
Assay for Cell Death: Chromium Release Assay of Cytotoxic Ability
Overview
Source: Frances V. Sjaastad1,2, Whitney Swanson2,3, and Thomas S. Griffith1,2,3,4
1 Microbiology, Immunology, and Cancer Biology Graduate Program, University of Minnesota, Minneapolis, MN 55455
2 Center for Immunology, University of Minnesota, Minneapolis, MN 55455
3 Department of Urology, University of Minnesota, Minneapolis, MN 55455
4 Masonic Cancer Center, University of Minnesota, Minneapolis, MN 55455
One of the main functions of the cells of the immune system is to remove target cells that have been infected with viruses or have undergone transformation into a tumor cell. In vitro assays for measuring the cytotoxic capacity of immune cells have been a staple in laboratories for many years. These assays have been used to determine the ability of T cells, NK cells, or any other immune cell to kill target cells in an antigen-specific or -nonspecific manner. Death ligands (e.g., Fas ligand or TRAIL), cytokines (e.g., IFNg or TNF), or cytotoxic granules (i.e., perforin/granzyme B) expressed by effector cells are some ways in which target cell death can be induced. With the explosion in tumor immunotherapy research in recent years, there is growing interest in finding agents to increase the cytotoxic activity of immune cells to improve patient outcomes. Conversely, some diseases are hallmarked by the overexuberant activity of immune cell cytotoxic activity, resulting in efforts to identify agents to temper these responses. Thus, having an assay in which the user can easily integrate any number of different effector cells, target cells, and/or response modifiers into the experimental design can serve as a valuable means of quickly assessing the cytotoxic capacity of effector cells and/or the responsiveness of the target cell.
These in vitro assays involve the mixing of different cell populations, as well as using a relatively low number of both effector and target cells. Thus, one necessity of the assay is to label the target cells in a manner that can easily be detected and quantitated, allowing the user to then determine the 'percent specific lysis' mediated by the effector cells. Radioactivity - especially, chromium 51 (51Cr) in the form of Na251CrO4- is an inexpensive way to rapidly and nonspecifically label cellular proteins within the target cells (1). The short labeling and total assay times reduces the potential for significant changes in the number and/or phenotype of the target cells, which could influence the outcome of the assay. Upon the loss of membrane integrity of the target cells as a result of the cytotoxic activity of the effector cells, the 51Cr-labelled cellular proteins within the target cells are released into the culture supernatant, becoming available for quantitation. As with any assay examining the function of immune cells in vitro, there are a number of important considerations to consider improving the performance of the experiment. One of the most critical features is to use healthy effector (for maximal cytotoxic activity) and target (for maximal responsiveness and minimal spontaneous death/51Cr release) cells. Effector and target cell contact is required (leading to the common use of round-bottom 96-well plates to encourage cell-cell contact) (2). Finally, data analysis is dependent on the inclusion of positive and negative control target cell populations.
The following protocol will outline the steps for performing a standard 51Cr release assay to measure the cytotoxic ability of a population of effector cells, though a nonradioactive version using Europium has recently been developed. 51Cr is a powerful γ-radiation emitter. Consequently, the use of this assay requires proper radiation safety training, dedicated laboratory space, a gamma counter, and disposal of radioactive samples.
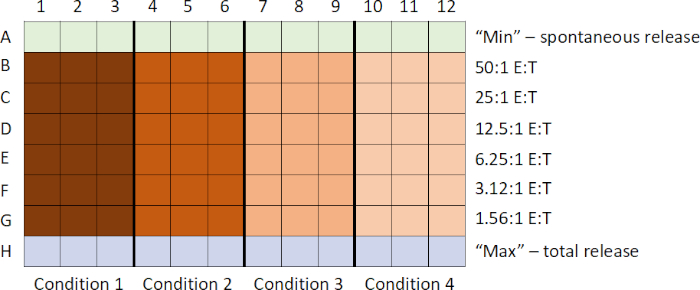
The general sequence of events in this assay are: 1) prepare 51Cr-labeled targets; 2) prepare effector cells and add to plate while target cells are labeling; 3) add labeled targets to plate; 4) incubate plate; 5) harvest supernatants; and 6) analyze data after running samples on counter. Samples are commonly prepared in triplicate, and then averaged to account for any subtle pipetting differences.
Proper PPE is important for this assay. Specifically, the user should wear a lab coat and gloves. Safety glasses may be required based on the laboratory or institution. There should be ample lead shielding for safe storage and use of the 51Cr during all steps. Finally, there should be dedicated lab space and equipment set aside for using 51Cr, including all the proper signage to indicate where samples with 51Cr are being kept and a Geiger counter equipped with gamma probe to survey the space for possible contamination.
In this lab exercise, we will determine the ability human peripheral blood mononuclear cells (PBMCs), (CpG stimulated vs. unstimulated) to kill melanoma cells, using human melanoma cell line WM793 as model and the chromium release assay.
Procedure
Procedure Overview
The typical 51Cr-release assay for measuring cell death involves the following steps:
- First, the target cells are labeled with Na2[51Cr]O4. This distinguishes them from the effector cells in the assay.
- While the target cells are labeling, the effector cells are collected and, using the serial dilution technique, a decreasing titration of the effector cells is generated in a round bottom 96-wel
Results
In this example, effector cells stimulated with CpG (Figure 1, black circles) killed the target cells more effectively, as the ratio of effector cells to target cells increased. This increase was not observed in the unstimulated PBMCs (white circles), indicating that CpG stimulation is necessary for the observed increase in target cell lysis.
Log in or to access full content. Learn more about your institution’s access to JoVE content here
Application and Summary
The assay described here has considerable flexibility, as a variety of effector and target cells can be used depending on the question being asked. For example, effector cell specificity can be determined by using different target cells or the mechanism of effector cell killing can be determined by using cells deficient in specific proteins or using protein specific inhibitors. A major problem with the 51Cr release assay is the potential for a high spontaneous release rates by the target cells. When cultured a
References
- Brunner, K. T., Mauel, J., Cerottini, J. C. and Chapuis. B. Quantitative assay of the lytic action of immune lymphoid cells on 51Cr-labelled allogeneic target cells in vitro; inhibition by isoantibody and by drugs. Immunology, 14 (2):181-196, (1968).
- Kemp, T. J., B. D. Elzey, and T. S. Griffith. Plasmacytoid dendritic cell-derived IFN-alpha induces TNF-related apoptosis-inducing ligand/Apo-2L-mediated antitumor activity by human monocytes following CpG oligodeoxynucleotide stimulation. The Journal of Immunology, 171 (1): 212-218, (2003).
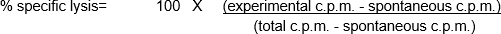
Skip to...
Videos from this collection:

Now Playing
Assay for Cell Death: Chromium Release Assay of Cytotoxic Ability
Immunology
151.5K Views

Flow Cytometry and Fluorescence-Activated Cell Sorting (FACS): Isolation of Splenic B Lymphocytes
Immunology
93.2K Views

Magnetic Activated Cell Sorting (MACS): Isolation of Thymic T Lymphocytes
Immunology
23.0K Views

ELISA Assays: Indirect, Sandwich, and Competitive
Immunology
239.2K Views

ELISPOT Assay: Detection of IFN-γ Secreting Splenocytes
Immunology
28.8K Views

Immunohistochemistry and Immunocytochemistry: Tissue Imaging via Light Microscopy
Immunology
79.1K Views

Antibody Generation: Producing Monoclonal Antibodies Using Hybridomas
Immunology
43.6K Views

Immunofluorescence Microscopy: Immunofluorescence Staining of Paraffin-Embedded Tissue Sections
Immunology
53.9K Views

Confocal Fluorescence Microscopy: A Technique to Determine the Localization of Proteins in Mouse Fibroblasts
Immunology
43.4K Views

Immunoprecipitation-Based Techniques: Purification of Endogenous Proteins Using Agarose Beads
Immunology
87.8K Views

Cell Cycle Analysis: Assessing CD4 and CD8 T Cell Proliferation After Stimulation Using CFSE Staining and Flow Cytometry
Immunology
24.3K Views

Adoptive Cell Transfer: Introducing Donor Mouse Splenocytes to a Host Mouse and Assessing Success via FACS
Immunology
22.5K Views
Copyright © 2025 MyJoVE Corporation. All rights reserved