
Method Article
Use of In Vivo Imaging to Screen for Morphogenesis Phenotypes in Candida albicans Mutant Strains During Active Infection in a Mammalian Host
In This Article
Summary
This manuscript describes a method for screening moderately sized Candida albicans mutant libraries for morphogenesis phenotypes during active infection in a mammalian host using non-invasive confocal microscopy.
Abstract
Candida albicans is an important human pathogen. Its ability to switch between morphologic forms is central to its pathogenesis; these morphologic changes are regulated by a complex signaling network controlled in response to environmental stimuli. These regulatory components have been highly studied, but almost all studies use a variety of in vitro stimuli to trigger filamentation. To determine how morphogenesis is regulated during the pathogenesis process, we developed an in vivo microscopy system to obtain high spatial resolution images of organisms undergoing hyphal formation within the mammalian host. The protocol presented here describes the use of this system to screen small collections of C. albicans mutant strains, allowing us to identify key regulators of morphogenesis as it occurs at the site of infection. Representative results are presented, demonstrating that some regulators of morphogenesis, such as the transcriptional regulator Efg1, have consistent phenotypes in vitro and in vivo, whereas other regulators, such as adenyl cyclase (Cyr1), have significantly different phenotypes in vivo compared to in vitro.
Introduction
Candida albicans is a common human fungal pathogen, causing mucocutaneous disease, disseminated disease, and localized tissue infections1. A key feature of C. albicans physiology is its complex polymorphic growth, which is linked to its role as both a commensal and a pathogen2,3,4. Under rich nutrient conditions in vitro at 30 °C, it typically grows as an ovoid budding yeast. A variety of environmental triggers, including nutrient deprivation, pH changes, growth at 37 °C, exposure to serum, and growth when embedded in agar, result in the transition to a polarized growth pattern, resulting in the formation of true hyphae and/or pseudohyphae5. The initiation of polarized growth and the resulting growth of filamentous organisms is referred to as morphogenesis.
Because of the importance of morphogenesis in the organism's virulence, the regulation of hyphal formation has been extensively studied6,7. There is a complex network of signaling pathways and transcriptional regulation that triggers morphogenesis. Despite the relationship of C. albicans morphogenesis with pathogenesis, most studies investigating morphogenesis have used in vitro stimuli to trigger hyphal formation. It is becoming increasingly clear that the various in vitro models of filamentation are not identical in terms of the individual regulatory pathways stimulated. Furthermore, no in vitro growth conditions correspond tightly with the complex environment of the host. Given the importance of C. albicans as a human pathogen, the goal of this protocol is to investigate its morphogenesis during active infection in a mammalian host using a system with moderate throughput, thus allowing an investigator to screen C. albicans mutant libraries.
To facilitate these investigations, an in vivo imaging system was developed that allows us to obtain high spatial resolution images of C. albicans cells during infection of the pinna of an anesthetized mouse using an inverted confocal microscope8,9,10. Because the skin of the pinna is quite thin, these images can be obtained without the need for tissue dissection. Thus, quantitative phenotype data can be measured at the site of active infections within the host tissue. The protocol described here involves the transformation of a reference strain and one or more mutant strains with different fluorescent protein expression cassettes11,12. The fluorescent protein-expressing strains are then mixed and co-injected intradermally. After the infection is established, confocal imaging is used to quantify both the frequency of filamentation and the length of the formed filaments. The data obtained from the mutant strains is normalized to that obtained from the reference strain, which is present in the same tissue region, thus providing an internal control. This system has allowed us to successfully screen several series of C. albicans mutant strains, many of which have morphogenesis defects in vitro9,10. Many of these strains readily filament in vivo, highlighting the importance of in vivo models for the investigation of morphogenesis.
Protocol
The studies in this protocol were approved by the University of Iowa Institutional Animal Care and Use Committee (IACUC). Refer to the CDC guidelines for equipment and procedures for working with BSL2 organisms13.
1. Preparing Candida albicans strains
- Identify an appropriate reference strain for use as a positive control. Ensure that this strain closely matches the experimental strains in terms of its lineage and genetic manipulations.
NOTE: For the representative experiments presented here, the mutants were created from the strains described in Homann, et al.14, which were constructed from SN152. These mutants are Arg-. Therefore, the reference strain selected was SN250, which was also created from SN152 and is also Arg-. Nutritional stressors are critical in the regulation of polarized growth in the yeast Saccharomyces cerevisiae15; they have also been implicated in filamentation in C. albicans and other fungi16,17,18. Therefore, the reference strain should be matched for auxotrophies with the experimental strains whenever possible to avoid potential confounding effects from nutritional stress. - Choose fluorescent protein expression constructs. When screening a variety of experimental strains, create a reference strain that expresses one fluorescent protein and use other fluorescent proteins to mark the mutant strains.
NOTE: The representative data presented here uses NEON for the reference strain and iRFP for the mutant strains. Any fluorescent protein can be used if it is highly expressed, relatively bright, and able to be excited/detected by the microscope being used. Control experiments comparing reference strains that express different fluorescent proteins have not demonstrated any impact of fluorescent protein expression on morphogenesis. - Transform the strains with fluorescent protein expression constructs.
NOTE: Many institutions require the use of Biological Safety Level 2 precautions for work with C. albicans. All work should follow local safety regulations. Regardless of local regulations, investigators working with C. albicans must be trained in the safe handling of the organism.- Transform the reference and experimental strains using standard lithium acetate protocols19.
NOTE: The experiments described here use the pENO1-NEON-NATR and pENO1-iRFP-NATR plasmids, generously provided by Dr. Robert Wheeler11,12. Plasmids were linearized using NotI9. - Select the transformants based on growth on nourseothricin or other relevant selection medium.
- Identify successful transformants. Pick a small dab of cells from each colony using a toothpick and then mix them into a 2.5 µL droplet of water on a microscope slide. Apply a coverslip and examine at 10x-40x magnification. Examine with a confocal imaging system (used for the rest of the protocol) or any standard wide-field fluorescence microscope. Appropriate transformants will have a bright signal with appropriate excitation and emission wavelengths.
NOTE: For the representative results, NEON expressing strains were visualized using an upright fluorescence microscope with a long pass filter set with a 472/30 nm bandpass excitation filter, a 520/35 nm bandpass emission filter, and a 495 nm single-edge dichroic beam splitter. Because iRFP is not visible by eye, iRFP expressing strains were visualized using the confocal microscopy system used for in vivo imaging, using a 638 nm laser for excitation and detecting emission light from 655-755 nm.- Alternatively, evaluate macroscopic colony fluorescence using fluorescence stereomicroscopy, hand-held fluorescence excitation systems, or fluorescence detection systems typically used for gels and Western blots.
- Create freezer stocks of selected transformants.
- Transform the reference and experimental strains using standard lithium acetate protocols19.
- Inoculate YPD (yeast extract peptone dextrose) solid media with a fluorescent protein-expressing reference and experimental strains from freezer stocks 3 days prior to injection, using a toothpick to transfer a dab of organisms from the freezer stock to the YPD solid media. Incubate at 30 °C for 2 days.
- For each strain, inoculate a flask containing 25 mL of YPD with C. albicans cells taken from several colonies 1 day prior to injection. Do this by using a toothpick to transfer a dab of organisms from one colony into the YPD; repeat several times to obtain cells from several different colonies. Incubate overnight at 30 °C in an orbital shaker incubator at 175 rpm.
NOTE: It is important to use multiple colonies as a source for the inoculum because C. albicans has a high frequency of spontaneous genetic alterations. Using multiple colonies when starting the inoculum culture minimizes the chance that all the organisms in the inoculum arise from a parent with significant spontaneous alterations. - On the day of injection:
- Centrifuge 1 mL of the culture for 2 min at 500 x g.
- Wash the culture three times with 1 mL of sterile Dulbecco's phosphate-buffered saline (dPBS). After the final wash, resuspend the pellet in 1 mL of sterile dPBS.
- Dilute a sample of the washed culture at 1:100 and count using a hemocytometer.
- Adjust the density of the washed culture to 1 x 108 organisms per mL using dPBS.
- For each set of strains to be injected, create the inoculum by mixing equal volumes of the reference strain and experimental strain(s). This maintains the density of the inoculum at 1 x 108 organisms per mL.
NOTE: The number of strains that can be evaluated per ear is limited by the ability of the microscopy system used to clearly discriminate the signal from each fluorescent protein. - Once the inoculum is prepared, proceed directly to the animal injections. Do not store the inoculum prior to use.
2. Animal preparation
- Obtain approval from the local Institutional Animal Care and Use Committee or the relevant local governing body.
- Obtain 6-12-week-old mice from a vendor or breeding program. House mice in the facility in which they will live throughout the experiment for at least 1 week prior to inoculation.
NOTE: For the representative results, 6-week-old female DBA2/N mice were used. - Feed animals chlorophyll-free chow for at least 7 days prior to inoculation.
3. Hair removal and inoculation
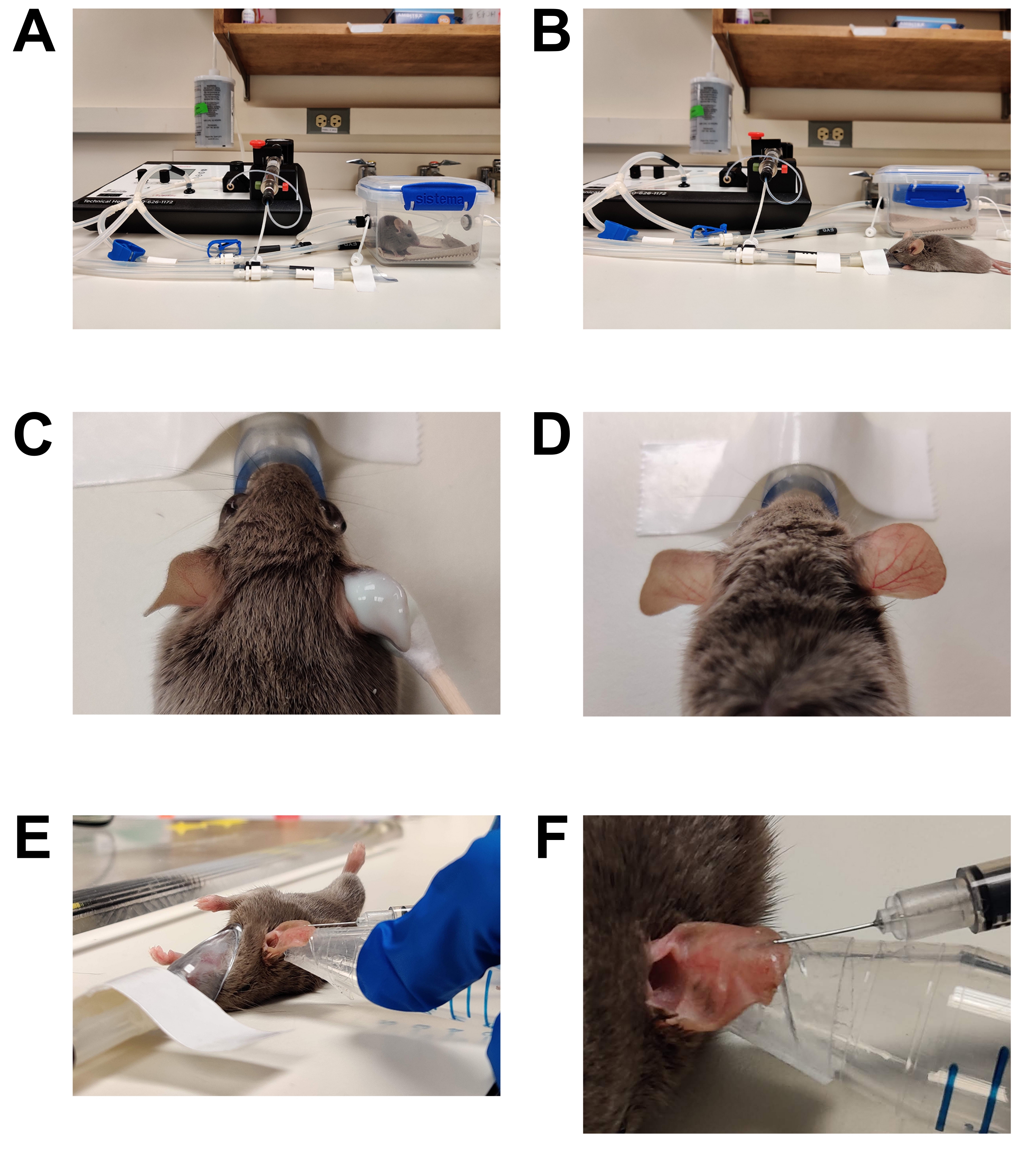
- Induce a surgical plane of anesthesia (Figure 1).
CAUTION: Inhaled anesthetic agents are hazardous materials and can cause eye or skin irritation as well as nervous system toxicity. All institutional policies and procedures and general laboratory safety practices for the use of inhaled anesthetics must be followed. Only individuals trained in the use of inhaled anesthetics may perform these steps. Standard practices include wearing gloves, a laboratory coat, eye protection, use of an anesthesia scavenger system, and careful recordkeeping as per institutional guidelines.- Place a mouse in a pre-warmed anesthesia induction chamber. Keep the animal in a warm environment throughout general anesthesia. Accomplish this using warming pads designed for this purpose and a warmed microscope stage. Do not use an over-the-counter heating pad as it can overheat and cause burns.
- Provide 2%-3% isoflurane to the induction chamber until the mouse has lost its righting reflex and respirations are slow and steady. Purge the induction chamber of the anesthesia and transfer the animal to a non-rebreather nose cone providing 1%-2% isoflurane.
- Validate the plane of anesthesia using the toe pinch reflex or other verification mechanisms. Throughout the experiment, monitor the animal's respiratory pattern and anesthetic plane, and adjust the anesthetic concentration as needed.
- Apply eye lubricant to prevent corneal desiccation.
- Hair removal (Figure 1C,D)
- Apply an over-the-counter depilatory cream liberally to the inner and outer surface of both ears using a cotton-tipped swab.
- After 2-3 min (or as per manufacturer's directions), gently wipe the ear with a dry gauze pad to remove all cream and hair. Wipe twice more with gauze pads saturated in sterile water to fully remove depilatory cream residue. Failure to remove all the depilatory cream will result in skin irritation/ inflammation.
- Injection (Figure 1E, F)
- Wipe the surface of the ear to be injected with a gauze pad saturated in 70% ethanol and allow to air dry.
- Mix the inoculum well by inverting multiple times or vortexing.
- Draw up 20-30 µL of inoculum into an insulin syringe. Holding the needle pointing up, tap the syringe gently to ensure that any air in the barrel is at the top. Carefully eject air and excess inoculum back into the inoculum tube or a waste tube so that the plunger is at the 10 µL mark.
- Wearing a thimble on a finger or thumb of the non-dominant hand, stabilize the ear by wrapping it across the thimble. Alternatively, apply over-the-counter double-sided skin tape (fashion tape) to a small conical or round-bottomed plastic tube and drape the ear across the tape. Take care not to dislodge the anesthesia nose cone while doing this. It can be helpful to tape the nosecone to the work surface.
NOTE: Either the inner or outer side of the ear can be injected based on the physical comfort of the investigator. For microscopy, be sure to place the mouse so that the side of the ear injected is facing toward the objective lens. - Keeping the syringe needle almost completely parallel to the skin and avoiding any large veins, insert the tip of the needle into the outermost layer of the skin until the bevel is just covered.
- Slowly inject the inoculum intradermally. A good intradermal injection will raise a small bubble in the skin. Keep the needle in place for 15-20 s prior to removing it from the ear to minimize leakage.
- If the lower side of the animal's ear becomes moist, the needle was too deep and passed through the ear entirely. In that case, repeat the injection in a different area of the ear.
- Repeat the process using the animal's other ear. This can be done with the same C. albicans strains for a replicate or with a different set of C. albicans strains.
- If not imaging immediately, place the animal in a warmed recovery chamber. Observe the animal until it has recovered from anesthesia and then return it to its housing cage.
- Following institutional protocols, clearly mark the cage with biohazard labels and indicate that animals in the cage are infected with Candida albicans.
- Complete all required recordkeeping related to animal anesthesia and any other required institutional practices.
- House the animal in animal facility conditions using Animal Biological Safety Level 2 precautions.
4. Quantify in vitro morphogenesis for comparison to in vivo results
- Using the same washed cultures that were used to prepare the inoculum, create a 1:50 dilution of organisms in RPMI1640 + 10% heat-inactivated fetal bovine serum and incubate at 37 °C with tumbling for 4 h. Alternatively, other media which stimulate morphogenesis in vitro can be used.
- Centrifuge the sample for 5 min at 500 x g and resuspend in 0.5 mL of dPBS.
- Dilute the sample at a ratio of 1:10, place 2.5 µL of the diluted sample on a microscope slide, and cover it with a coverslip.
- Examine the sample using a fluorescence microscope. Count at least 100 cells, recording the number of yeast and filamentous cells for each strain. In the representative results presented here, a filamentous cell is defined as any cell with a length of more than twice the length of the mother cell.
NOTE: Quantification of in vitro morphogenesis can be done on the same day as inoculation of the animals. It is possible to initiate the in vitro morphogenesis assay while preparing the inoculum for injection and perform the inoculation of the animals during the 4 h incubation period. If all animal procedures are completed before the end of the 4 h incubation period, proceed directly to examination of the in vitro stimulated cells. Alternatively, cells can be resuspended in 3.7% formaldehyde in dPBS (in step 4.2) and stored at 4 °C for several days. The fixed organisms can then be quantified as time allows. Inoculation of the animals must not be delayed for the in vitro quantification assay.
5. Preparation for in vivo imaging
- Prepare the microscope (Figure 2A).
- Turn on all the microscopy equipment and start the imaging software.
- If available, load any pre-determined imaging settings.
- Activate the lasers and detectors needed to detect the fluorescent proteins being used.
- Turn on the heated microscope stage and allow it to warm to 37 °C.
NOTE: It is possible to use a microscope with a fully enclosed environmental chamber rather than a heated stage. - Set a z-axis reference point with the plane of focus is at the top surface of the coverslip. This can be done by taping a coverslip in place over the stage opening and putting a droplet of water on the cover slip. Use a lower magnification (10x) “dry” objective to focus on the edge of the water droplet and then set the z-axis reference point. Use a piece of toweling to absorb the water from the coverslip before removing it from the stage.
- Rotate the very long working distance objective into place and place a droplet of immersion fluid on the lens. Lower the lens to avoid possible damage when placing the coverslip.
- Place a #1.5 coverslip over the stage opening and tape it in place. Ensure that the tape fully covers all edges of the coverslip to prevent any fluid from wicking below the coverslip into the microscope. Raise the objective to the z-axis reference point so that the immersion fluid is in contact with both the objective lens and the coverslip.
- Prepare multiple pieces of pressure-sensitive tape so they are ready to use when positioning the nosecone and the mouse.
- Turn on all the microscopy equipment and start the imaging software.
- Induce general anesthesia in the mouse to be imaged as above (step 3.1).
- Position the mouse (Figure 2B-D).
- Secure an anesthesia nose cone on the microscope stage with the nosecone positioned so that it will cover the animal's nose fully when the animal is in position for imaging. This can be accomplished more easily with two investigators. Have the first investigator place the anesthetized mouse's nose into the nosecone and move the mouse into position for imaging while continuing to hold the nosecone over the animal's nose. Have the other investigator tape the nosecone and tubing into place so it sits stably over the mouse's nose. Once the nosecone is taped in place, leave it there for the remainder of the imaging session.
- After establishing the best place to place the nosecone, note its position. For subsequent imaging sessions, the nosecone can be secured to the stage prior to bringing an animal to the microscope.
- Place a drop of sterile water on the coverslip above the objective lens.
- Position the mouse on the microscope stage. Ensure the ear is on top of the water droplet and flat against the coverslip.
- Use a second (top) coverslip to flatten the ear.
- Place the edge of the coverslip parallel to the mouse's body, with the edge touching the mouse where the ear meets the head.
- Lower the free edge of the coverslip to the microscope stage with a hinge motion. As the coverslip comes against the microscope stage, it will flatten out the ear. Take care not to create folds or ridges in the ear.
- Tape the top coverslip in place securely so that it holds just enough pressure to keep the ear flat. Take care not to catch the mouse's hair or whiskers in the tape.
- Unless a microscope with an environmental chamber is used, loosely cover the mouse's body with a sterile drape to maintain a normothermic environment.
- Identify a field of interest.
- Make sure that the objective is at the z-axis reference point.
- Using white light/wide-field imaging, adjust the focus plane into the ear tissue. A good strategy is to focus on the blood vessels-if one can see red blood cells moving in the blood vessels, the plane of focus is within the tissue.
- If the microscope is equipped with wide-field fluorescence capability, use it to identify a field of interest. If not, use confocal imaging. In general, using wide-field microscopy to identify areas of interest is faster and requires less irradiation of the ear tissue.
- Using a filter cube for detection of the fluorescent protein expressed in the reference strain, identify a field of view that has a fluorescent signal from the reference strain. Bear in mind that out-of-focus light will likely prevent the ability to focus on individual organisms. The goal of this step is to identify an area of interest for confocal imaging.
- Change to a filter cube that will detect the fluorescent protein expressed by the experimental strain(s) and verify their presence in the selected field of view.
6. Imaging
- Determine the settings.
- While the imaging software is in live confocal mode, examine the field of interest as the focal plane is moved through the z-axis. Choose a z-axis plane with a strong signal from all the fluorescent proteins being used.
- Adjust the laser power and/or imaging speed to obtain a strong enough signal such that morphology can be determined for all cells in the field of view. To avoid tissue damage, use the lowest possible laser power.
NOTE: As with all imaging, there is a balance between laser power, acquisition speed, and resolution. Identify settings that clearly identify organism morphology while balancing speed and laser power to minimize irradiation of the ear tissue. Because imaging is occurring through the outer dermis, higher laser power is required for excitation than is typically necessary for confocal imaging of traditional samples mounted on slides. Fortunately, the level of spatial resolution required for the analysis of morphology is not extreme. Thus, obtaining images with a sufficient signal to determine organism morphology without causing tissue damage is readily achievable. - Once these parameters are established, use these throughout the imaging session to serve as a starting point for subsequent imaging sessions. It is thus helpful to save the imaging settings.
NOTE: Individual areas of infection may be shallower or deeper within the tissue. Deeper areas may require an increase in laser power. Because this assay relies on the spatial distribution of signal rather than its intensity, it is acceptable to change image settings between fields as needed.
- Obtain the images.
- Choose a field of view that has clear filament formation in the reference strain and where most organisms are spread out enough that their morphology can be determined.
- Set the top and bottom planes of focus for a z-stack. It is not necessary to cover the entire depth of the infected area, but bear in mind that organisms at the top or bottom of the imaged volume are typically excluded from the analysis.
- Acquire z-stack images, pseudo color each channel to distinguish each strain, and overlay the channels. Save the images.
- Repeat for other fields of view. Morphogenesis can vary from location to location; therefore, acquiring and analyzing at least three fields from each ear is important.
7. Manual two-dimensional analysis: Frequency of filamentation
- Use imaging software to perform a maximum projection of the z-stack into a two-dimensional image. Instructions provided here are for FIJI/Image J.
- Open microscopy images using ImageJ software.
- If necessary, apply a pseudo-color to each channel to allow straightforward identification of each C. albicans strain. For this, click on Image > Lookup Table > LUT Color and select the chosen pseudo-color.
- Convert the stack file to a maximum intensity projection two-dimensional image:
- Select the z-stack file. Click on Image > Stacks > Z Projection.
- Select the top and bottom plane and select the projection type Max Intensity.
- Count each organism seen in the maximum projection images by strain type (distinguished by channel color) and morphology.
- Organisms that are significantly overlapping or areas with very high organism density will be difficult to count accurately. Exclude these from the count, but take care not to introduce bias against filamentous forms, which are more likely to overlap.
- Filamentous forms projecting straight into or out of the z-stack will appear as small round objects in a maximum projection. Likewise, organisms that are cut off by the border of the image may appear to be yeast because the filament is out of the field of view. Thus, two-dimensional analysis will always over-estimate the percentage of yeast forms. As this will occur equally with the reference and experimental strain(s), always compare experimental results to that of a reference strain.
- Perform statistical comparisons of the results as dictated by the experimental design.
8. Manual two-dimensional analysis: Filament length
- For C. albicans, aberrant filament formation can occur because: a) fewer "mother" yeast cells undergo morphogenesis, b) filaments grow at a slower rate, or c) filamentous growth is initiated but not maintained. To evaluate these possibilities, quantify the curve path length of each filament in the max projection image as a surrogate for the true three-dimensional length (discussed below).
- When a bud is developing on a mother cell, it is not possible to tell if it will become a filament or a yeast. To ensure that only filamentous cells are included in this analysis, measure only organisms in which the daughter cell is at least twice the length of the mother cell.
- Open the max projection image created in step 7.
- On the ImageJ toolset, right-click on the Straight/Segmented line tool and choose the option Segmented Line. The segmented line option allows the user to measure the filament length along a curved path, a necessity given the plasticity of C. albicans filaments.
- Measure the filament length from the bud neck to the growing end of the filament. Left-click on the bud neck; the pointer will change to a small square. Trace the filament along its length, clicking the center of the filament each time there is a curve, turn, or change of the long axis of the filament. Double click at the growing tip of the filament.
- Press Control + M. This will open a pop-up window that tabulates the Area, Mean, Min, Max, and Length measurements. After measuring each filament, press Control + M again to add the current measurement to the table of measurements.
- When all filaments have been measured, copy and paste the length measurements into a data analysis file.
- Perform statistical analysis to evaluate the distribution of filament lengths in the reference strain and the mutant strain.
9. Manual three-dimensional analysis
- To obtain a more precise measurement of morphogenesis that avoids the overestimation of yeast forms and may allow discrimination between pseudohyphae and hyphae, manually scroll up and down through the z-stack while evaluating the morphology of each organism in three dimensions.
- Alternatively, create a three-dimensional image of each z-stack and analyze the shape of each organism while rotating the image.
10. Automated analysis
- Using the imaging software, automate the enumeration of organisms and their morphology in two or three dimensions.
NOTE: Some algorithms for the discrimination of morphology types can introduce bias. Thus, automation strategies need to be carefully validated in relation to the experimental design. Well-designed and validated automated image analysis can increase the throughput of the analysis step.
Results
The results presented here are based on previously published reports9,10. The goal of this analysis is to quantitatively evaluate the ability of mutant C. albicans strains to undergo morphogenesis during active infections. The typical parameters that distinguish pseudohyphae from hyphae can be difficult to evaluate in organisms growing in three dimensions in a complex in vivo environment. This is particularly true when looking at the two-dimensional cross-sections created by confocal imaging. Therefore, this screening analysis is focused on identifying organisms growing as filamentous versus yeast. For follow-up studies using a more in-depth analysis, including three-dimensional reconstructions, this method could be adapted to discriminate yeast, hyphae, and pseudohyphae.
The expression of a fluorescent protein in a reference or mutant strain of C. albicans allows straightforward detection of strain morphology in vivo (Figure 3 and Figure 4). In general, quantitative light microscopy analysis is best performed when few to none of the pixels in the image are saturated. For this protocol, however, a saturation of the image often simplifies the analysis. Fluorescent protein localization is not uniform throughout the cell and is often higher in the mother yeast than in filaments. Fortunately, for the investigation of morphogenesis, the spatial distribution of the signal, rather than its intensity, defines the outcome. Therefore, obtaining images in which many pixels are saturated improves the ability to quantify morphogenesis in this assay.
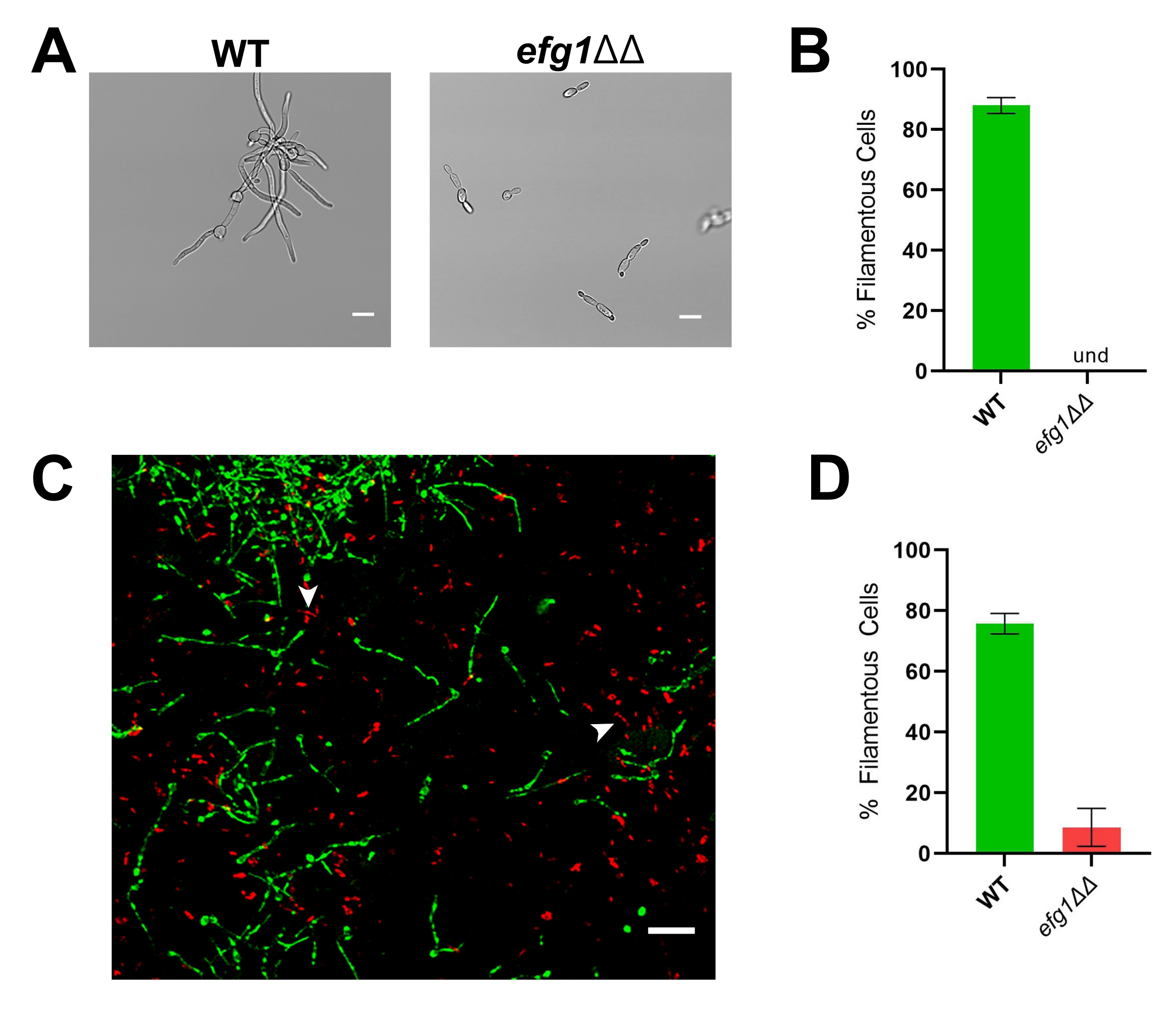
To illustrate the importance of evaluating morphogenesis in vivo, representative results are presented for the reference strain (SN250) and two mutants: one lacking the transcription factor Efg1 and the other lacking adenylyl cyclase Cyr1. In vitro, neither of these strains grow as filaments20,21. When grown in vitro in RPMI medium supplemented with 10% serum, the reference strain rapidly forms filaments, whereas the efg1ΔΔ and cyr1ΔΔ strains do not (Figure 3 and Figure 4). Under these conditions, the efg1ΔΔ mutant exhibits somewhat polarized growth, with the daughter cells being slightly elongated compared to the mother cells. This emphasizes the importance of using a clear definition of filamentation. Any such definition is by default arbitrary, but it is necessary for consistent evaluation of the phenotype. For these studies, a filamentous pattern of growth is defined as an organism with the longest dimension of a daughter cell more than twice that of the mother cell. Using this definition, the elongated efg1ΔΔ cells are not filamentous.
Consistent with its in vitro phenotype, the efg1ΔΔ exhibits a significant filamentation defect in vivo: approximately 9% of efg1ΔΔ cells grew as filaments in vivo (Figure 3). In contrast, 53% of the cyr1ΔΔ mutant cells grew as filaments in vivo (Figure 4). Although the number of cyr1ΔΔ mutant cells undergoing filamentation in vivo was significantly lower than the reference strain, the ability of the cyr1ΔΔ mutant to form filaments in vivo represents a substantial change from its complete lack of morphogenesis in vitro. Visually, the filaments formed by the cyr1ΔΔ mutant appeared to be shorter than the reference strain. To evaluate this quantitatively, the curve path length of the filamentous cells was measured using a two-dimensional projection of the in vivo images (Figure 4E). The median length of cyr1ΔΔ filaments was 29% shorter than filaments of the reference strain.

Figure 1: Anesthesia and inoculation. (A) Induction of anesthesia using an induction chamber. (B) Anesthesia is maintained using a nose cone, allowing the mouse to be repositioned as needed. (C) Hair removal cream is applied using a cotton-tipped applicator. Eye lubrication gel has been applied to protect the eyes during anesthesia. (D) Effective hair removal of the right ear. Compare to the untreated left ear. (E) Intradermal injection of C. albicans into the mouse ear. The ear is held in place using the tip of a conical tube wrapped with double-sided skin tape (fashion tape). (F) Close up of intradermal injection. A pale bubble is formed in the skin, which is a sign of a successful intradermal placement. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Preparation for imaging. (A) Microscope stage prepared for imaging. The anesthesia nose cone is secured in place. A coverslip is taped to the stage covering the lens opening. Additional pieces of tape are available. The heated stage is pre-warmed to 37 °C (not shown). (B) Placement of the anesthetized mouse into the anesthesia nose cone. (C) The mouse is rotated slightly so that the side of the ear that was inoculated is facing toward the bottom coverslip/objective lens. The ear is then flattened against the bottom coverslip and secured in place by placing a second coverslip on top of the ear. (D) The top coverslip is taped to the stage to secure the ear in place relative to the microscope stage. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: In vitro and in vivo morphology of the efg1ΔΔ mutant strain. (A) Widefield image of WT and efg1ΔΔ mutant strains after in vitro induction of filamentation by growing cells in RPMI + 10% serum at 37 °C for 4 h. Scale bars represent 10 µm. Image contrast was adjusted using photo editing software to facilitate viewing. (B) Percentage of filamentation in vitro observed in the WT and efg1ΔΔ mutant strains. Und = undetectable (no filaments were detected). Bar height represents the median percentage of filamentous cells from three independent experiments wherein at least 100 cells were quantified. The error bars indicate standard deviation (results compared by student's t-test, p < 0.001). (C) Confocal micrograph of WT (green) and the efg1ΔΔ mutant (red) growing in vivo 24 h post-inoculation. Arrows indicate examples of efg1ΔΔ mutant cells which meet our definition of "filamentous". Scale bar represents 50 µm. (D) Percentage of filamentation in vivo observed in the WT and efg1ΔΔ mutant strains. Bar height represents the median percentage of filamentous cells from two independent experiments. The error bars indicate standard deviation (results compared by student's t-test, p < 0.001). Please click here to view a larger version of this figure.
{kind=link}

Figure 4: In vitro and in vivo morphology of the cyr1ΔΔ mutant strain. (A) Widefield image of the cyr1ΔΔ mutant strain after in vitro induction of filamentation by growing cells in RPMI + 10% serum at 37 °C for 4 h. Scale bar represents 10 µm. Image contrast was adjusted using photo editing software to facilitate viewing. (B) Percentage of filamentation in vitro observed in the WT and cyr1ΔΔ mutant strains. Und = undetectable (no filaments were detected). Bar height represents the median percentage of filamentous cells from three independent experiments wherein at least 100 cells were quantified. The error bars indicate standard deviation (results compared by student's t-test, p < 0.001). (C) Confocal micrograph of WT (green) and the cyr1ΔΔ mutant (red) growing in vivo 24 h post-inoculation. Scale bar represents 50 µm. (D) Percentage of filamentation in vivo observed in the WT and cyr1ΔΔ mutant strains. Bar height represents the median percentage of filamentous cells from two independent experiments. The error bars indicate standard deviation (results compared by student's t-test, p < 0.001). (E) Distribution of filament length in vivo, as measured by curve path length in a two-dimensional projection of the z-stack. Each dot represents the length of one filament. Cells growing as yeast were not included in this analysis. Bar indicates median filament length. The distribution of lengths is significantly different when analyzed using a Mann-Whitney U test (p < 0.001). Please click here to view a larger version of this figure.
{kind=link}
Discussion
This model utilizes confocal microscopy to obtain images of C. albicans organisms as they grow within the tissue of a mammalian host, thus allowing us to evaluate morphogenesis phenotypes during active infection. The process of morphogenesis is central to C. albicans pathogenesis and has been widely studied using a variety of in vitro assays2,3,4. However, no in vitro assay can fully model the complex biochemical and structural environment of the host.
The protocol described here is focused on the use of this in vivo imaging system to screen a series/library of C. albicans mutants to identify the genes involved in morphogenesis during infection. The use of C. albicans strains expressing different fluorescent proteins allows us to quantify in vivo morphogenesis of C. albicans mutant strains compared to that of a reference strain. Comparing morphogenesis in the mutant to the reference strain within the same area of infection ensures that the organisms are exposed to identical environments. This allows for quantitative measurement of the percentage of cells undergoing filamentation, as well as the extent of filamentation. Normalization of the measurements of the mutant strain(s) to those of the reference strain allows us to better compare the performance of one mutant to another.
The representative results presented here demonstrate the potential for a significant discrepancy between in vitro and in vivo phenotypes. The C. albicans efg1ΔΔ mutant strain is often used as a negative control for morphogenesis assays as it fails to filament in almost all in vitro conditions20. Although the in vivo results were very similar to the in vitro results, even this severely hampered strain did occasionally form filaments in the host tissue environment (Figure 3). This emphasizes the strength of the host environment in triggering morphogenesis.
In contrast, the cyr1ΔΔ mutant strain demonstrates a substantial discordance between in vitro and in vivo growth; although none of the mutant cells undergo filamentation in vitro, approximately half of the cells grow as filaments in vivo (Figure 4)10,21. Interestingly, these filaments were significantly shorter than those formed by the reference strain, suggesting that CYR1 contributes to either the rate of filament growth or the ability to maintain a filamentous phenotype. To facilitate the analysis of filament length, the curve path length of the filaments was measured using a two-dimensional projection of the images. In two-dimensional projections of filaments growing in three dimensions, any filament growing on an axis that is not parallel to the xy plane will project as shorter than its true length. As this foreshortening also occurs for the reference strain, evaluating the distribution of filament lengths in a two-dimensional projection still allows a quantitative comparison between the reference and mutant strains. Analysis of filament length in two rather than three dimensions requires less intensive image analysis; thus, it can be performed relatively quickly on a typical desktop computer. Using this simpler analysis allows for the inclusion of filament length distribution as part of a screening protocol, giving a more nuanced understanding of the ability of each mutant to undergo morphogenesis without causing substantial delays in throughput.
The representative studies presented here were performed using DBA2/N mice, which have a defect in their complement system causing a failure to recruit neutrophils to the site of C. albicans infection22. The goal of these studies was to investigate mechanisms of the regulation of C. albicans filamentation within the host tissue. Therefore DBA2/N mice were used to avoid confounding the results because of an individual strain's susceptibility or resistance to neutrophils. As the neutrophil anti-C. albicans response can impact filamentation23, neutrophil recruitment to the site of infection could impact the results of a morphogenesis assay. If a strain is capable of filamenting in vivo but is strongly inhibited from filamentation when neutrophils are present, filamentation would be detected in DBA2/N mice but would be unlikely to be seen when using mice with intact neutrophil chemotaxis. Thus, the strain of the mouse used as a host is an important factor when using this protocol.
The observation that the efg1ΔΔ mutant strain fails to filament in vivo is unlikely to be related to host neutrophil responses, because this strain also fails to filament in vitro. The filamentation observed in vivo with the cyr1ΔΔ strain is discordant with its failure to filamentation in vitro. Data from the zebrafish model of C. albicans infection indicate that responding neutrophils are important in the prevention of morphogenesis24. Therefore, the use of DBA2/N mice, which lack neutrophil responses, is unlikely to account for the increase in filamentation of the cyr1ΔΔ in vivo compared to in vitro. Nevertheless, the in vivo environment is clearly impacting the morphogenesis of the cyr1ΔΔ strain; thus, further investigation of this strain may provide important information about the regulation of C. albicans morphogenesis during active infections. The protocol described here is designed as a screening assay to identify strains such as the cyr1ΔΔ strain to be used in future studies.
The use of a low-flow gas anesthesia system is very helpful for this protocol (Figure 1A,B). During the initial development of this protocol, mice were anesthetized using an injectable anesthetic cocktail of ketamine mixed with xylazine. While it was possible to perform limited imaging with that anesthetic method, the duration of anesthesia was unpredictable, requiring imaging sessions to be ended quickly to avoid the mouse recovering from anesthesia during imaging. Traditional inhaled anesthetic systems are bulky and require high rates of flow of anesthetic gasses, often requiring them to be used within a fume hood. Thus, traditional inhaled anesthetic systems would be very difficult to use with the space constraints of a confocal microscope without inadvertently exposing the investigators to the anesthetic agents. The use of a low-flow inhaled anesthetic system allows consistent anesthesia of the animal while maintaining a safe environment for the investigator. The low-flow nosecone allows easy positioning of the animal for both inoculation and microscopy. The small-caliber, low-volume delivery tubing allows the use of relatively long tubes, which enables the anesthesia machine to be placed at a sufficient distance so as not to interfere with microscopy.
The chlorophyll present in typical mouse chow leads to significant tissue autofluorescence25. This creates substantial noise in the images, making it difficult to obtain high-quality, high-spatial resolution images. When animals were fed chlorophyll-free chow for 7 days prior to imaging, background from autofluorescence was substantially decreased in tissue, but chlorophyll deposited in the hair continued to be problematic. Removing the hair on the pinnae using an over-the-counter chemical depilatory cream is effective at minimizing autofluorescence in the hair (Figure 1C,D). Thus, the combination of chlorophyll-free chow and adequate hair removal substantially decreased autofluorescence and improved image quality dramatically. Because the hair is removed from the ear prior to imaging, the color of the animal's hair does not impact this system. This protocol has been used successfully to study C. albicans infections in BALB/c (white), C57BL/6 (black), and DBA2/N (brown) mice. The protocol can also be used with C57BL/6 knockout mice that are deficient in various host genes; this will allow future investigations into how the mammalian host immune system impacts filamentation. One feature of this model not discussed in this protocol is that, because this imaging system is non-invasive, the same animal can be imaged repeatedly over several days, allowing to follow the progress of individual infection over time. This feature will likely play a key role in future studies on the host-pathogen interaction.
In summary, this protocol results in high-spatial resolution images of C. albicans growing in the tissue of a live mammalian host, allowing accurate evaluation of morphogenesis in mutant strains8,9,10. The results presented here demonstrate how this protocol can be used to screen a library of C. albicans mutants. Of the C. albicans mutants tested to date, a large portion of mutants with known defects in morphogenesis in vitro readily undergo filamentation in vivo9,10. This highlights the importance of including an in vivo system such as this one in experiments designed to elucidate the mechanisms of C. albicans pathogenesis.
Disclosures
The authors have no conflicts of interest to disclose.
Acknowledgements
This work was supported by NIH grant 1R01AI33409 and the Department of Pediatrics, Carver College of Medicine, University of Iowa.
Materials
| Name | Company | Catalog Number | Comments |
| #1.5 coverslips | Thermo-Fisher | 20811 | large enough to cover the universal stage opening |
| 0.1 mL Insulin syringes | EXELint | 26018 | Can use syringes that are 5/16"–1/2" long and 29–32 G |
| 3.7% formaldehyde in dPBS | Sigma-Aldrich | SHBJ5734 | |
| 70% Ethanol/30% water | Decon Laboratories | A05061001A | |
| Alcohol prep pads | Covidien | 5110 | Alternative: gauze pads soaked in 70% isopropyl alcohol |
| C.albicans reference strain and experimental strains | SN250 | FGSC Online Catalog | The specific C. albicans strain varies with experiment and the investigators goals. We have used strains derived from SC5314 as well as other clinical isolates. |
| Chlorophyl free mouse chow | Envigo | 2920x | |
| Computer | Dell | Optiplex 7050 | Computer that can run imaging software for acquisition and for analysis of images. A variety of imaging software is available and varies with the specific microscope and user system. |
| Cotton tip applicator | Pro Advantage | 76200 | |
| DBA2/N (6-12 week old mice) | BALB/c and C57/BL6 mice can also be used. The latter allow for the use of widely available knockout mouse models as well as mouse models in which individual cell types, such as phagocytes, are identified by their expression of fluorescent proteins. | ||
| Double sided tape designed to hold fabric to skin (fashion tape) | local pharmacy or grocery store | Double sided adhesive tape designed for keeping clothing in place over human skin. This is typically available over the counter in pharmacies and variety stores. It is important to use this type of tape as it is designed for gentle adherence to skin. Examples: https://www.amazon.com/Womens-Fashion-Clothing-Transparent-Suitable/dp/B08S3TWR3H/ref=sr_1_40?crid=2UWFL8FMFAKGM&keywords =fashion+tape&qid=1649174406&sprefix= fashion+tape%2Caps%2C70&sr=8-40 https://www.amazon.com/Fearless-Tape-Sensitive-Clothing-Transparent/dp/B07QY8V5XT/ref=sr_1_26?crid=2UWFL8FMFAKGM&keywords =fashion+tape&qid=1649174320&sprefix= fashion+tape%2Caps%2C70&sr=8-26 https://www.amazon.com/Hollywood-Fashion-Secrets-Tape-Floral/dp/B009RX77MK/ref=sr_1_29?crid=2UWFL8FMFAKGM&keywords =fashion+tape&qid=1649174406&sprefix= fashion+tape%2Caps%2C70&sr=8-29 | |
| Dulbecco's phosphate buffered saline | Gibco / Thermo-Fisher | 14190-144 | Must be sterile; open a new container for every experiment |
| Fetal bovine serum | Gibco / Thermo-Fisher | 26140-079 | |
| Gauze pad | Pro Advantage | P157112 | |
| Gel eye lurbicant | local pharmacy or grocery store | ||
| ImageJ or FIJI analysis software | NIH | ImageJ (FIJI) | |
| Isoflurane | Akorn | J119005 | |
| Leica DMi8 (SP8 platform) with Leica 11506375 objective lens | Leica | DMi8 (SP8) | The objective lens (Leica 11506375) used here is a 25x water immersion lens to allow us to have a high NA (0.95) while approximating the refractive index of the ear tissue. The microscope (Leica DMi8 (SP8 platform) has 488 nm and 638 nm diode laser lines and is equipped with filter-free spectral detection with computer controlled adjustable bandwidth for detection of emission light. The stage must have enough clearance to allow the objective to reach the bottom coverslip without hitting the stage. |
| Low-flow anesthesia system or traditional anesthesia vaporizer | Kent Scientific International | SomnoSuite | |
| Nair hair remover lotion | local pharmacy or grocery store | Over the counter depilatory cream | |
| Nourseothricin | Jena Bioscience | AB-101L | |
| pENO1-NEON-NATR pENO1-iRFP-NATR plasmids | Fluorescent protein expression transformation constructs generously given to us by Dr. Robert Wheeler (Seman, et al., 2018, Infection and Immunity; Bergeron, et al., 2017, Infection and Immunity) | ||
| Pressure sensitive laboratory tape | Tape & Label Graphic Systems Inc | 1007910 | |
| RPMI1640 cell culture medium | Gibco / Thermo-Fisher | 11875-093 | |
| Thimble, plastic 15 mL conical tube, or Falcon 5 mL round bottom polystyrene tubes | Falcon | 352196 | To safely hold the animals ear during injectinos |
References
- Lopes, J. P., Lionakis, M. S. Pathogenesis and virulence of Candida albicans. Virulence. 13 (1), 89-121 (2022).
- Saville, S. P., Lazzell, A. L., Monteagudo, C., Lopez-Ribot, J. L. Engineered control of cell morphology in vivo reveals distinct roles for yeast and filamentous forms of Candida albicans during infection. Eukaryotic Cell. 2 (5), 1053-1060 (2003).
- Arita, G. S., et al. Cell wall associated proteins involved in filamentation with impact on the virulence of Candida albicans. Microbiological Research. 258, 126996(2022).
- Rai, L. S., Wijlick, L. V., Bougnoux, M. E., Bachellier-Bassi, S., d'Enfert, C. Regulators of commensal and pathogenic life-styles of an opportunistic fungus-Candida albicans. Yeast. 38 (4), 243-250 (2021).
- Sudbery, P. E. Growth of Candida albicans hyphae. Nature Reviews Microbiology. 9 (10), 737-748 (2011).
- Basso, V., d'Enfert, C., Znaidi, S., Bachellier-Bassi, S. From genes to networks: The regulatory circuitry controlling candida albicans morphogenesis. Current Topics in Microbiology and Immunology. 422, 61-99 (2019).
- Mancera, E., et al. Evolution of the complex transcription network controlling biofilm formation in Candida species. Elife. 10, 64682(2021).
- Mitra, S., Dolan, K., Foster, T. H., Wellington, M. Imaging morphogenesis of Candida albicans during infection in a live animal. Journal of Biomedical Optics. 15 (1), 010504(2010).
- Wakade, R. S., Huang, M., Mitchell, A. P., Wellington, M., Krysan, D. J. Intravital imaging of Candida albicans identifies differential in vitro and in vivo filamentation phenotypes for transcription factor deletion mutants. mSphere. 6 (3), 0043621(2021).
- Wakade, R. S., Kramara, J., Wellington, M., Krysan, D. J. Candida albicans filamentation does not require the cAMP-PKA pathway in vivo. mBio. 13 (3), 0085122(2022).
- Bergeron, A. C., et al. Candida albicans and Pseudomonas aeruginosa interact to enhance virulence of mucosal infection in transparent zebrafish. Infection and Immunity. 85 (11), 00475(2017).
- Seman, B. G., et al. Yeast and filaments have specialized, independent activities in a zebrafish model of Candida albicans infection. Infection and Immunity. 86 (10), 00415-00418 (2018).
- Centers for Disease Control and Prevention. Biosafety in Microbiological and Biomedical Laboratories (BMBL). 6th edition. , Available from: https://www.cdc.gov/labs/BMBL.html (2020).
- Homann, O. R., Dea, J., Noble, S. M., Johnson, A. D. A phenotypic profile of the Candida albicans regulatory network. Plos Genetics. 5 (12), 1000783(2009).
- Cullen, P. J., Sprague, G. F. The regulation of filamentous growth in yeast. Genetics. 190 (1), 23-49 (2012).
- Herrero, A. B., et al. Control of filament formation in Candida albicans by polyamine levels. Infection and Immunity. 67 (9), 4870-4878 (1999).
- Ahmad Hussin, N., et al. Biotin auxotrophy and biotin enhanced germ tube formation in Candida albicans. Microorganisms. 4 (3), 37(2016).
- Nantel, A., et al. Transcription profiling of Candida albicans cells undergoing the yeast-to-hyphal transition. Molecular Biology of the Cell. 13 (10), 3452-3465 (2002).
- Noble, S. M., Johnson, A. D. Strains and strategies for large-scale gene deletion studies of the diploid human fungal pathogen Candida albicans. Eukaryotic Cell. 4 (2), 298-309 (2005).
- Glazier, V. E. EFG1, Everyone's favorite gene in Candida albicans: A comprehensive literature review. Frontiers in Cellular Infection and Microbiology. 12, 855229(2022).
- Huang, G., Huang, Q., Wei, Y., Wang, Y., Du, H. Multiple roles and diverse regulation of the Ras/cAMP/protein kinase A pathway in Candida albicans. Molecular Microbiology. 111 (1), 6-16 (2019).
- Saville, S. P., Lazzell, A. L., Chaturvedi, A. K., Monteagudo, C., Lopez-Ribot, J. L. Use of a genetically engineered strain to evaluate the pathogenic potential of yeast cell and filamentous forms during Candida albicans systemic infection in immunodeficient mice. Infection and Immunity. 76 (1), 97-102 (2008).
- Brothers, K. M., Newman, Z. R., Wheeler, R. T. Live imaging of disseminated candidiasis in zebrafish reveals role of phagocyte oxidase in limiting filamentous growth. Eukaryotic Cell. 10 (7), 932-944 (2011).
- Brothers, K. M., et al. NADPH oxidase-driven phagocyte recruitment controls Candida albicans filamentous growth and prevents mortality. PLoS Pathogens. 9 (10), 1003634(2013).
- Holmes, H., Kennedy, J. C., Pottier, R., Rossi, R., Weagle, G. A recipe for the preparation of a rodent food that eliminates chlorophyll-based tissue fluorescence. Journal of Photochemistry and Photobiology. B: Biology. 29 (2-3), 199(1995).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved