of 2x lytic detection reagent to each well containing 100 µL of cells. For 384-well format, add 40 µL of 2x lytic detection reagent to each well containing 40 µL of cells. Mix the plate on a microplate vortex mixer for 10-20 min at 350 rpm.
- Measure luminescence on a luminometer capable of reading luminescence in a 96- or 384-well plate.
- Optional cell viability multiplexing
NOTE: This step is performed using a commercially available CellTiter-Fluor (CTF) kit (see Table of Materials).- 30-40 min prior to the desired endpoint measurement, prepare a 6x cell viability detection reagent solution by adding 10 µL of the substrate to 2 mL of the assay Buffer. Prepare enough 6x reagent for each well to be assayed, including extra volume for pipetting error (i.e., number of wells + 10%).
- Add the prepared reagent to wells. For 96-well format add 20 µL of 6x reagent to each well already containing a 100 µL volume. For 384-well format add 8 µL of 6x reagent to each well containing 40 µL of cells. Mix briefly on a microplate vortex mixer, then incubate the plate for 30 min in a 37 °C incubator.
- At the desired endpoint of measurement (i.e., 6 or 24 h post treatment, step 1.2.3), measure fluorescence on an instrument capable of reading fluorescence (380-400nmEx/505nmEm) in 96- or 384- well format.
- Prepare 2x lytic detection reagent by adding 20 µL of lytic substrate and 10 µL of LgBiT protein per 1 mL of lytic buffer. Prepare enough 2x detection reagent for number of wells to be assayed, including extra volume to account for pipetting error (e.g., number of wells + 10%).
- Add the prepared lytic detection reagent to wells. For 96-well format add 120 µL of 2x lytic detection reagent to each well already containing a 120 µL volume. For 384-well format add 48 µL of 2x lytic detection reagent to each well already containing a 48 µL volume. Mix the plate on a microplate vortex mixer for 10-20 min.
- Measure luminescence on a luminometer capable of reading luminescence in 96- or 384- well plates.
- Quantification of degradation and cell viability
- Average the relative light units (RLU) from the DMSO control at the measured time point. Use this value as the baseline protein level of the target to calculate fractional degradation by normalizing all other treatments tested at this same time point to this value. For example, if the average RLU for the DMSO control wells at 6 h was 10,000, and the RLU for a given compound treatment at 6 h was 5,000, the fractional degradation would be calculated as 5,000 ÷ 10,000 = 0.5 (Equation 1).
Equation 1:
- Determine the percentage degradation from Fractional RLU:
Equation 2:
- Plot fractional RLU or % degradation at specific time points to rank the activity of compounds.
- Optionally, analyze relative fluoresecence unit (RFU) data for the cell viability assay measurement by comparing the values from all treatments to the DMSO control. If a significant decline in RFU is observed for any treatment relative to the DMSO control, the degradation data can be additionally normalized to the cell viability assay data to determine changes in protein level relative to losses in cell viability.
- Average the relative light units (RLU) from the DMSO control at the measured time point. Use this value as the baseline protein level of the target to calculate fractional degradation by normalizing all other treatments tested at this same time point to this value. For example, if the average RLU for the DMSO control wells at 6 h was 10,000, and the RLU for a given compound treatment at 6 h was 5,000, the fractional degradation would be calculated as 5,000 ÷ 10,000 = 0.5 (Equation 1).
2. Real-time kinetic degradation of HiBiT CRISPR target proteins and optional cell viability luminescence assay
NOTE: The ability to perform kinetic screening and degradation requires LgBiT protein co-expression in the cell, which has been described previously18,19,63. This can be achieved via transient transfection of a LgBiT vector, use of BacMam LgBiT, or by performing HiBiT CRISPR insertion into a LgBiT stable cell line.
- Plating of adherent cell lines.
- Remove medium from cell flask by aspiration, wash cells with DPBS, dissociate cells with 0.05% trypsin-EDTA, and allow cells to dissociate from the flask bottom. For suspension cell lines, proceed to section 2.2.
- Neutralize trypsin using serum-containing cell culture medium, mix to collect and resuspend cells, and transfer cell suspension to a conical tube.
- Spin down cells at 125 x g for 5 min. Discard the cell culture medium and resuspend in an equal volume of fresh cell culture medium.
- Plate cells into assay plates with a minimum of triplicate wells per experimental and control condition. For 96-well format count to estimate cell density, adjust density to 2 x 105 cells/mL in assay medium and dispense 100 µL (20,000 cells) per well in a 96-well plate. For 384-well format count to estimate cell density, adjust density to 4.44 x 105 cells/mL in the assay medium and dispense 18 µL (8,000 cells) per well.
- Incubate plates at 37 °C, 5% CO2 overnight or in conditions which are optimal for their growth.
- Plating of suspension cells
- Adjust cell density to 2.22 x 105 cells/mL in CO2-independent medium supplemented with 10% FBS and 1x Endurazine (1:100 dilution of the stock reagent).
- Plate cells into assay plates with a minimum of 3 wells per experimental and control condition. For 96-well format dispense 90 µL (20,000 cells) per well. For 384-well format dispense 36 µL (8,000 cells) per well.
NOTE: For suspension cell lines which have low signal to background (S:B) luminescence, e.g., when working with CRISPR pools rather than clones, it is possible to increase the luminescence by increasing the number of cells plated, up to 100,000 cells/well in 96-well format, or 40,000 cells/well in 384-well format.
- Kinetic degradation assays using HiBiT CRISPR cells expressing LgBiT
- For suspension cells already containing Endurazine, which was included at the plating step in 2.2., proceed directly to step 2.3.3. For adherent cell lines prepare Nano-Glo Endurazine solution. For 96-well format, prepare a 1x solution of Endurazine by diluting stock reagent 1:100 into CO2-independent medium supplemented with 10% FBS. For 384-well format, prepare a 2x solution of Endurazine by diluting stock reagent 1:50 into CO2-independent medium supplemented with 10% FBS.
- Add Endurazine solution to each well of adherent cells. For 96-well format aspirate medium and add 90 µL of 1x Endurazine solution. For 384-well format, add 18 µL of 2x Endurazine solution to 18 µL of cells. Do not aspirate medium as the degradation assay is performed in a 50:50 mixture of culture medium and CO2 independent medium in 384-well format.
- Incubate suspension or adherent cell plates containing Endurazine for 2.5 h in an incubator at 37 °C and 5% CO2 to allow luminescence to equilibrate.
- Prepare a 10x concentration of test PROTAC titration in CO2-independent medium and add 10 µL to each well of 96-well plate or 4 µL for 384-well plate. For compounds with unknown efficacy, a final concentration of 1-10 µM at the highest point is recommended as a starting point.
- Collect kinetic measurements of luminescence in luminometer pre-equilibrated to 37 °C for a period between 0-48 h. The time increments of measurement can be customized for each experiment, but a recommended initial experiment would be luminescence measurements every 5-15 min for 24 h or the desired amount of time.
- Optional cell viability same-well multiplex analysis after final kinetic measurement
NOTE: This assay is performed with a commercially available CellTiter-Glo (CTG) kit (see Table of Materials).- Equilibrate CTG reagent to room temperature.
- Following degradation measurement at the last time point of the kinetic analysis, add 100 µL (96-well plate) or 40 µL (384-well plate) of the reagent per well of the plate, and mix on a plate shaker at 500-700 rpm (96-well plate) or a microplate vortex mixer (384-well plate) for 5 min.
- Incubate the plate at room temperature for 30 min to allow for cell lysis and quenching of HiBiT signal.
- Measure total luminescence on a luminometer by following manufacturer’s recommendation.
- Quantification of kinetic degradation profiles
- Using the kinetic luminescence measurements collected, normalize the raw RLUs for each PROTAC concentration to the replicate averaged DMSO condition at every time point to account for changes in free furimazine concentration over time. Calculate Fractional RLU using Equation 1.
Equation 1: - From the degradation curves, fit a single-component exponential decay model using Equation 2 to the initial degradation portion of each curve to the point where the data reaches a plateau.
NOTE: It may be helpful to exclude from the fit the first few data points as there may be a brief lag before degradation is observed.
Equation 2:
- From Equation 2, determine the parameter ƛ, which represents the degradation rate constant and the Plateau, which represents the lowest amount of protein remaining.
- Calculate Dmax, which is the maximum fractional amount of degraded protein and is calculated as 1-Plateau.
- Plot Dmax for each concentration of PROTAC to determine a time-independent degradation potency curve.
- Determine the Dmax50 value for the plot in 2.3.5 to analyze efficacy of compounds.
NOTE: To determine a DC50 at a specific time point, plot the calculated percent degradation for each concentration at the chosen time. This can be specified as DC50 t=4 h or DC50 t=12 h.
- Using the kinetic luminescence measurements collected, normalize the raw RLUs for each PROTAC concentration to the replicate averaged DMSO condition at every time point to account for changes in free furimazine concentration over time. Calculate Fractional RLU using Equation 1.
Results
To demonstrate single concentration endpoint lytic degradation analysis, several CDK target proteins; CDK2, CDK4, CDK7, and CDK10 were endogenously tagged with HiBiT at their C-terminus in HEK293 cells and treated with a 1 µM concentration of the pan-kinase Cereblon-based PROTAC, TL12-18654 (Figure 1A). The level of CDK protein was measured at different time points and the fractional RLU relative to the DMSO control was determined (Figure 1A). Each CDK protein showed different extents of degradation in response to the compound treatment and the various time points (Figure 1A). To understand how CDK proteins compared to each other directly in terms of protein loss, the fractional RLUs in Figure 1A were calculated as total % degradation and plotted for each time point in Figure 1B. This shows that even at early time points, 2 or 4 h, some of the CDK family members show high levels of degradation which continue to trend upwards over time (Figure 1B).
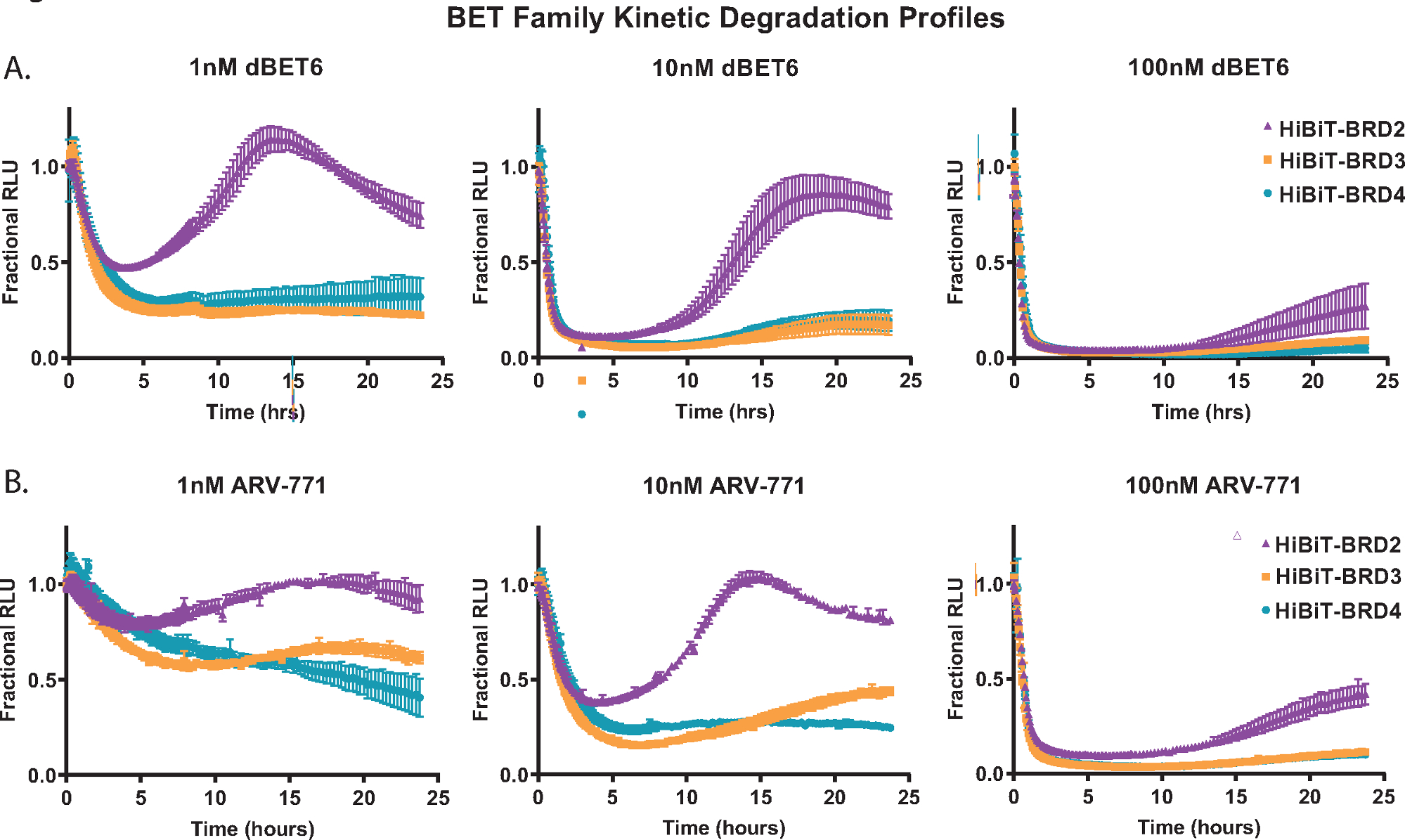
To demonstrate kinetic degradation analysis, each of the BET family member proteins; BRD2, BRD3, and BRD4 were endogenously tagged with HiBiT at their N-terminus in HEK293 cells stably expressing the LgBiT protein18. These were then treated with three different concentrations of the pan-BET PROTACs; the Cereblon-based dBET650 (Figure 2A) and the VHL-based ARV-77141 (Figure 2B). Kinetic measurements were collected over a 24 h period, and from the profiles at each concentration, the differences in BET family member response are readily apparent. The ability of BRD2 to initiate a more rapid recovery response post-degradation compound treatment (Figure 2A,B) has been previously observed with other pan-BET PROTACs and is likely due to a transcriptional feedback response competitive to the degradation process18.
Both endpoint and kinetic analysis can be done with full compound dose response treatments. Shown in Figure 3 are kinetic dose response degradation profiles of treatment of Ikaros/IKZF1-HiBiT CRISPR Jurkat cells stably expressing LgBiT protein with four different molecular glue compounds2,26,55,57; lenalidomide (Figure 3A), iberdomide (CC-220) (Figure 3B), thalidomide (Figure 3C), and pomalidomide (Figure 3D). These degraders show significant differences in degradation response amongst the compounds as well as across the concentration series (Figure 3).
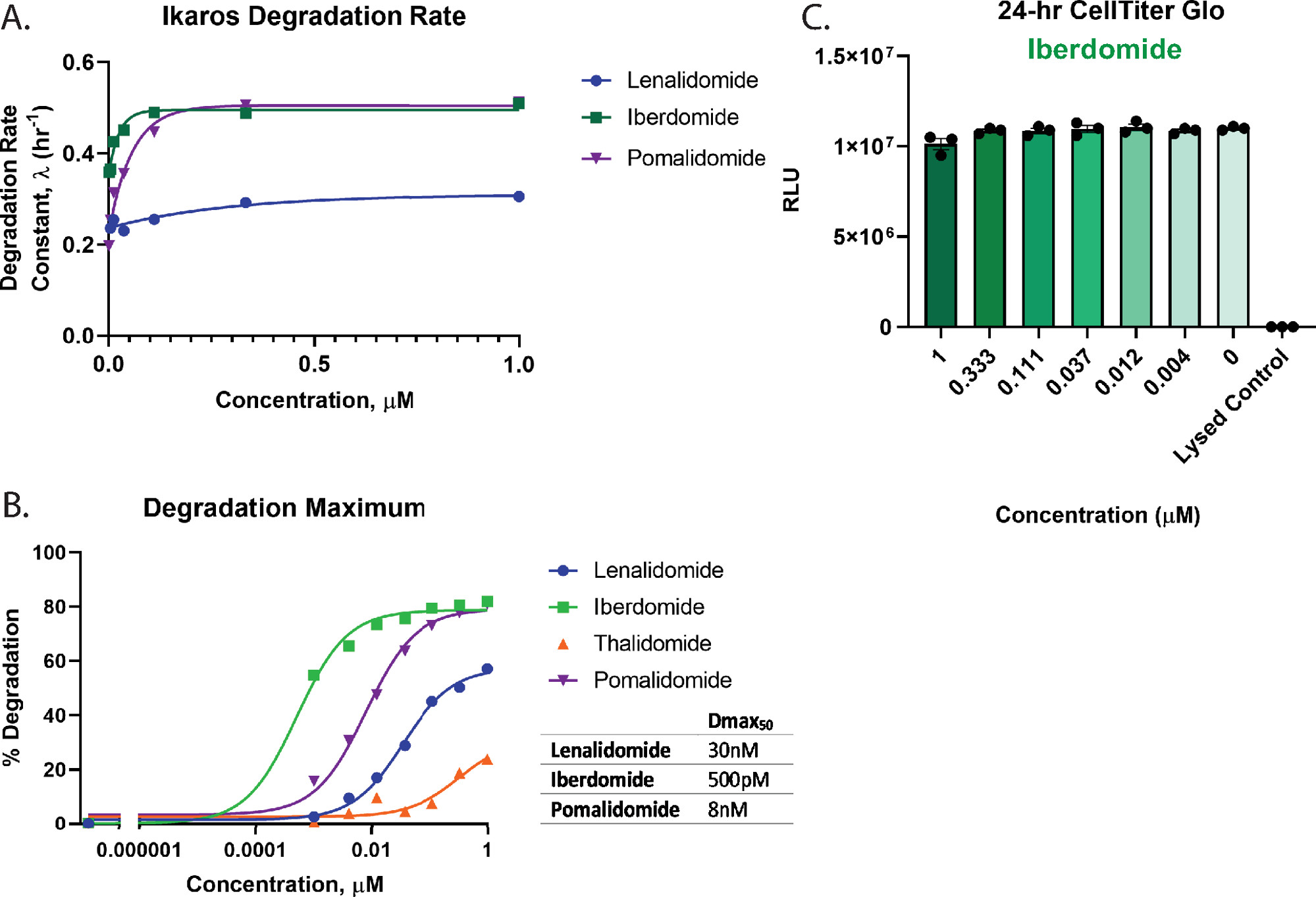
To quantitatively assess degradation and rank order the compounds in Figure 3 the dose response profiles were used to calculate key degradation parameters including the degradation rate (Figure 4A), Dmax (Figure 4B), and Dmax50 values (Figure 4B). These analyses show that iberdomide (CC-220) and pomalidomide have very similar rapid initial degradation rates (Figure 4A), yet iberdomide (CC-220) has the highest potency as has been previously seen in orthogonal studies55,57 (Figure 4B). Since Iberdomide exhibits such high potency, and all concentrations tested show greater than 50% degradation, the Dmax50 value obtained for Iberdomide represents an estimate based on the limitation in accurately fitting the data. From the graphs in Figure 3C,D and Figure 4B, neither lenalidomide nor thalidomide degrade the Ikaros/IKZF1 target to completion at the highest concentrations tested. Due to very little degradation observed with thalidomide, the degradation traces could not be accurately fit to an exponential decay model, therefore, degradation rate was not quantified for this treatment. For the most potent degrader, iberdomide (CC-220)55,57 (Figure 4B). Cell viability multiplex assays showed no loss in cell viability for the concentrations tested (Figure 4C).

Figure 1: CDK endpoint degradation and toxicity with pan-kinase PROTAC, TL12-18654. (A) Select panel of endogenous CDK target proteins fused with HiBiT on the C-terminus via CRISPR/Cas9 and assessed for degradation with 1 µM TL12-186 PROTAC54 at 2 h, 4 h, 8 h, and 24 h treatment. Values are represented as Fractional RLU relative to a DMSO control measured at each time point. Error bars represent SD of the mean of 3 technical replicates. (B) Percent degradation of panel of CDK target proteins calculated from (A) representing the amount of degradation of each family member observed at 2, 4, 8, and 24 h time points. Error bars represent SD of the mean of 3 technical replicates. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Profiling kinetic degradation selectivity of BET family members with BET degraders, dBET650 and ARV-77141. Kinetic degradation profiles of endogenous BET family members, BRD2, BRD3, and BRD4, tagged with HiBiT on the N-terminus via CRISPR/Cas9 with treatment of single concentrations of 1 nM (left), 10 nM (middle), or 100 nM (right) dBET650 (A) or ARV-77141 (B) PROTACs. Values are represented as Fractional RLU calculated from a DMSO control at each kinetic time point. Error bars represent SD of the mean of 4 technical replicates. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Live cell kinetic degradation dose response profiles of Ikaros/IKZF1-HiBiT with a molecular glue panel2,26,55,57. Jurkat cells stably expressing the LgBiT protein were engineered using CRISPR/Cas9 to tag the C-terminus of Ikaros/IKZF1 with the HiBiT peptide. Cells were treated with an 8 point dose response concentration series including DMSO of four different molecular glue compounds2,26,55,57: (A) lenalidomide, (B) iberdomide (CC-220), (C) thalidomide, or (D) pomalidomide. Luminescence was measured every 5 min for a total of 19.5 h. Relative light unit (RLU) data from (A-D) were converted to fractional RLU as described in Step 2.4.1 and graphed as a function of time. Error bars represent SD of 3 technical replicates. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Calculation of degradation rate and Dmax50 for Ikaros/IKZF1-HiBiT, and multiplexing cell health assays. Kinetic degradation data from Figure 3 were used to calculate quantitative degradation parameters. (A) Degradation rates and (B) degradation maximum values (Dmax) are graphed at each drug concentration for the indicated molecular glue compounds2,26,55,57. (B) Dmax50 values for each compound were calculated using a dose-response model with constrained Hill slope of 1, which can be used to rank order degradation compounds for a target. (C) Cell viability assays with the iberdomide (CC-220)55,57 degradation dose response from Figure 3B was performed as an endpoint measurement upon completion of the kinetic degradation measurements. Error bars represent SD of 3 technical replicates. Please click here to view a larger version of this figure.
{kind=link}
Discussion
We present here two methods of screening degradation compound activity in either endpoint lytic format or live-cell kinetic mode. These approaches are based upon the same luminescent measurement principles yet provide different levels of detail and understanding. The choice of either approach will likely be dependent upon screening goals and size of compound library. For large compound screening decks or primary screens to observe any detectable degradation, endpoint lytic screening offers sensitive and efficient high-throughput compatibility where other endpoint approaches, such as western blot or mass spectrometry can be either impractical or difficult to adapt14. A starting point for these screens could be performed with a limited number of concentrations and timepoints. Recommended initial concentrations to test are in the range of 100 nM-10 µM, to account for initial degraders having low potency, poor permeability, or in some cases with highly potent compounds, the presence of a hook effect. It is further recommended that a minimum of two different time points should be tested to establish early onset degradation at 4-6 h and latent or sustained degradation at 18-24 h. Compounds that exhibit high degradation potency and on-target mechanism are readily observed within a 4-6 h timeframe, whereas degradation or apparent protein loss observed only at later time points could be due to a variety of mechanisms. It is highly recommended to monitor cell viability at both early and late time points, such that protein loss can be uncoupled from loss due to cell death. Similar to any type of luminescent or fluorescent assay, there is a potential for compounds within libraries to interfere or inhibit signal, therefore, orthogonal follow-up experiments with lead compounds using unrelated fusions or alternative approaches to monitoring protein level will be important for assessing that loss of RLU in these assays is associated directly with target protein degradation.
The ability to screen in live cell kinetic format over extended periods of time relies heavily on the assay signal to background (S:B). Factors that contribute to S:B include the expression level of the target protein itself, which can span several orders of magnitude, the efficiency of LgBiT expression in the cell line chosen for peptide insertion, and the availability of the tagged target for complementation in its various native complexes. We have established a general cutoff requirement consisting of a S:B of 15 to successfully measure degradation in kinetic mode with either Endurazine or Vivazine. The S:B is determined by measuring the baseline signal of the HiBiT-edited cells co-expressing LgBiT relative to unedited parental cells expressing LgBiT alone in the presence of either Endurazine or Vivazine live cell substrates. Vivazine will produce a higher luminescent signal but will decay faster than Endurazine and may limit signal acquisition to 24 hours or less. Furthermore, S:B can also be highly dependent on whether CRISPR pools or clones are used. For targets in cell lines which are more amenable and have high efficiency for CRISPR/Cas9 engineering, a heterogeneous CRISPR pool population of edited cells may have sufficient S:B for kinetic analysis. For targets in more difficult cell lines where less efficient genomic integration via CRISPR results in pools with low S:B, isolating CRISPR clones might be necessary to enrich edited populations and achieve a sufficiently high S:B for kinetic analysis. For any of these scenarios, if S:B is less than 15 with either Endurazine or Vivazine substrates, endpoint lytic screening is advised.
For better understanding and characterization of compounds, including determination of a degradation profile with quantitative parameters, real-time kinetic analysis in live cells is the recommended screening approach14,18. Like endpoint analysis discussed above, initial kinetic screening can be done with a limited number of concentrations in the range of 100nM-10µM in high-throughput fashion. In 384-well format, over 100 compounds can readily be screened in triplicate at one concentration on a single plate. The resulting degradation profiles will provide guidance not only on the extent of degradation observed, but the rate of degradation, the duration of degradation, and potential recovery of the protein14,18 (Figure 2 and Figure 3). The shapes of the degradation profile also yield valuable information. Specific and potent degraders often show an initial rapid loss of the target protein to a plateau in a matter of hours18,53, whereas other mechanisms such as transcriptional feedback or compound toxicity typically result in more linear loss of the protein over time. These details and nuances are missed with endpoint lytic analysis, and with real-time analysis over 24-48 hours, one does not have to predict the time to capture the true Dmax within sets of new or unknown compounds.
Real-time kinetics also allow for efficient dose response screening to better understand compound efficacy, how compound concentration impacts initial degradation rate, and offers possibilities to rank compounds based on more than one parameter. Classical measurements of degradation potency involve DC50 calculations at a specific point in time based on apparent degradation maxima. In contrast, our kinetic approach to evaluate potency incorporates the true degradation maximum at each concentration regardless of when it occurs in time18. We call this measurement of kinetic degradation potency, the Dmax5018. Analysis in this fashion accounts for compounds that may initiate degradation more slowly at lower concentrations and therefore take a longer time post-treatment to reach their Dmax. It can be especially informative to rank compounds on both degradation rate and Dmax. For the most potent degraders this will further differentiate slow, but potent degraders from those which are both fast and potent. Together, both lytic and live cell kinetic screening utilizing HiBiT CRISPR cell lines are powerful approaches which yield a more comprehensive picture of targeted protein degradation, compound function, and enable the screening process from initial activity assessment to downstream chemical optimization through enhancement of key degradation parameters.
Disclosures
Promega Corporation is the commercial owner by assignment of patents of the HiBiT and NanoLuc technologies and applications.
Acknowledgements
K.M.R, S.D.M, M.U. and D.L.D are all employees of Promega Corporation
Materials
| Name | Company | Catalog Number | Comments |
| CellTiter-Glo 2.0 reagent | Promega | G9241 | Cell Viability luminescent assay |
| CellTiter-Fluor Cell Viability Assay | Promega | G6080 | Cell Viability fluorescent assay |
| CO2-independent medium | ThermoFisher | 18045-088 | Cell culture |
| DMSO | Sigma Aldrich | D2650 | For compound dilution and control |
| DPBS | Gibco | 14190 | Cell culture |
| Fetal Bovine Serum | Seradigm | 89510-194 | Cell culture |
| HEK293 LgBiT stable cell line | Promega | N2672 | For complementation with HiBiT to generate luminescence |
| HiBiT CRISPR mammalian cell line | Promega | https://www.promega.com/crispr-tpd | |
| Hygromycin B solution | Gibco | 10-687-010 | Cell culture |
| LgBiT BacMam | Promega | CS1956C01 | For complementation with HiBiT to generate luminescence |
| LgBiT Expression Vector | Promega | N2681 | For complementation with HiBiT to generate luminescence |
| Luminometer Plate Reader | Luminomenter capable of measuring luminescence and fluorescence (e.g. GloMax Discover System, Promega GM3000) | ||
| NanoGlo Endurazine live cell substrate | Promega | N2570 | Kinetic HiBiT reagent |
| NanoGlo Vivazine live cell substrate | Promega | N2580 | Kinetic HiBiT reagent |
| NanoGlo HiBiT Lytic Detection system | Promega | N3030 | Enpoint lytic HiBiT reagent |
| Opti-MEM Reduced Serum Medium, no phenol red (ThermoFisher) | ThermoFisher | 11058-021 | Cell culture |
| Tissue culture plates, white, 96 well plate | Costar | 3917 | Cell culture |
| Tissue culture plates, white, 384 well plate | Corning | 3570 | Cell culture |
| Trypsin/EDTA | Gibco | 25300 | Cell culture |
References
- Burslem, G. M., Crews, C. M. Proteolysis-targeting chimeras as therapeutics and tools for biological discovery. Cell. 181 (1), 102-114 (2020).
- Chamberlain, P. P., Hamann, L. G. Development of targeted protein degradation therapeutics. Nature Chemical Biology. 15 (10), 937-944 (2019).
- Churcher, I. Protac-induced protein degradation in drug discovery: Breaking the rules or just making new ones. Journal of Medicinal Chemistry. 61 (2), 444-452 (2018).
- Ciulli, A., Farnaby, W. Protein degradation for drug discovery. Drug Discovery Today: Technologies. 31, 1-3 (2019).
- Crews, C. M. Inducing protein degradation as a therapeutic strategy. Journal of Medicinal Chemistry. 61 (2), 403-404 (2018).
- Cromm, P. M., Crews, C. M. Targeted protein degradation: from chemical biology to Drug Discovery. Cell Chemical Biology. 24 (9), 1181-1190 (2017).
- Deshaies, R. J. Protein degradation: Prime time for PROTACs. Nature Chemical Biology. 11 (9), 634-635 (2015).
- Lai, A. C., Crews, C. M. Induced protein degradation: an emerging drug discovery paradigm. Nature Reviews Drug Discovery. 16 (2), 101-114 (2017).
- Ottis, P., Crews, C. M. Proteolysis-targeting chimeras: Induced protein degradation as a therapeutic strategy. ACS Chemical Biology. 12 (4), 892-898 (2017).
- Wu, T., et al. Targeted protein degradation as a powerful research tool in basic biology and drug target discovery. Nature Structural and Molecular Biology. 27, 605-614 (2020).
- Hanan, E. J., et al. Monomeric targeted protein degraders. Journal of Medicinal Chemistry. , (2020).
- Collins, I., Wang, H., Caldwell, J. J., Chopra, R. Chemical approaches to targeted protein degradation through modulation of the ubiquitin-proteasome pathway. Biochemical Journal. 474 (7), 1127-1147 (2017).
- Carmony, K. C., Kim, K. B. PROTAC-induced proteolytic targeting. Methods in Molecular Biology. 832, 627-638 (2012).
- Daniels, D. L., Riching, K. M., Urh, M. Monitoring and deciphering protein degradation pathways inside cells. Drug Discovery Today: Technologies. 31, 61-68 (2019).
- Gu, S., Cui, D., Chen, X., Xiong, X., Zhao, Y. PROTACs: An emerging targeting technique for protein degradation in drug discovery. Bioessays. 40 (4), 1700247 (2018).
- Neklesa, T. K., Winkler, J. D., Crews, C. M. Targeted protein degradation by PROTACs. Pharmacology and Therapy. 174, 138-144 (2017).
- Raina, K., Crews, C. M. Targeted protein knockdown using small molecule degraders. Current Opinion in Chemical Biology. 39, 46-53 (2017).
- Riching, K. M., et al. Quantitative live-cell kinetic degradation and mechanistic profiling of PROTAC mode of action. ACS Chemical Biology. 13 (9), 2758-2770 (2018).
- Schwinn, M. K., et al. CRISPR-mediated tagging of endogenous proteins with a luminescent peptide. ACS Chemical Biology. 13 (2), 467-474 (2018).
- Schwinn, M. K., Steffen, L. S., Zimmerman, K., Wood, K. V., Machleidt, T. A simple and scalable strategy for analysis of endogenous protein dynamics. Science Reports. 10 (1), 8953 (2020).
- Bensimon, A., et al. Targeted degradation of SLC transporters reveals amenability of multi-pass transmembrane proteins to ligand-induced proteolysis. Cell Chemical Biology. 27 (6), 728-739 (2020).
- Bondeson, D. P., et al. Lessons in PROTAC design from selective degradation with a promiscuous warhead. Cell Chemical Biology. 25 (1), 78-87 (2018).
- Buckley, D. L., et al. HaloPROTACS: Use of small molecule PROTACs to induce degradation of HaloTag fusion proteins. ACS Chemical Biology. 10 (8), 1831-1837 (2015).
- Bulatov, E., Ciulli, A. Targeting Cullin-RING E3 ubiquitin ligases for drug discovery: structure, assembly and small-molecule modulation. Biochemical Journal. 467 (3), 365-386 (2015).
- Burslem, G. M., et al. The advantages of targeted protein degradation over inhibition: An RTK case study. Cell Chemical Biology. 25 (1), 67-77 (2018).
- Chamberlain, P. P., et al. Evolution of cereblon-mediated protein degradation as a therapeutic modality. ACS Medicinal Chemistry Letters. 10 (12), 1592-1602 (2019).
- Crew, A. P., et al. Identification and characterization of Von Hippel-Lindau-recruiting Proteolysis Targeting Chimeras (PROTACs) of TANK-Binding Kinase 1. Journal of Medicinal Chemistry. 61 (2), 583-598 (2018).
- DeMars, K. M., Yang, C., Castro-Rivera, C. I., Candelario-Jalil, E. Selective degradation of BET proteins with dBET1, a proteolysis-targeting chimera, potently reduces pro-inflammatory responses in lipopolysaccharide-activated microglia. Biochemical and Biophysical Research Communication. 497 (1), 410-415 (2018).
- Erb, M. A., et al. Transcription control by the ENL YEATS domain in acute leukaemia. Nature. 543 (7644), 270-274 (2017).
- Farnaby, W., et al. BAF complex vulnerabilities in cancer demonstrated via structure-based PROTAC design. Nature Chemical Biology. 15 (7), 672-680 (2019).
- Gadd, M. S., et al. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nature Chemical Biology. 13 (5), 514-521 (2017).
- Gechijian, L. N., et al. Functional TRIM24 degrader via conjugation of ineffectual bromodomain and VHL ligands. Nature Chemical Biology. 14 (4), 405-412 (2018).
- Gustafson, J. L., et al. Small-Molecule-Mediated Degradation of the Androgen Receptor through Hydrophobic Tagging. Angewandte Chemie International Edition England. 54 (33), 9659-9662 (2015).
- Kerres, N., et al. Chemically induced degradation of the oncogenic transcription factor BCL6. Cell Reports. 20 (12), 2860-2875 (2017).
- Lohbeck, J., Miller, A. K. Practical synthesis of a phthalimide-based Cereblon ligand to enable PROTAC development. Bioorganic and Medicinal Chemistry Letters. 26 (21), 5260-5262 (2016).
- Lu, J., et al. Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chemical Biology. 22 (6), 755-763 (2015).
- Lu, M., et al. Discovery of a Keap1-dependent peptide PROTAC to knockdown Tau by ubiquitination-proteasome degradation pathway. Eurupean Journal of Medicinal Chemistry. 146, 251-259 (2018).
- Nabet, B., et al. The dTAG system for immediate and target-specific protein degradation. Nature Chemical Biology. 14 (5), 431-441 (2018).
- Nowak, R. P., et al. Plasticity in binding confers selectivity in ligand-induced protein degradation. Nature Chemical Biology. 14, 706-714 (2018).
- Powell, C. E., et al. Chemically induced degradation of Anaplastic Lymphoma Kinase (ALK). Journal of Medicinal Chemistry. 61 (9), 4249-4255 (2018).
- Raina, K., et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proceedings of National Academy of Science U. S. A. 113 (26), 7124-7129 (2016).
- Sakamoto, K. M., et al. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proceeding National Academy Science. 98 (15), 8554-8559 (2001).
- Sakamoto, K. M., et al. Development of Protacs to target cancer-promoting proteins for ubiquitination and degradation. Molecular Cell Proteomics. 2 (12), 1350-1358 (2003).
- Schiedel, M., et al. Chemically induced degradation of Sirtuin 2 (Sirt2) by a proteolysis targeting chimera (PROTAC) based on sirtuin rearranging ligands (SirReals). Journal of Medicinal Chemistry. 61 (2), 482-491 (2018).
- Slabicki, M., et al. The CDK inhibitor CR8 acts as a molecular glue degrader that depletes cyclin K. Nature. , (2020).
- Smith, B. E., et al. Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nature Communications. 10 (1), 131 (2019).
- Sun, B., et al. BET protein proteolysis targeting chimera (PROTAC) exerts potent lethal activity against mantle cell lymphoma cells. Leukemia. 32 (2), 343-352 (2018).
- Tong, B., et al. A Nimbolide-based kinase degrader preferentially degrades oncogenic BCR-ABL. ACS Chemical Biology. 15 (7), 1788-1794 (2020).
- Winter, G. E., et al. Drug Development. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science. 348 (6241), 1376-1381 (2015).
- Winter, G. E., et al. BET Bromodomain proteins function as master transcription elongation factors independent of CDK9 recruitment. Molecular Cell. 67 (1), 5-18 (2017).
- Zengerle, M., Chan, K. H., Ciulli, A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chemical Biology. 10 (8), 1770-1777 (2015).
- Zhang, C., et al. Proteolysis targeting chimeras (PROTACs) of anaplastic lymphoma kinase (ALK). European Journal of Medicinal Chemistry. 151, 304-314 (2018).
- Zoppi, V., et al. Iterative design and optimization of initially inactive proteolysis targeting chimeras (PROTACs) identify VZ185 as a potent, fast, and selective von Hippel-Lindau (VHL) based dual degrader probe of BRD9 and BRD7. Journal of Medicinal Chemistry. 62 (2), 699-726 (2019).
- Huang, H. T., et al. A chemoproteomic approach to query the degradable kinome using a multi-kinase degrader. Cell Chemical Biology. 25 (1), 88-99 (2018).
- Bjorklund, C. C., et al. Iberdomide (CC-220) is a potent cereblon E3 ligase modulator with antitumor and immunostimulatory activities in lenalidomide- and pomalidomide-resistant multiple myeloma cells with dysregulated CRBN. Leukemia. 34 (4), 1197-1201 (2020).
- Matyskiela, M. E., et al. SALL4 mediates teratogenicity as a thalidomide-dependent cereblon substrate. Nature Chemical Biology. 14 (10), 981-987 (2018).
- Matyskiela, M. E., et al. A cereblon modulator (CC-220) with improved degradation of Ikaros and Aiolos. Journal of Medicinal Chemistry. 61 (2), 535-542 (2018).
- Bussiere, D. E., et al. Structural basis of indisulam-mediated RBM39 recruitment to DCAF15 E3 ligase complex. Nature Chemical Biology. 16 (1), 15-23 (2020).
- Du, X., et al. Structural basis and kinetic pathway of RBM39 recruitment to DCAF15 by a sulfonamide molecular glue E7820. Structure. 27 (11), 1625-1633 (2019).
- Ting, T. C., et al. Aryl sulfonamides degrade RBM39 and RBM23 by recruitment to CRL4-DCAF15. Cell Reports. 29 (6), 1499-1510 (2019).
- Hughes, S. J., Ciulli, A. Molecular recognition of ternary complexes: a new dimension in the structure-guided design of chemical degraders. Essays in Biochemistry. 61 (5), 505-516 (2017).
- Schapira, M., Calabrese, M. F., Bullock, A. N., Crews, C. M. Targeted protein degradation: expanding the toolbox. Nature Review Drug Discovery. 18 (12), 949-963 (2019).
- Dixon, A. S., et al. NanoLuc complementation reporter optimized for accurate measurement of protein interactions in cells. ACS Chemical Biology. 11 (2), 400-408 (2016).
- Gilan, O., et al. Selective targeting of BD1 and BD2 of the BET proteins in cancer and immuno-inflammation. Science. 368 (6489), 387-394 (2020).
- Oh-Hashi, K., Furuta, E., Fujimura, K., Hirata, Y. Application of a novel HiBiT peptide tag for monitoring ATF4 protein expression in Neuro2a cells. Biochemical Biophysical Report. 12, 40-45 (2017).
- Ottis, P., et al. Cellular resistance mechanisms to targeted protein degradation converge toward impairment of the engaged ubiquitin transfer pathway. ACS Chemical Biology. 14 (10), 2215-2223 (2019).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved