Method Article
Synthesis and Characterization of Multi-Modal Phase-Change Porphyrin Droplets
In This Article
Summary
In this protocol, methods for synthesizing and characterizing multi-modal phase-change porphyrin droplets are outlined.
Abstract
Phase-change droplets are a class of ultrasound contrast agents that can convert into echogenic microbubbles in situ with the application of sufficient acoustic energy. Droplets are smaller and more stable than their microbubble counterparts. However, traditional ultrasound contrast agents are not trackable beyond acoustic feedback measurements, which makes quantifying contrast agent bio-distribution or accumulation ex vivo difficult. Researchers may have to rely on fluorescent or optically absorbent companion diagnostic particles to infer bio-distribution. The purpose of this protocol is to detail steps for creating multi-modal phase-change porphyrin droplets using a condensation method. Porphyrins are fluorescent molecules with distinct absorbance bands that can be conjugated onto lipids and incorporated into droplets to extend droplet versatility, enabling more robust bio-distribution while retaining acoustic properties. Seven formulations with varying porphyrin-lipid and base lipid contents were made to investigate microbubble and droplet size distributions. Characterizations suited to porphyrin-containing structures are also described in the protocol to demonstrate their analytic versatility in-solution. Sizing showed that the post-condensed mean diameters were 1.72 to 2.38 times smaller than precursor populations. Absorbance characterization showed intact assemblies had a Q-band peak of 700 nm while disrupted samples had an absorbance peak at 671 nm. Fluorescence characterization showed intact 30% porphyrin-lipid assemblies were fluorescently quenched (>97%), with fluorescence recovery achieved upon disruption. Acoustic vaporization showed that porphyrin droplets were non-echogenic at lower pressures and could be converted into echogenic microbubbles with sufficient pressures. These characterizations show the potential for porphyrin droplets to eliminate the need for absorbance or fluorescence-based companion diagnostic strategies to quantify ultrasound contrast agent bio-distribution for delivery or therapeutic applications in vivo or ex vivo.
Introduction
Ultrasound imaging is a non-invasive, non-ionizing form of medical imaging that utilizes acoustic waves. While ultrasound scanners are more portable and can provide real-time images, ultrasound imaging can suffer from low contrast, making it difficult for sonographers to reliably distinguish similarly echogenic pathological features. To counteract this limitation, microbubbles can be injected into the host to improve vascular contrast. Microbubbles are micron-sized gas filled contrast agents that are highly echogenic to acoustic waves and can provide enhanced vessel contrast1,2. The shells and gas cores of microbubbles can be tailored for different applications, such as imaging, thrombolysis, cell membrane permeabilization, or transient vascular opening2.
A drawback of microbubbles is their short circulation half-lives. For example, clinically available perflutren lipid microspheres only have a half-life of 1.3 minutes3. For long imaging sessions, multiple injections of microbubbles are needed. Another drawback of microbubbles is their large diameters. While perflutren lipid microspheres are around 1 to 3 µm in diameter, small enough to circulate in vasculature, they are too large to extravasate and passively accumulate into tissues of interest, such as tumors4. One strategy to overcome these limitations is to condense the gas-core microbubbles into smaller, liquid-core droplets5,6. While droplets are not echogenic in their liquid state, they can be vaporized into microbubbles upon exposure to ultrasound with sufficiently high peak negative pressure, regaining their ability to provide contrast. This allows for the droplet to take advantage of the more favorable pharmacokinetics of a small liquid-core, while retaining the ability to provide contrast when insonated and without changing the chemical composition4,7.
Decafluorobutane is an ideal perfluorocarbon compound for phase-shifting between gaseous and liquid states5,6,7. Decafluorobutane allows for condensation of microbubbles into droplets with temperature reduction alone, whereas less dense perfluorocarbons require additional pressurization5. This gentle method minimizes destruction of bubbles during condensation7,8,9. As their cores are liquid, droplets are non-echogenic and invisible to ultrasound. However, with the application of sufficient acoustic or thermal energy, the liquid cores can vaporize back into a gaseous state, generating echogenic microbubbles8. This vaporization allows for control of when and where to generate microbubbles.
While droplets are useful for passive accumulation, in situ vaporization, or improving cell permeability4, droplets (and their fragments) cannot be imaged or quantified ex vivo. Therefore, quantifiable companion diagnostic agent, such as fluorescent4,10,11, magnetic particles12, optically absorbent agents13, are utilized as an analogue to gauge droplet delivery to tissues of interest. For example, Helfield et al. used a co-injection of fluorescent nano-beads for histology image quantification of mouse organs as droplets could not be detected fluorescently4. The disadvantage of companion diagnostic agents is the trackable component may act independently from the droplet depending on its individual pharmacokinetic profile.
Fortunately, the shell of microbubbles and droplets can be customized. For example, Huynh et al. demonstrated ultrasound contrast agents with porphyrin-lipid shells, creating multi-modal microbubbles14. Porphyrins are a class of organic compounds with an aromatic macrocylic structure14,15. They are optically absorbent, fluorescent, and can be chelated to a wide variety of metals for radiotherapy, radionuclide-based imaging, or trace metal-based quantification14. One example of porphyrin is pyropheophorbide (Pyro). By conjugating Pyro onto lipids, incorporating Pyro-lipids in microbubbles or droplets allow them to be imaged and tracked through multiple modalities: acoustically, fluorescently, and through absorbance14. This multi-modal contrast agent could be used to track and quantify accumulation. This could eliminate the need for companion diagnostic agents as the quantifiable component is now conjugated onto the shell, enabling more accurate delivery quantification16.
Herein, a protocol for creating multi-modal phase-change porphyrin droplets is outlined. As ultrasound contrasts agents can be used as a platform for drug delivery to tissues of interest, such as tumors2,4, extending their detectability beyond ultrasound could prove useful for delivery efficacy quantification. The purpose of these droplets is to provide trackable ultrasound contrast agents capable of passive accumulation in vivo, in situ vaporization and acoustics, and with the potential to quantify bio-distribution or accumulation from ex vivo organs without the reliance on secondary sensors. Characterization methods are also outlined to showcase porphyrin droplets' potential as bio-distribution sensors. The effects of Pyro-lipid loading in the shell (0% to 50% by molar ratio) are also discussed.
Protocol
1. Dehydrated lipid films
- Calculate the masses of each of the shell components needed (see Supplemental File "Lipid Formula Sheet").
NOTE: For this protocol, the shell composition will be: 10 molar % 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-5000] ammonium salt (DSPE-PEG5K), x molar % pyropheophorbide conjugated 1-stearoyl-2-hydroxy-sn-glycero-3-phosphocholine (Pyro-SPC), and (90 - x) molar % 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC). The amount of Pyro-SPC will be varied across 7 shell compositions (x = 0, 1, 10, 20, 30, 40, 50). Check the molecular weight of DSPE-PEG5K on the stock bottle.- Scale the protocol to any lipid volume with a minimum volume of 1 mL. For this protocol, a total lipid volume of 10 mL with a total lipid concentration of 1 mg per mL was used for all formulations. The excipient solution will be: 10% propylene glycol, 10% glycerol, and 80% phosphate buffer saline (PBS, 1X, 7.4 pH) (% v/v/v) (see Step 2.3 and "Lipid Formula Sheet").
NOTE: Synthesis protocol of Pyro-lipids is outlined in the Supplemental File "Other Protocols and Data" Steps S1 to S1.19, which was modified from the work done by Zheng et al.15.
- Scale the protocol to any lipid volume with a minimum volume of 1 mL. For this protocol, a total lipid volume of 10 mL with a total lipid concentration of 1 mg per mL was used for all formulations. The excipient solution will be: 10% propylene glycol, 10% glycerol, and 80% phosphate buffer saline (PBS, 1X, 7.4 pH) (% v/v/v) (see Step 2.3 and "Lipid Formula Sheet").
- Based on the calculated masses (see Step 1.1 and "Lipid Formula Sheet"), weigh out each of the non-Pyro-lipids and transfer to a sufficiently sized borosilicate glass vial with a screw-on cap.
- Cap the vial, label the cap and vial, and cover the bottom and walls of the lipid vial with aluminum foil. This vial will be referred to as the "Lipid Vial" for the rest of the protocol. Store the Lipid Vial in a cool, dry, dark area.
- If the formulation contains Pyro-SPC, dissolve 10 mg of Pyro-SPC dry film (see "Other Protocols and Data") into 1 mL of chloroform. Vortex for 5 s, measure the absorbance, and calculate the appropriate volume to add to the Lipid Vial.
CAUTION: Chloroform is a health hazard, irritant, and toxic. Wear a protective lab coat, eye protection, gloves, and avoid breathing fumes.
NOTE: As Pyro-SPC is light sensitive, reduce the lights in the working area if possible when handling Pyro-SPC. Keep Pyro-SPC sealed and covered when not in use.- On the ultraviolet-visible spectrophotometer, set it to measure absorbance from a wavelength range of 800 nm to 300 nm with 0.5 nm increments, and measure a baseline with 2000 µL of pure methanol in a compatible 1 cm path length cuvette.
CAUTION: Methanol is a health hazard, irritant, toxic, and flammable. Wear a protective lab coat, eye protection, gloves, and avoid breathing fumes. Keep away from sparks and heat. - Add 2 µL of the Pyro-SPC in chloroform into 2000 µL of methanol, and vortex for 30 s. Transfer it into a clean, compatible 1 cm cuvette, and measure the absorbance. Adjust this dilution factor if the absorbance peak at 667 nm out of the ultraviolet-visible spectrophotometer's absorbance range.
NOTE: Whenever transferring chloroform or methanol, use a clean glass syringe or a positive-displacement pipette rather than a mechanical pipette for better accuracy. - Repeat Step 1.4.2 two more times to get triplicate absorbance values.
- Average the absorbance peak values at 667 nm and use the following equation to calculate the volume of Pyro-SPC in chloroform needed for the Lipid Vial14,15:

where V is the volume of Pyro-SPC in chloroform needed, m is the required mass of Pyro-SPC (see Step 1.1 and "Lipid Formula Sheet"), M is the molecular weight of Pyro-SPC at 1040.317 g·mol-1, A is the averaged absorbance at 667 nm, d is the dilution factor based on the methanol and Pyro-SPC in chloroform volumes (Step 1.4.2), L is the cuvette path length at 1 cm, and ε is 667 nm molar attenuation coefficient of Pyro-SPC at 45000 L·mol-1·cm-1.
NOTE: The denominator of the equation is the Beer-Lambert Law, which relates the concentration of an analyte in solution to the measured optical absorbance over a distance. - Add the calculated volume of Pyro-SPC in chloroform from the previous step to the Lipid Vial using a clean glass syringe (Figure 1A) and then cap and cover the vial.
NOTE: Figure 1 only shows the 30% Pyro-lipid formulation only. - If there is any Pyro-SPC in chloroform remaining: In a fume hood, uncap the Pyro-SPC + chloroform vial from Step 1.4, partially tilt the vial to its side and continuously flow nitrogen gas as gently as possible into the Pyro-SPC/chloroform vial. Rotate the vial to dry out the chloroform using the nitrogen gas flow and to evenly coat the Pyro-SPC onto the interior wall of the vial as it dries. Ensure no splashes are made and none of the solution falls out.
- Once the Pyro-SPC lipid film appears dry and coated onto the wall of the vial, turn off the nitrogen gas flow. Cap the vial, seal the vial neck with wax film, and store the vial at -20 °C and in the dark.
- On the ultraviolet-visible spectrophotometer, set it to measure absorbance from a wavelength range of 800 nm to 300 nm with 0.5 nm increments, and measure a baseline with 2000 µL of pure methanol in a compatible 1 cm path length cuvette.
- Prepare a solution of 90% chloroform and 10% methanol (% v/v), and add 5 mL of it to the Lipid Vial. Cap the Lipid Vial, and gently swirl to homogenize the contents (Figure 1B).
NOTE: If the formulation contains phosphatidic acid lipids (such as 1,2-distearoyl-sn-glycero-3-phosphate sodium salt (DSPA)), add: 60% chloroform, 32% methanol, and 8% ultrapure water (% v/v/v) to the Lipid Vial instead. More intense swirling may be necessary to fully dissolve the lipid contents. - In a fume hood, uncap the Lipid Vial, partially tilt the Lipid Vial to its side and continuously flow nitrogen gas as gently as possible into the Lipid Vial. Rotate the Lipid Vial to dry out the solution using the nitrogen gas flow and evenly coat the lipid film onto the interior wall of the Lipid Vial as it dries. Ensure no splashes are made and none of the solution falls out.
- Once the lipids appear dry and coated onto the interior walls of the lipid vial (Figure 1C), turn off the nitrogen gas flow, cover the bottom and wall of the Lipid Vial with aluminum foil, and cover the top opening with aluminum foil poked with a few holes with a needle for venting (Figure 1D).
- Label and place the covered Lipid Vial inside a vacuum desiccator and allow the lipids to dry further for at least 24 h but no more than 72 h.
NOTE: The protocol can be resumed later, after 24 to 72 h.
2. Lipid Hydration
- Fill a bath sonicatorwith water and heat it to 70 °C. Turn on the sonication to help mix the water.
CAUTION: The water and sonicator are at high temperatures. Avoid touching the water and the sonicator. Wear eye protection, a protective lab coat, and protective gloves. - Once the bath sonicator has reached 70 °C, remove the Lipid Vial from the vacuum desiccator. Reduce the lights in the working area if possible.
- Prepare an excipient solution of 10% propylene glycol, 10% glycerol, 80% PBS (% v/v/v) and add 10 mL of it to the Lipid Vial (see Step 1.1.1 and "Lipid Formula Sheet").
NOTE: If using a standard air-displacement pipette, use care when handling viscous solvents like propylene glycol and glycerol. Aspirate and plunge the volume slowly and wait for the residual volume to reach the bottom of the pipette tip. Ensure liquid does not cling to the outside of the pipette tip when transferring volumes by moving slowly. - Cap the Lipid Vial, remove the aluminum cover, and gently swirl the vial in the bath sonicator for 15 min while the sonication is on to dissolve the lipids. Make sure the Lipid Vial's neck is above the water. Occasionally check if the vial cap is securely closed.
- Occasionally, remove the Lipid Vial from the water bath. Briefly hold it to the light to check if the contents are fully dissolved (Figure 1E).
- If the Lipid Vial contents are not homogenizing, remove the Lipid Vial from the bath sonicator. Secure the cap, swirl more aggressively, and return it to the bath sonicator.
- Remove the Lipid Vial from the bath sonicator, and turn off the bath sonicator. Dry the Lipid Vial exterior with paper towels, and re-label the Lipid Vial.
- Cover the Lipid Vial with aluminum foil, and cool the Lipid Vial at room temperature in a cool, dark, dry area for 10 minutes.
- Aliquot the Lipid Vial contents: about 2 mL of the lipid solution in 3 mL borosilicate glass clear serum vials (7 mm inner mouth diameter, 13 mm outer mouth diameter).
NOTE: Some protocols may use 1.5 mL of lipid solution in the 3 mL vial7. - Cap the serum vials with lyophilization-style gray chlorobutyl rubber stoppers (7 mm inner mouth diameter, 13 mm outer mouth diameter) and secure the rubber stopper with tear-off aluminum seals (13 mm outer mouth diameter) and a crimper (Figure 1F).
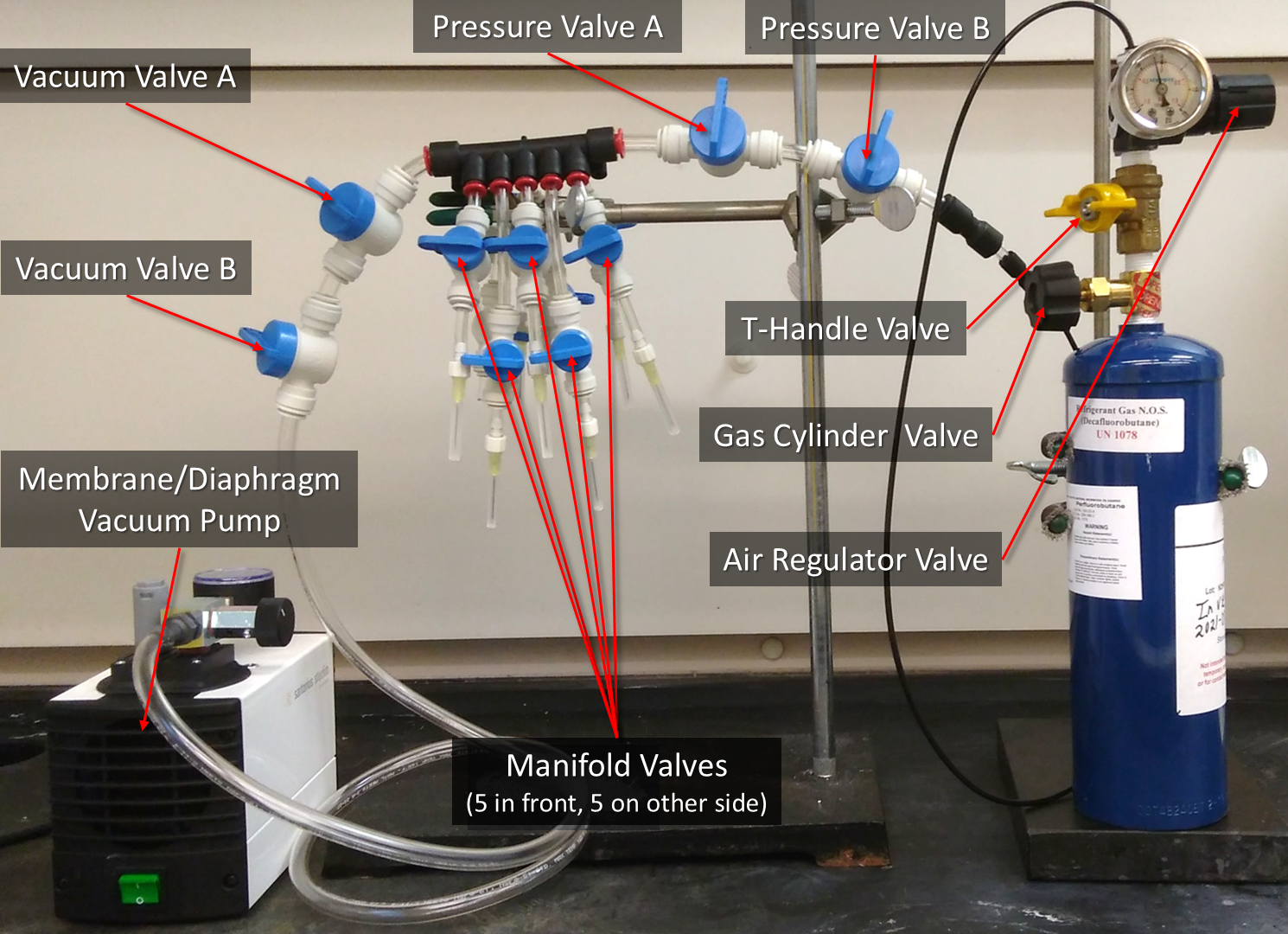
- Vacuum, degas, and re-pressurize the lipid solution in each of the serum vials4,5,7 (Figure 2).
NOTE: Refer to "Other Protocols and Data" for instructions on how to assemble the Gas Exchanger and more details.- Close every valve between and including Pressure Valve A and Gas Cylinder Valve (Figure 2). Connect the serum vials to the manifold needles, then open the corresponding Manifold Valves, then open Vacuum Valve A and Vacuum Valve B, and vacuum at -90 kPa (-13 psi, -900 mbar) for 5 min to remove atmospheric air. DO NOT vacuum out any of the liquid (see "Other Protocols and Data" Steps S3 to S3.1.5).
CAUTION: The vacuum pump can burst if handled incorrectly. Do not use the vacuum pump with organic, acidic, or basic chemicals. - With the vacuum still on, hold a serum vial (to prevent it from swinging) and tap it rapidly with a pen or marker to degas. Keep tapping until no bubbles form and there are no bubbles in the vial. DO NOT let any liquid be vacuumed out. Pause tapping if necessary. Repeat for all connected serum vials. After degassing all serum vials, CLOSE Vacuum Valve A and Vacuum Valve B and TURN OFF the vacuum pump (see "Other Protocols and Data" Steps S3.2 to S3.2.3).
- With the serum vial still connected to the needle and with the vacuum pump turned off, slowly turn the Gas Cylinder Valve 1/16 to 1/8 (about 22.5 to 45 ° of full revolution) counterclockwise to partially open, then open the T-Handle Valve, and SLOWLY turn the Air Regulator Valve clockwise to 3 psi (20.7 kPa) gauge. Then, open Pressure Valve A and Pressure Valve B (see "Other Protocols and Data" Steps S3.3 to S3.3.21).
CAUTION: The decafluorobutane gas cylinder is under pressure and can explode if heated. Keep way from heat and impact. Decafluorobutane gas may cause oxygen displacement and suffocation. Wear proper eye protection and handle in a fume hood. If handled incorrectly, it is possible to vacuum the gas cylinder, which can cause rapid de-pressurization and implosion. Opening the Gas Cylinder Valve more than 1/8 turn can damage the Air Regulator. - After 30 s of pressurization (gauge should still read 3 psi (20.7 kPa)), close all Manifold Valves with serum vials, disconnect the serum vials, sheath the needles, and CLOSE the Gas Cylinder Valve.
- Relieve the built-up pressure by partially opening a single Manifold Valve until Air Regulator gauge needle goes to its resting position. Then close everything between and including the Manifold Valves and the T-Handle.
- Close every valve between and including Pressure Valve A and Gas Cylinder Valve (Figure 2). Connect the serum vials to the manifold needles, then open the corresponding Manifold Valves, then open Vacuum Valve A and Vacuum Valve B, and vacuum at -90 kPa (-13 psi, -900 mbar) for 5 min to remove atmospheric air. DO NOT vacuum out any of the liquid (see "Other Protocols and Data" Steps S3 to S3.1.5).
- Label serum vials and store them at 4 °C and in the dark. Ensure all Gas Exchanger valves are closed and the vacuum pump is turned off afterwards.
NOTE: The serum should be stable for up to 4 months in this state. At this step, the protocol can be resumed later, after 4 months at most.
3. Decafluorobutane vials
- With clean, empty 3 mL borosilicate glass clear serum vials (7 mm inner mouth diameter, 13 mm outer mouth diameter), cap them with lyophilization-style gray chlorobutyl rubber stoppers (7 mm inner mouth diameter, 13 mm outer mouth diameter) and secure the rubber stopper with tear-off aluminum seals (13 mm outer mouth diameter) and a crimper4,7,8.
- Follow Step 2.9.1 to vacuum the atmospheric air (see "Other Protocols and Data" Steps S3.1 to S3.1.5).
- Skip the degassing and follow Steps 2.9.3 to 2.9.5 to re-pressurize the vial (see "Other Protocols and Data" Steps S3.3 to S3.3.21).
- Label the decafluorobutane vials and store them at 4 °C and in the dark. Ensure all Gas Exchanger valves are closed and the vacuum pump is turned off afterwards.
NOTE: Serum vials filled with decafluorobutane gas will be needed for the droplet condensation. They should be stable for up to 4 months in this state. At this step, the protocol can be resumed later, after 4 months at most.
4. Droplet formation
- Remove a hydrated lipid solution in the serum vial (from Step 2.10) from the fridge.
- Using a decapper, remove the aluminum seal on the serum vial and transfer 1 mL of the lipid solution to a 1.85 mL borosilicate glass sample vial (with a phenolic screw cap) by letting the lipid solution flow down the interior wall. Do not create bubbles.
- If there is any remaining lipid solution in the serum vial, follow Steps 2.9 to 2.10 to degas and re-pressurize the serum vial for storage (see "Other Protocols and Data" Steps S3 to S3.3.21).
- With the 1.85 mL sample vial, gently flow in decafluorobutane gas into the sample vial headspace using the Gas Exchanger (see Figure 2 for the specific valve names).
- Ensure that all the valves on the Gas Exchanger are properly closed and the pump is turned off.
CAUTION: If done incorrectly, it is possible to vacuum out the gas cylinder causing rapid decompression and implosion. - Open one manifold valve and carefully unsheathe the corresponding needle from the manifold.
CAUTION: Sharp object, avoid contact/piercing. - Open Pressure Valve A and Pressure Valve B and turn the Gas Cylinder Valve 1/16 to 1/8 (about 22.5 to 45 ° of full revolution) counterclockwise to partially open and then open the T-Handle Valve.
CAUTION: Do not open the Gas Cylinder Valve more than this as it could cause damage to the Air Regulator. - Uncap the sample vial with the lipid solution and move it so the Manifold needle is above the liquid-air interface inside the vial. Hold the vial there.
- SLOWLY turn the Air Regulator Valve clockwise until the Air Regulator gauge needle moves slightly from its resting position and perfluorocarbon gas is gently flowing out of the manifold needle. Let the perfluorocarbon gas gently flow into the vial headspace for 30 s. Do not create bubbles. Adjust the Air Regulator Valve if necessary.
NOTE: The liquid-air interface should be slightly perturbed by the decafluorobutane gas flow. As the system is now open, the Air Regulator gauge cannot properly read pressure. - After 30 s, carefully and quickly cap the sample vial without moving the vial too much.
- Close the Gas Cylinder Valve (clockwise), the T-Handle Valve, Air Regulator Valve (counterclockwise), Pressure Valve A, Pressure Valve B, and Manifold Valve.
- Carefully sheath the needle.
- Label the sample vial and seal the neck with wax film going clockwise (Figure 3A and 3B).
NOTE: Figures 3B to 3F only show the 30% Pyro-lipid formulation.
- Ensure that all the valves on the Gas Exchanger are properly closed and the pump is turned off.
- Store the sample vial in the dark and at 4 °C for at least 10 min or up to 24 h.
NOTE: At this step, the protocol can be resumed later, 24 h at most. - Place about 100 g of dry ice (carbon dioxide) in an insulated container and place regular ice in another insulated container. Retrieve prepared decafluorobutane serum vials mentioned in Step 3, two 3.81 cm (1.5 inch) 20-gauge needles, a 1 mL plastic syringe (unstick the plunger before use), a 200 mL container, metal tongs, and a thermometer (-20 to 100 °C).
NOTE: If only making microbubbles, then there is no need for isopropanol, dry ice, and ice. - Place the sample vial with the lipid solution in the mechanical agitator and agitate for 45 s (Figure 3C).
- After the mechanical agitation, stand the sample vial right side-up, shielded from light, and start a 15 min countdown to cool down the vial and size-select the microbubbles8,17.
- When the 15 min countdown has reached 10 minutes (5 minutes left on the countdown), fill a container with about 200 mL of isopropanol and cool it to -20 °C with dry ice using metal tongs.
NOTE: The target temperature is -15 to -17 °C but the isopropanol will warm up while handling the microbubbles.
CAUTION: Isopropanol is flammable. Keep away from heat and sparks. Dry ice can cause skin damage. Handle with tongs. Wear gloves, eye protection, and a protective lab coat.
- When the 15 min countdown has reached 10 minutes (5 minutes left on the countdown), fill a container with about 200 mL of isopropanol and cool it to -20 °C with dry ice using metal tongs.
- After the microbubbles have been size-selected for 15 min, look for the size-selected partition inside the sample vial (Figure 3D).
- Keeping the sample vial right side-up, carefully uncap the sample vial, and withdraw about 0.7 mL of the bottom partition with a 1.5 inch 20-gauge needle attached to a 1 mL plastic syringe. Ensure none of the top partition is withdrawn. Do not flick the syringe to remove air pockets.
- Insert a different 20-gauge needle into a decafluorobutane serum vial (keeping the needle near the top of the serum vial) to vent and then insert the needle/syringe with the size-selected microbubbles.
- Slowly transfer the size-selected microbubbles. Tilt the vial and angle the syringe to let the liquid slide down the interior wall of the decafluorobutane serum vial.
- Once all the size-selected microbubble solution has been transferred, remove the needle with the syringe but keep the venting needle in to relieve negative pressure (Figure 3E).
- If making only size-selected microbubbles, stop here. Keep the venting needle in and near the top. Keep the vial in the dark and at room temperature.
- Add small amounts of dry ice or room temperature isopropanol to the isopropanol bath to ensure the bath temperature is between -15 to -17 °C.
- With the 20-gauge venting needle inserted near the top of the serum vial, place the serum vial in the isopropanol bath, keeping the microbubble level below the level of the isopropanol but the vial neck above it, and intermittently swirl the serum vial for 2 min to condense the microbubbles.
NOTE: This step was modified from the work done by Sheeran et al.6- Do no swirl the serum vial continuously in the isopropanol and do not let the solution freeze. Swirl for about 5 s, and lift the serum vial out of the isopropanol. Check for ice nucleation, and resume swirling in isopropanol. If there is ice formation, swirl the serum vial in the air until it dissipates.
- After the 2-minute condensation, remove the serum vial from the isopropanol bath and remove the venting needle.
NOTE: The microbubbles should have been condensed into droplets, as indicated by the change in translucency (Figure 3E versus Figure 3F for the 30% Pyro-lipid formulation). - Wipe the serum vial, label it, and place it on regular ice in a dark, insulated container until ready for use. Unopened (intact aluminum seal) droplets should be stable in this state for up to 6 h as long as the melted ice gets replaced as needed.
- When ready to use, remove the aluminum seal with the decapper. Keep droplets (even open vials) on ice and in dark while not in use. Keep microbubbles in dark and at room temperature.
5. Morphological and Optical Characterization
- Prepare 1% Triton by: adding 5 mL of Triton X-100 into 500 mL of PBS (1x, pH 7.4) and stir with a magnetic stir bar until homogenous14.
NOTE: Triton X-100 very viscous. If using a standard air-displacement pipette, use care when handling. Aspirate and plunge the volume slowly and wait for the residual volume to reach the bottom of the pipette. Ensure liquid does not cling to the outside of the pipette tip when transferring volumes by moving slowly. - Prepare droplets (Step 4). If microbubbles are also being characterized, collect a small volume of the size-selected microbubbles prior to condensation (Step 4.9).
- Size the microbubbles or droplets on a Coulter Counter (CC) from 0.2 to 6 µm to obtain size-distributions and concentrations (Figure 4).
- Fill a clean 20 mL cuvette with 10 mL of CC electrolyte that was filtered through 0.2 µm pore polyethersulfone membrane filter. Measure it on the CC with three runs to get a baseline.
- In the same CC electrolyte, add 5 µL of microbubble or droplet sample and gently mix.
NOTE: 2 to 20 µL of sample can be added depending on how concentrated the sample is. - Run the sample on the CC (3 runs), subtract the averaged baseline, and calculate the size distribution and concentration (number per mL).
- Measure the droplet absorbance with UV-Vis spectroscopy (Figure 5).
NOTE: Figure 5 only shows the 30% Pyro-lipid formulation only.- On the UV-Vis spectrophotometer, set the absorbance measurement as 800 nm to 300 nm wavelengths with 0.5 nm increments and enable baseline correction.
- Using a clean 1 cm path length cuvette filled with PBS, perform a baseline measurement. Ensure the volume is high enough to intersect the spectroscope beam path.
- Dilute the Pyro-lipid droplets into PBS (recommend 2 µL to 500 µL of droplets into 2000 µL of diluent) and mix by pipetting.
NOTE: DO NOT vortex or else the assemblies will be destroyed. - Transfer the diluted droplets into a cleaned cuvette and measure the absorbance. Alter the dilutions if necessary.
- Repeat Steps 5.4.1 to 5.4.4 but use 1% Triton instead of PBS. After dilution, transfer sample to a sealable/capped vial and vortex for 30 s before measuring.
- Measure the fluorescence of microbubbles or droplets (Figure 6).
NOTE: Figure 6 only shows the 30% Pyro-lipid formulation only.- On the fluorescence spectrophotometer, set the excitation wavelength as 410 nm and the emission wavelength range from 600 to 750 nm with 1 nm increments.
- Measure the fluorescence of the PBS diluent to get a baseline using a cuvette that is compatible with the fluorescence spectrophotometer.
- Dilute the Pyro-lipid microbubbles or droplets into PBS (recommend 0.5 µL to 10 µL of droplets into 2000 µL of diluent) and mix by pipetting.
NOTE: DO NOT vortex or else the assemblies will be destroyed. - Transfer the diluted sample to the cleaned, baselined cuvette and measure the fluorescence. Alter the dilutions if necessary and avoid signal saturation.
- Repeat Steps 5.5.1 to 5.5.4 but use 1% Triton instead of PBS. After diluting in 1% Triton, transfer the diluted sample to a sealable/capped vial and vortex for 30 s before measuring. Add enough volume to ensure bubbles generated from vortexing are above the laser path.
NOTE: The fluorescence signal of the sample in Triton will be much higher than in PBS due to fluorescence unquenching (Figure 6).
6. Vaporization imaging
- Fill an appropriately sized water tank with deionized water and let it rest for 24 hours to equilibrate the gases in the water with the atmosphere.
- Prepare droplets and keep on ice and in dark until use.
- Make a flow phantom tube from 2% agar as described by Pellow et al.18 Submerge the phantom into a water tank heated to 37 °C.
- Warm up PBS to 37 °C and flow through the phantom.
- With a pre-clinical ultrasound system and 21 MHz linear array transducer (see Table of Materials), align the view to the flow phantom, set it to B-mode imaging, set the output pressures, and capture videos or images to acquire baselines at each pressure.
- Dilute 20 µL of the droplet into 50 mL of 37 °C PBS and mix gently. Transfer the solution into a 30 mL plastic syringe and push the solution through the agar phantom.
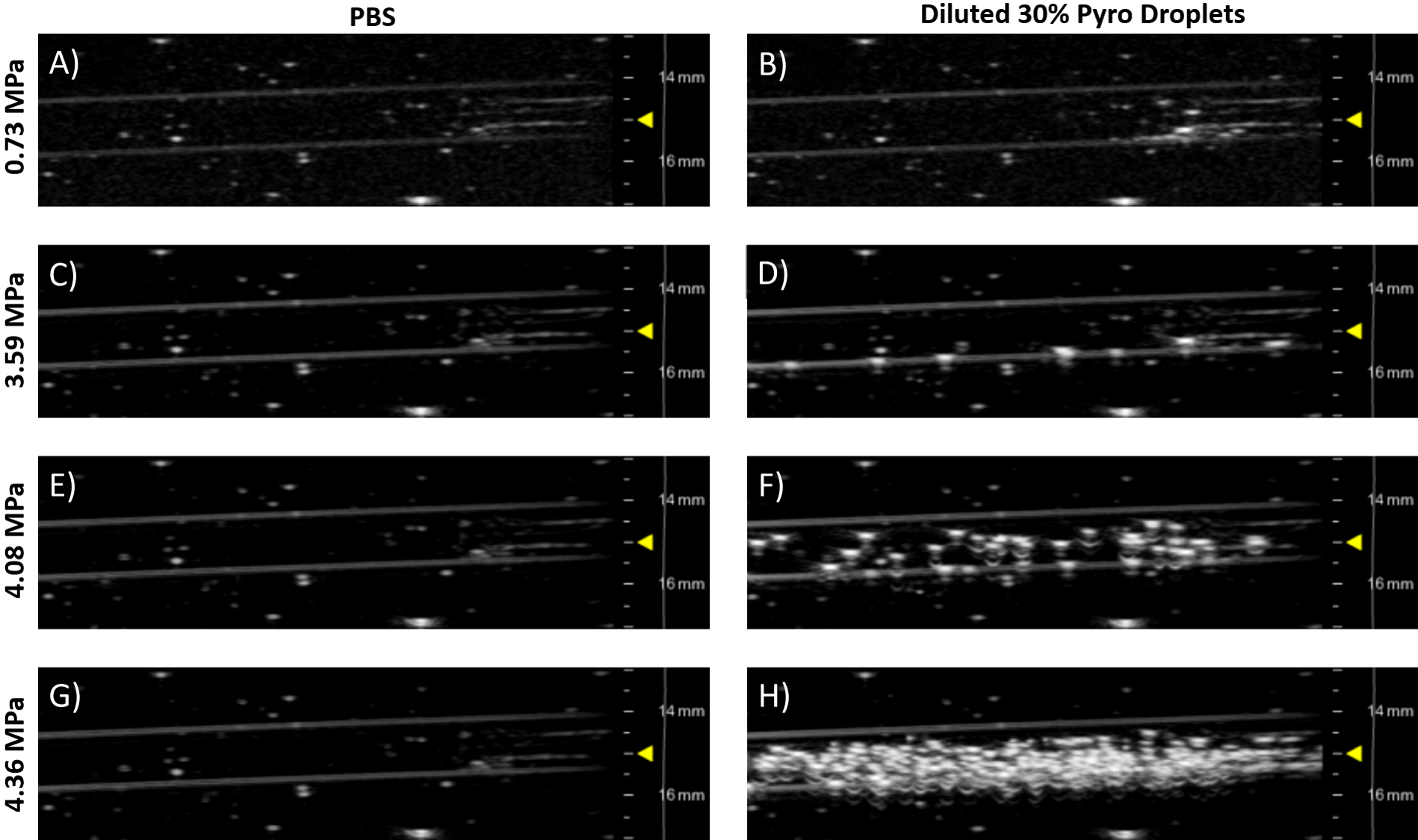
- Keeping the same alignment as Step 6.5, increase the output pressures until vaporization is observed (bright speckles in the phantom, see Figure 7).
NOTE: Figure 7 shows the 30% Pyro-lipid droplet sample. This commercially available 21 MHz linear array transducer is capable of both imaging and vaporizing droplets.
Results
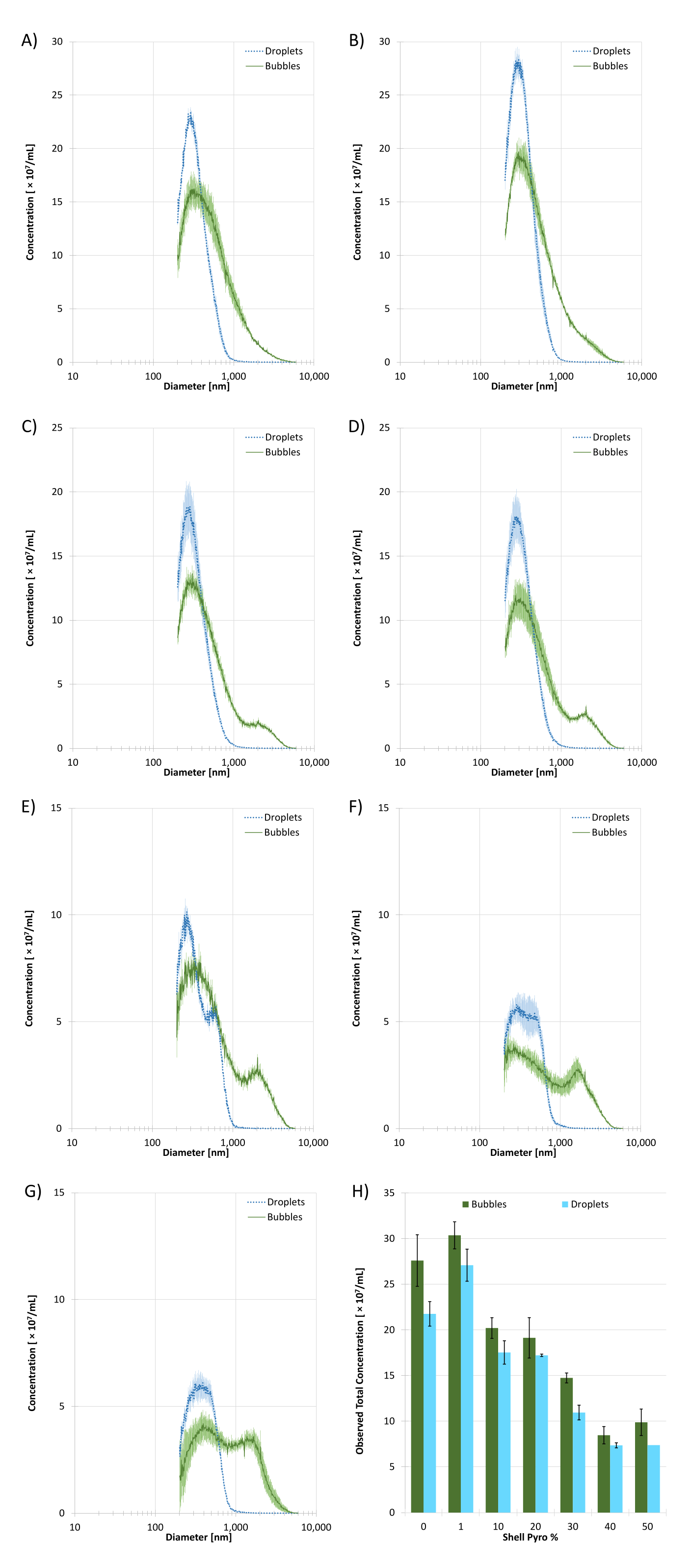
Pre-condensed, size-selected microbubble samples (n = 3) and post-condensed droplet samples (n = 3) were sized on a Coulter Counter (CC) with a 10 µm aperture. A limitation of the 10 µm aperture is it cannot measure particles smaller than 200 nm, which can bias the mean size and concentration. Figure 4 shows the sizing data for each of the Pyro-lipid content formulations. Table 1 shows statistics based on the sizing data. Using a ratio of pre- and post-condensed mean diameters, the results showed that the 0% Pyro-lipid formulation had the smallest mean diameter shift at 1.72 ± 0.02. The 50% Pyro-lipid formulation had the greatest mean diameter at 2.38 ± 0.08. The 1% Pyro-lipid droplet sample had the highest observed concentration at (2.71 ± 0.13) × 1010 /mL while the 40% Pyro-lipid droplet sample had the lowest observed concentration at (7.36 ± 0.81) × 109 /mL. Sizing data showed the 10% Pyro-lipid droplet sample had the smallest peak dimeter at 261 ± 13 nm while the 50% Pyro-lipid droplet sample had the largest at 390 ± 55 nm. Generally, as the Pyro-lipid content increased, the concentration decreased and the mean diameter increased. As the post-condensed samples are based on the precursor microbubble sample, the trend occurred for both types of ultrasound contrast agents. As the Pyro-lipid content increased, a microbubble subpopulation (with a peak size at approximately 2000 µm) started to form. This secondary peak was not present in the 0% Pyro-lipid microbubble sample and most apparent in the 40% and 50% Pyro-lipid populations.
Figure 5 shows representative absorbance measurements of the 30% Pyro-lipid droplet sample. The peak of the intact sample in PBS was 700 nm while the disrupted sample in Triton shifted the peak to 671 nm. This showed that the intact assembles have different optical properties compared to the individual, unassembled lipid components.
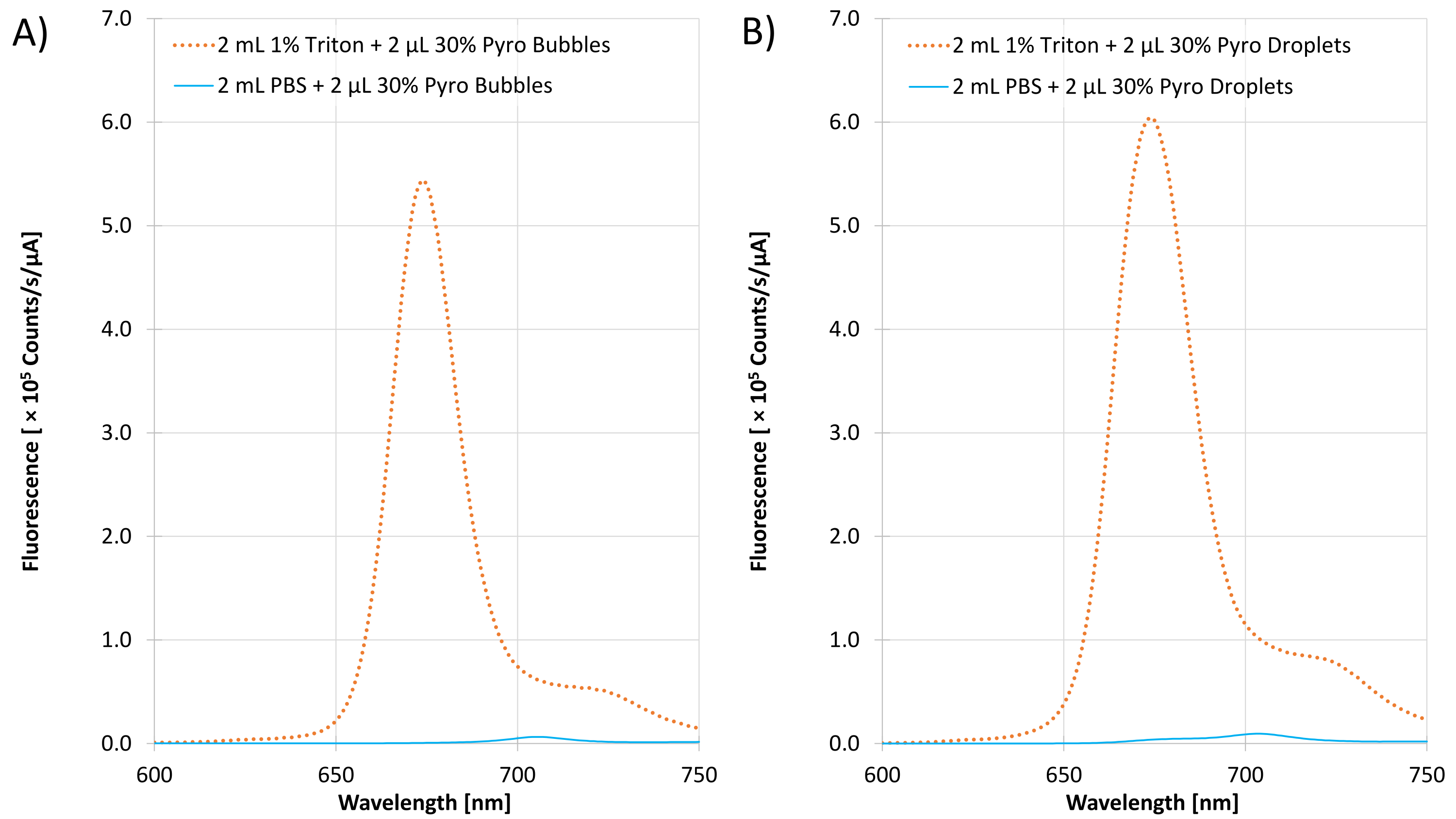
Figure 6A shows representative fluorescence measurements of the pre-condensed microbubble sample while Figure 6B shows post-condensed droplet sample with 30% Pyro-lipid. The intact sample in PBS had a fluorescence peak at 704 nm while the disrupted form had a peak at 674 nm. Subtracting the disrupted area under the curve with the intact area under the curve, and dividing the difference by the disrupted area under the curve gives the quenching efficiency, which works out to be 98.61% and 98.07% for the 30% Pyro-lipid microbubble sample and droplet sample, respectively.
To demonstrate droplets converting to microbubbles, diluted droplets were imaged and vaporized in a 37 °C flow phantom with an ultrasound system. Figure 7 shows representative ultrasound images of the 30% Pyro-lipid droplet sample imaged at different pressures. At low pressures (Figure 7A), there was very little signal, only background signal from air bubbles stuck from the agar synthesis. This is because droplets are non-echogenic and do not scatter ultrasound. At a slightly high power, a few microbubbles were generated (Figure 7B) as shown by the appearance of bright speckles. As the pressure increased, more microbubbles were generated (Figure 7C and 7D). This also demonstrated that the droplets will not spontaneously vaporize at 37 °C.

Figure 1: Images of the steps to form the 30% Pyro-lipid solution. A) Lipid powder plus Pyro-SPC in chloroform. B) Dissolving solution added. C) Lipid film dried and coated onto interior wall of vial. D) Lipid vial wrapped in aluminum foil (exterior foil taped for reuse). E) Hydrated lipid solution. F) Lipid solution in serum vial. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: The 10-manifold gas exchanger. The valves referenced in the protocol are labelled. See Supplemental File "Other Protocols and Data" for instructions on how to assemble the Gas Exchanger. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: A) The lipid solutions of the 7 formulations (0% to 50% Pyro-SPC) in sample vials. Figures B to D show images of the steps taken to make 30% Pyro-lipid droplets. B) 30% Pyro-lipid solution in a sample vial. C) Post-agitation. D) 15 min size-selected. E) Bottom partition transferred to decafluorobutane vial. D) Post-condensation. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Coulter Counter (CC) sizing data of the size-selected microbubble and droplet samples with different Pyro-lipid shell content (n = 3). The solid green lines represent microbubbles and the dotted cyan lines represent droplets. A) 0% Pyro-SPC. B) 1% Pyro-SPC. C) 10% Pyro-SPC. D) 20% Pyro-SPC. E) 30% Pyro-SPC. F) 40% Pyro-SPC. G) 50% Pyro-SPC. H) The total observed concentrations of microbubble and droplet samples from the CC based on Pyro-SPC content in the shell. All error bars indicate standard deviation. All measurements were performed using a 10 µm aperture which has a size range of 200 nm to 6000 nm. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Representative ultraviolet-visible (UV-Vis) spectroscopy absorbance measurements from 300 to 800 nm of the post-condensed 30% Pyro-lipid droplet sample diluted in PBS and in 1% Triton. Please click here to view a larger version of this figure.
{kind=link}

Figure 6: Representative fluorescence emission from 600 to 750 nm excited at 410 nm. A) Size-selected, pre-condensed 30% Pyro-lipid microbubble sample in PBS and in 1% Triton. B) Post-condensed 30% Pyro-lipid droplet sample in PBS and in 1% Triton. Please click here to view a larger version of this figure.
{kind=link}

Figure 7: Representative ultrasound images of a 37 °C agar flow phantom taken with a pre-clinical 21 MHz linear array transducer in B-mode (see Table of Materials). The left column (Figures A, C, E,& G) shows PBS controls. The right column (Figures B, D, F,& H) shows 20 µL of post-condensed 30% Pyro-lipid droplet sample diluted into 50 mL of 37 °C PBS. Each row represents free-field peak negative pressures, which were estimated from the work done by Sheeran et al.8 The yellow triangles indicate focus depth. Please click here to view a larger version of this figure.
{kind=link}
| Method | Agent | Pyro Shell % | 0 | 1 | 10 | 20 | 30 | 40 | 50 |
| CC | Bubbles | Conc. [/mL] | (2.76 ± 0.28) × 10^10 | (3.04 ± 0.15) × 10^10 | (2.02 ± 0.11) × 10^10 | (1.91 ± 0.22) × 10^10 | (1.47 ± 0.05) × 10^10 | (8.47 ± 0.95) × 10^9 | (9.89 ± 0.15) × 10^9 |
| CC | Bubbles | Peak [nm] | 329 ± 6 | 297 ± 15 | 305 ± 21 | 273 ± 14 | 310 ± 40 | 266 ± 33 | 393 ± 89 |
| CC | Bubbles | Mean [nm] | 609 ± 2 | 603 ± 15 | 635 ± 6 | 690 ± 8 | 812 ± 1 | 935 ± 22 | 950 ± 55 |
| CC | Bubbles | Median [nm] | 450 ± 6 | 421 ± 6 | 414 ± 6 | 432 ± 5 | 490 ± 2 | 596 ± 37 | 695 ± 41 |
| CC | Droplets | Conc. [/mL] | (2.18 ± 0.07) × 10^10 | (2.71 ± 0.13) × 10^10 | (1.75 ± 0.18) × 10^10 | (1.72 ± 0.13) × 10^10 | (1.09 ± 0.01) × 10^10 | (7.36 ± 0.81) × 10^9 | (7.38 ± 0.28) × 10^9 |
| CC | Droplets | Peak [nm] | 292 ± 0 | 297 ± 17 | 261 ± 13 | 280 ± 9 | 268 ± 17 | 287 ± 38 | 390 ± 55 |
| CC | Droplets | Mean [nm] | 353 ± 5 | 350 ± 5 | 347 ± 1 | 347 ± 4 | 397 ± 1 | 399 ± 6 | 400 ± 7 |
| CC | Droplets | Median [nm] | 318 ± 4 | 318 ± 4 | 310 ± 1 | 315 ± 2 | 340 ± 0 | 367 ± 5 | 370 ± 9 |
Table 1: Sizing data statistics of the microbubble and droplet samples with different Pyro-SPC content from Coulter Counter (CC) (n = 3). All errors indicate standard deviation.
Supplementary Information - Lipid Formula Sheet: Please click here to download this File.
Supplementary Information - Other Protocols and Data: Please click here to download this File.
Discussion
After adding all the lipid components together (Steps 1.2 and 1.4.5, Figure 1A), a solution of chloroform and methanol (and water if phosphatidic acid lipids like DSPA are present) was added to ensure the Pyro-lipid and non-Pyro lipid components were fully homogenized (Step 1.5, Figure 1B). To prevent the formation of lipid vesicles with heterogeneous lipid composition, the dissolved lipids were dried and coated onto the interior of the wall of the vial as a thin film (Figure 1C). The coating (Step 1.6) also makes the hydration (Step 2.1 to 2.4) easier as it increases the surface area of the dried film. The drying (Step 1.6, Figure 1C) and vacuuming (Step 1.8, Figure 1D) were done to ensure the chloroform and methanol were fully evaporated as these chemicals can disrupt the formation of microbubbles. While the protocol can be scaled down to make lipid solution volumes as low as 1 mL, larger volumes can reduce vial-to-vial variation. While this may run the risk of degrading the Pyro-SPC while not in use, the storage condition of the lipid solution (Step 2.9 to 2.10) was meant to reduce that risk. The degassing step with the gas exchanger (Step 2.9.2, Figure 1F and Figure 2) serves to eliminate as much oxygen as possible to prevent oxidization. It is not recommended storing lipid solutions containing porphyrin-lipids while atmospheric gases are still dissolved in the solution (Figure 1E).
In step 2.10, the lipid solution is in a serum vial with a pressurized headspace, similar to how the clinically approved ultrasound contrast agent perflutren lipid microspheres are sold (similar to Figure 1F). Internal work has shown stable microbubbles could not be generated via mechanical agitation with the presence of Pyro-lipids if the cap was a soft material like the rubber stopper. Therefore, the lipid solution was transferred to a sample vial with a non-rubber phenolic cap (Steps 4.1 to 4.4, Figure 3A and 3B). When the decafluorobutane gas was flowed into the sample vial (Steps 4.1 to 4.4), the denser decafluorobutane should displace the atmospheric air in the sample vial headspace. Currently, it is unknown why Pyro-lipids are unable to form microbubbles with rubber stoppers. With no Pyro-lipids, stable microbubbles can be made directly in the serum vials with rubber stoppers4,7. Thus, it is recommended using the Gas Exchanger to degas and re-pressurize the serum vial then agitate the serum vial itself for non-Pyro-lipid formulations4,5,6,7 (see "Other Protocols and Data"). The advantage of being able to mechanically agitate in serum vial is the headspace can be pressurized and size-selection can be done by inverting the serum vial upside-down8. In this protocol, the 0% Pyro-lipid formulation was transferred to a sample vial (Steps 4.1 to 4.4) to be consistent with the formulations that did contain Pyro-lipids. Additionally, longer acyl lipid chain lengths result in more stable droplets due to better van der Waals interactions19. The lipid shell composition was chosen based on what was commercially available, 18-acyl chain length for all lipid types. DSPE-PEG5K was incorporated in all the formulation (Step 1.1) as the presence of the polyethylene glycol chains prevents coalescence of structures via repulsive steric forces19. During lipid hydration, the bath sonicator bath was set to 70 °C (Step 2.1) as high enough to fully disperse the 18-acyl chain length lipid film18. For longer acyl chain lengths, higher temperatures will be required.
Higher Pyro-lipid loading would increase the concentration of optically absorbing and fluorescing components, which may be desired for certain applications that benefit from maximized porphyrin loading. However, as the Pyro-lipid content increased, the observable droplet concentration decreased and the diameters increased (Figure 4 and Table 1). This illustrates a trade-off between optical fluorescence and absorbance properties versus droplet concentration and diameter. To researchers that must prioritize small diameters for in vivo accumulation through small leaky vessels or if a high concentration of droplets needs to be injected, increasing Pyro-lipid loading may not be worth the increase in droplet dimeter or decrease in droplet concentration. If high droplet concentrations and/or small droplet diameters are paramount, similarly sized companion diagnostic agents should be considered instead of Pyro-lipids. While 1% Pyro-lipid droplets did not result in a decrease in concentration or increase in size, 1% Pyro-lipid loading may be too low to be reasonably detectable from tissue background fluorescently. However, the flexibility of porphyrin moiety provides multiple options for functionalization which will impart alternative means of quantification more suitable for low-concentration applications. For example, Pyro-lipids can be chelated with copper-64 for positon emission tomography imaging and gamma counting20, or with palladium for trace-metal quantification using mass spectrometry, or with manganese for magnetic resonance imaging14.
While some experiments may only require a small volume of the droplet solution, 1 mL of the lipid solution is needed to fill the 1.85 mL sample vial. Goertz et al. demonstrated that changes to handling, headspace pressure, liquid-to-gas ratio, and even the vial shape can all affect microbubble populations17. Vial temperature during agitation and size-selection can also influence the size distribution. Therefore, for the methods optimized by the end-user, it is critical to be as consistent as possible when making droplets. Unopened droplets may be frozen (-20 °C) and thawed later for future use but this will affect size populations.
The agitation procedure that activates a lipid solution into microbubbles does not produce a morphologically homogeneously population (Step 4.6); rather, the sample is filled with microbubbles, multilamellar vesicles, liposomes, and micelles18,21,22. While microbubble sizes span the micron and nanometer range, the other structures are largely below 800 nm 23. The sizing techniques used do not distinguish between these various structures, and thus the post-agitated microbubble samples (Step 4.6, Figure 3C) and the post-condensed droplet samples (Step 4.14, Figure 3F) must be assumed as mixtures. The ultrasound-insensitive assemblies (multilamellar vesicles, liposomes, and micelles) are likely conserved post-condensation and will not change size as they do not have phase-changeable cores. Since the Coulter Counter cannot distinguish between these different supramolecular assemblies, the shift in population size following condensation should be interpreted with the assumption that some proportion of the nanoscale structures are inconvertible and contribute to the observed population in that size region. Additionally, these structures contribute to the spectroscopic and fluorescent signatures of these samples14. The fluorescence and absorbance signatures of micelles, liposome/vesicles, and droplets are all similar, including their degree of fluorescence quenching14. Thus, it is important to consider that there is a mixture of assemblies in Figures 3C to 3F, Figure 4, the PBS diluted sample in Figure 5, and the PBS diluted sample in Figure 6.
After size-selecting and prior to condensation (Step 4.9), it is possible to eliminate the non-bubble assemblies by centrifuging the microbubble sample to separate the buoyant bubbles from the non-buoyant assemblies as described by Feshitan et al.21 The degree of separation can be controlled by adjusting the spin force and duration. However, experiments of microbubble condensation of such size-isolated samples revealed that using the larger precursor microbubble populations that are selected using size isolation procedures yielded larger droplets (see "Other Protocols and Data" Step S5 for post-spun bubble and droplet sizing). Since an intended application of droplets produced with this protocol is a platform for passive extravasation and accumulation due to their small size compared to microbubbles4,8, droplet populations that are as small as possible were desired. Thus, this protocol used post-agitated microbubble samples that were not size-isolated via centrifugation, even if that meant ultrasound-insensitive micelles, liposomes, and vesicles were present in the final solution. This does imply that quantification procedures for bio-distribution will derive signal for all of the injected structures and are not limited to just the droplets. However, since these similarly-sized structures most likely accumulate via a passive mechanism that is primarily dictated by size, it is not suspected that this should change the main inferences that can be made if this platform is to be utilized in vivo, although all these aspects should be individually considered depending on the context in which the platform may be used. Tests using experimental arms with and without ultrasound can be performed to ensure that it is the ultrasound-sensitive droplets that are responsible for any changes in bio-distribution, as only the perfluorocarbon core assemblies in the solution will respond to ultrasound.
After agitation, the vial was rested for 15 minutes and a partition was observed in the vial (Figure 3C versus 3D). Size-selection via buoyancy is a simple method of eliminating the larger structures/bubbles from an activated microbubble solution8,17. In this case, particles with diameters greater than 5 µm were mostly removed after size-selection (Figure 4). The extent of size-selection can be tuned by controlling the duration of the size-selection17. Sheeran et al. has shown that not size-selecting can result in generated microbubbles that occlude vasculature8.
Perfluorocarbons have the advantage of being biologically inert7. While decafluorobutane's boiling point is -1.7 °C, above body temperature, the droplets do not immediately vaporize when exposed to 37 °C (Figure 7B). As the droplets are meta-stable at 37 °C, additional acoustic energy is needed to vaporize the droplets to microbubbles7,9. Poproski et al. has demonstrated porphyrin droplets condensed via pressurization22. This is a viable and even essential method when using less dense perfluorocarbons but high pressures may destroy some bubbles in the process. Octafluoropropane (C3F8) has a boiling point of -36.7 °C, so both cooling and pressurization is needed for droplet condensation. However, the lighter perfluorocarbon leads to less stable droplets. Dodecafluoropentane (C5F12) can lead to more stable droplets with a boiling point of 28 °C. However, it is a liquid at room temperature and will need stronger acoustic energies to vaporize. Thus, choice of the containing gas of the ultrasound contrast agent should consider the conditions of its intended biological application in addition to the parameters of its fabrication. In this protocol, the isopropanol bath for condensation was set to -15 to -17 °C (Step 4.7.1 and Step 4.13) while other protocols used -10 °C 5,6. Even with a common decafluorobutane core, the condensation temperature may vary depending on excipient composition, total lipid concentration, and lipid shell composition. If using other formulations, optimization may be required to ensure proper droplet condensation without causing the solution to freeze.
As the droplets are smaller and more stable than their microbubble precursor7, they can take better advantage of passive accumulation mechanisms to extravasate into certain tissues of interest, such as the enhanced permeability and retention effect of certain tumor types4,24. With fluorescent, optically absorbent, and acoustic methods of detection14, it is possible to use a single formulation to quantify uptake. Additionally, this platform can be used to investigate whether the acoustic vaporization of droplets can improve delivered agent fraction beyond passive levels16. To quantify agent bio-distribution in tissues and organs of interest after injection, a known amount of Pyro-lipid droplets should be injected into the animal, ultrasound may or may not be applied depending on the control set, the animal should be sacrificed a pre-specified time-point, and the organs should be removed and weighed. The organs should be homogenized, filtered, diluted in surfactant (detergent) to decellularize the tissue, and quantified with fluorescence or UV-Vis spectroscopy to obtain injected dose percentages per organ mass based on the Pyro signals. For Step 5.4.5 (Figure 5) and Step 5.5.5 (Figure 6), Triton X-100 surfactant (detergent) was used to disrupt the samples as it is non-fluorescent at 410 nm and its absorbance wavelengths do not overlap with Pyro's.
Microbubbles were not characterized with UV-Vis absorbance. As the UV-Vis spectroscope's laser source is parallel with the detector, any large bubbles could scatter light away from the detector, making them appear more optically absorbent14. Unlike the UV-Vis spectrophotometer, the fluorescence spectrophotometer's detector is/should be perpendicular to the laser source to prevent the source from interfering with the detector. UV-Vis was used to quantify the absorbance of the intact and disrupted droplet samples (Step 5.4, Figure 5). 300 to 800 nm was chosen as the absorbance wavelengths as the two main absorbance bands of pyro-lipid, the Soret band (340 to 500 nm) and the Q-band (640 to 730 nm), fall within this range14. When assembled into a droplet (or other supramolecular structures), the Q-band peak of Pyro-lipid is red-shifted from 671 nm to 700 nm (Figure 5). When this supramolecular structure is disrupted by a surfactant like Triton, the peak shifts back to 671 nm14,15. Based on this shift, it is possible to tell whether the Pyro-lipids are in an assembled state or in a disrupted state. The ratio of the two peaks can be used to estimate the decay of the assemblies over time.
For the fluorescence measurements (Step 5.5, Figure 6), an excitation wavelength of 410 nm was chosen as it corresponds to the Soret band peak for unassembled Pyro-lipid14. An emission wavelength range from 600 to 800 nm was selected as the peaks of the intact assemblies in PBS and disrupted Pyro-lipids in Triton are contained within this range. The shift and increase in fluorescence (Figure 6) between the intact (704 nm in PBS) and disrupted (674 nm in Triton) samples occurred because of structure-induced quenching. In the assembled form, the Pyro-lipid molecules were packed closely together so generated photons were absorbed by nearby Pyro-lipid molecules. This is evident in the intact versus disrupted quenching efficiency. Thus, it is necessary to dilute samples in surfactant (detergent) like 1% Triton X-100 to relieve quenching and maximize signal for bio-distribution quantification14.
For simplicity, the same linear array ultrasound transducer was used to both vaporize and image (Steps 6.5 and 6.7, Figure 7). This ultrasound transducer (Table of Materials) was capable of reaching the necessary peak negative pressures needed to vaporize droplets8. Filling a tank with deionized water from a tap generates gases that become dissolved in the water (Step 6.1). To minimize interference from the dissolved gases in the water with vaporization and imaging, the water was rested for 24 h in the tank to allow the gases in the water to equilibrate with the atmosphere (Step 6.1). Alternatively, the deionized water can be degassed with a sufficiently sized, sealable container connected to a sufficiently powerful vacuum. The ultrasound images demonstrated the microbubbles were successfully condensed as the droplets were unobservable/non-echogenic at low pressures (Figure 7B). It was only at higher output pressures that the droplets vaporized into observable, echogenic microbubbles (Figure 7D, 7F, 7H). While the post-condensed droplet sample contains micelles and liposomes/vesicles, these assemblies are non-echogenic and only droplets can vaporize into echogenic microbubbles. A PBS control was flowed through the phantom to establish baseline images (Figures 7A, 7C, 7E, 7G). As the pressure increased in the PBS, no contrast was generated. This indicated that the high pressures from the transducer could not produce spontaneous cavitation in a water-based medium alone, and thus all other generated contrast could be attributed to the ultrasound contrast agent employed. If the output pressure is too high, generated microbubbles can be destroyed. By incrementally increasing the pressure and observing the generated contrast, the optimal pressure can be found8. The circulation half-life of the droplets can be determined in a similar way by vaporizing the droplets are certain time intervals and observing the contrast generated over time7.
In summary, multi-modal phase-change droplets with varying Pyro-lipid content were created with the condensation method. Sizing showed that there was a trade-off between Pyro-lipid loading and microbubble/droplet concentration. Characterizations showed that there were differences in intact and disrupted forms in both absorbance and fluorescence. Ultrasound imaging showed droplets were non-echogenic at 37 °C and were vaporizable into echogenic microbubbles at sufficient pressures. Characterizations also showed the potential for Pyro-lipid droplets to replace companion diagnostic agents for droplet bio-distribution or accumulation tests. Future work will investigate in-solution vaporization thresholds, in-solution stability, and in vivo circulation durations in nude mice.
Disclosures
The authors have nothing to disclose.
Acknowledgements
The authors would like to thank Dr. Brandon Helfield for helping build the gas exchanger and Dr. Miffy Hok Yan Cheng for technical discussions. The authors would like to thank the following funding sources: Ontario Graduate Scholarship, Canadian Institutes of Health Research, Terry Fox Research Institute, and Princess Margaret Cancer Foundation.
Materials
| Name | Company | Catalog Number | Comments |
| 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-5000] (ammonium salt) | Avanti Polar Lipids | 880220 | Also known as "DSPE-PEG5K" |
| 1-stearoyl-2-hydroxy-sn-glycero-3-phosphocholine | Avanti Polar Lipids | 855775 | Also known as "DSPC" |
| Aluminum Foil | Any brand | ||
| Aluminum Seals, Tear-Off | VWR | 16171-840 | Standard Aluminum, 13 mm outer diameter |
| Bath Sonicator | Any brand | Capable of sonicating and heating up to 70 °C, | |
| Bio-Stor Screw Cap Vials | National Scientific | BS20NABP | Plastic, 2 mL Skirted, with O-ring |
| Borosilicate glass clear serum vials | VWR | 16171-285 | 3 mL, 7 mm inner mouth diameter, 13 mm outer mouth diameter |
| Borosilicate Glass Sample Vial with Phenolic Screw Cap | VWR | 66011-020 | 1.85 mL, Short Form Style, 12 mm outer diameter, 35 mm height, 8-425 cap size |
| Borosilicate Glass Vial with Screw-On cap | Any brand | Sizes will depend on desired volumes | |
| Chloroform | Any brand | ||
| Coulter Counter Elctrolyte Diluent | Any brand | Compatible with Coulter Counter | |
| Decafluorobutane (C4F10) | FluoroMed | 355-25-9 | |
| Deionized Water | Any brand | ||
| Dry Ice (Carbon Dioxide) | Any brand | ||
| Dynamic Light Scattering (DLS) Particle Analyzer | Any brand | Capable of temperature control | |
| E-Z Crimper, 13 mm | Wheaton | W225302 | 13 mm Standard Aluminum Seals |
| E-Z Decapper, 13 mm | Wheaton | W225352 | 13 mm Standard Aluminum Seals |
| Fluorescent Spectrophotometer | Any brand | Capable of 400 to 600 excitation and 300 to 800 nm emission detection, detector perpendicular to laser source | |
| Fluorescent Spectrophotometer Compatible Cuvette | Any brand | Can hold at least 2 mL, capable of 300 to 800 nm, all four sides are optical windows | |
| Gas Exchanger | Made in-house | Refer to Supplementary Information - "Other Protocols and Data" for assembly instructions. | |
| Glass syringes | Any brand | Sizes will depend on desired volumes | |
| GLWR Custom Aperture Tube 10 um | Beckman Coulter | B42812 | 10 µm aperture, compatible with Beckman Coulter MultiSizer 4e |
| Glycerol | Any brand | ||
| Insulated Styrofaom containers with lids | Any brand | ||
| Isopropanol | Any brand | ||
| Lyophilization-Style Rubber Stoppers | VWR | 71000-060 | 7 mm inner mouth diameter, 13 mm outer mouth diameter, 2-leg, Chlorobutyl |
| Membrane Diaphram Vacuum Pump | Sartorius Stedim | 16694-1-60-06 | Adjustable pressure |
| Metal Tongs | Any brand | ||
| Methanol | Any brand | ||
| MS250 21 MHz Linear Array Ultrasound Transducer | VisualSonics | 21 MHz, Capable of B-mode and non-linear imaging. | |
| MultiSizer 4e | Beckman Coulter | Capable of sizing from 0.2µm to 6 µm | |
| Nalgene Rapid-Flow Sterile Single Use Vacuum Filter Units | Thermo Scientific | 567-0010 | Polyethersulfone (PES) membrane, 0.1μm pore size, 1000 mL volume. As Isoton II is non-sterile, can use Filter units multiple times |
| Needles, Conventional | BD | 305176 | 20 gauge, 1.5 inch length |
| Nitrogen Gas | Any brand | Make sure there are regulator valves and tubes to direct the flow. Setup will be dependend on brand and source. | |
| Parafilm | Any brand | Called "wax film" in the protocol. | |
| Phosphate Buffered Saline (PBS) | Any brand | 1X, 7.4 pH | |
| Pipette | Any brand | Sizes will depend on desired volumes | |
| Pipette Tips | Any brand | Sizes will depend on desired volumes | |
| Plastic Syringes | Any brand | 1 mL, 3 mL, and 30 mL. With Luer Lock connections | |
| Polyethersulfone (PES) Membrane Filter | Any brand | 0.2 µm pore size | |
| Propylene Glycol | Any brand | ||
| Pyropheophorbide conjugated 1-stearoyl-2-hydroxy-sn-glycero-3-phosphocholine | Made in-house | Also known as "Pyro-SPC". Refer to "Supplementary Information - Other Protocols and Data" for synthesis. | |
| Thermometer | Any brand | (-20 to 100 °C) | |
| Triton X-100 | Any brand | Also known as "2-[4-(2,4,4-trimethylpentan-2-yl)phenoxy]ethanol" | |
| Ultrapure Water | Any brand | Type 1 Purity | |
| Ultraviolet–Visible (UV-Vis) Spectrophotometer | Any brand | Capable of absorbance from 300 to 800 nm, at least 0.5 nm resolution | |
| Ultraviolet–Visible (UV-Vis) Spectrophotometer Compatible Cuvette, 1 cm Path Length | Any brand | Can hold at least 2 mL, capable of 300 to 800 nm | |
| Vacuum Desiccator | Any brand | ||
| Vevo 2100 Ultrasound Imaging Platform | VisualSonics | Pre-clinical ultrasound imaging system | |
| Vialmix | Bristol-Myers-Squibb | Called "mechanical agitator" in the protocol. Agitates for 45 s. | |
| Vortex Mixer | Any brand |
References
- Gramiak, R., Shah, P. M. Echocardiography of the aortic root. Investigative Radiology. 3 (5), 356-366 (1968).
- Ferrara, K., Pollard, R., Borden, M. Ultrasound microbubble contrast agents: fundamentals and application to gene and drug delivery. Annual Review of Biomedical Engineering. 9, 415-447 (2007).
- Bing, K. F., Howles, G. P., Qi, Y., Palmeri, M. L., Nightingale, K. R. Blood-brain barrier (BBB) disruption using a diagnostic ultrasound scanner and Definity in mice. Ultrasound in Medicine and Biology. 35 (8), 1298-1308 (2009).
- Helfield, B. L., et al. Investigating the accumulation of submicron phase-change droplets in tumors. Ultrasound in Medicine and Biology. 49 (10), 2891 (2020).
- Sheeran, P. S., Luois, S. H., Mullin, L. B., Matsunaga, T. O., Dayton, P. A. Design of ultrasonically-activatable nanoparticles using low boiling point perfluorocarbons. Biomaterials. 33 (11), 3262-3296 (2012).
- Sheeran, P. S., et al. Methods of generating submicrometer phase-shift perfluorocarbon droplets for applications in medical ultrasonography. IEEE Transactions on Ultrasonics, Ferroelectrics, and Frequency Control. 64 (1), 252-263 (2016).
- Yoo, K., et al. Impact of encapsulation on in vitro and in vivo performance of volatile nanoscale phase-shift perfluorocarbon droplets. Ultrasound in Medicine and Biology. 44 (8), 1836-1852 (2018).
- Sheeran, P. S., et al. Image-guided ultrasound characterization of volatile sub-micron phase-shift droplets in the 20-40 MHz frequency range. Ultrasound in Medicine and Biology. 42 (3), 795-807 (2016).
- Sheeran, P. S., et al. More than bubbles: creating phase-shift droplets from commercially available ultrasound contrast agents. Ultrasound in Medicine and Biology. 43 (2), 531-540 (2017).
- Chen, C. C., et al. Targeted drug delivery with focused ultrasound-induced blood-brain barrier opening using acoustically-activated nanodroplets. Journal of Controlled Release. 172 (3), 795-804 (2013).
- Wu, S. Y., et al. Focused ultrasound-facilitated brain drug delivery using optimized nanodroplets. Physics in Medicine and Biology. 63 (3), 035002 (2018).
- Lee, J. Y., et al. Ultrasound-enhanced siRNA delivery using magnetic nanoparticle-loaded chitosan-deoxycholic acid nanodroplets. Advanced Healthcare Materials. 6 (8), 1601246 (2017).
- Cao, Y., et al. Drug release from phase-changeable nanodroplets triggered by low-intensity focused ultrasound. Theranostics. 8 (5), 1327-1339 (2018).
- Huynh, E., Jin, C. S., Wilson, B. C., Zheng, G. Aggregate enhanced trimodal porphyrin shell microbubbles for ultrasound, photoacoustic, and fluorescence imaging. Bioconjugate Chemistry. 25 (4), 796-801 (2014).
- Zheng, G., et al. Low-density lipoprotein reconstituted by pyropheophorbide cholesteryl oleate as target-specific photosensitizer. Bioconjugate Chemistry. 13 (3), 392-396 (2002).
- Dhaliwal, A., Zheng, G. Improving accessibility of EPR-insensitive tumor phenotypes using EPR-adaptive strategies: Designing a new perspective in nanomedicine delivery. Theranostics. 9 (26), 8091-8108 (2019).
- Goertz, D. E., de Jong, N., vander Steen, A. F. Attenuation and size distribution measurements of Definity and manipulated Definity populations. Ultrasound in Medicine and Biology. 33 (9), 1376 (2007).
- Pellow, C., Acconcia, C., Zheng, G., Goertz, D. E. Threshold-dependent nonlinear scattering from porphyrin nanobubbles for vascular and extravascular applications. Physics in Medicine and Biology. 63 (21), 215001 (2018).
- Kwan, J. J., Borden, M. A. Lipid monolayer collapse and microbubble stability. Advances in Colloid and Interface Science. 183, 82-99 (2012).
- Overchuk, M., et al. Subtherapeutic Photodynamic Treatment Facilitates Tumor Nanomedicine Delivery and Overcomes Desmoplasia. Nano Letters. 21 (1), 344-352 (2021).
- Feshitan, J., Chen, C. C., Kwan, J. J., Borden, M. A. Microbubble size isolation by differential centrifugation. Journal of Colloid and Interface Science. 329 (2), 316-324 (2009).
- Paproski, R. J., et al. Porphyrin nanodroplets: Sub-micrometer ultrasound and photoacoustic contrast imaging agents. Small. 12 (3), 371-380 (2016).
- Periyasamy, P. C., Leijten, J. C. H., Dijkstra, P. J., Karperien, M., Post, J. N. Nanomaterials for the local and targeted delivery of osteoarthritis drugs. Journal of Nanomaterials. 2021, 1-13 (2012).
- Matsumura, Y., Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Research. 46 (12), 6387-6392 (1986).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved