
需要订阅 JoVE 才能查看此. 登录或开始免费试用。
Method Article
生成CRISPR / Cas9介导的单等位基因缺失来研究小鼠胚胎干细胞功能增强
摘要
Experimental validation of enhancer activity is best approached by loss-of-function analysis. Presented here is an efficient protocol that uses CRISPR/Cas9 mediated deletion to study allele-specific regulation of gene transcription in F1 ES cells which contain a hybrid genome (Mus musculus129 x Mus castaneus).
摘要
Enhancers control cell identity by regulating tissue-specific gene expression in a position and orientation independent manner. These enhancers are often located distally from the regulated gene in intergenic regions or even within the body of another gene. The position independent nature of enhancer activity makes it difficult to match enhancers with the genes they regulate. Deletion of an enhancer region provides direct evidence for enhancer activity and is the gold standard to reveal an enhancer's role in endogenous gene transcription. Conventional homologous recombination based deletion methods have been surpassed by recent advances in genome editing technology which enable rapid and precisely located changes to the genomes of numerous model organisms. CRISPR/Cas9 mediated genome editing can be used to manipulate the genome in many cell types and organisms rapidly and cost effectively, due to the ease with which Cas9 can be targeted to the genome by a guide RNA from a bespoke expression plasmid. Homozygous deletion of essential gene regulatory elements might lead to lethality or alter cellular phenotype whereas monoallelic deletion of transcriptional enhancers allows for the study of cis-regulation of gene expression without this confounding issue. Presented here is a protocol for CRISPR/Cas9 mediated deletion in F1 mouse embryonic stem (ES) cells (Mus musculus129 x Mus castaneus). Monoallelic deletion, screening and expression analysis is facilitated by single nucleotide polymorphisms (SNP) between the two alleles which occur on average every 125 bp in these cells.
引言
转录调控元件由于异常的基因表达2是发展1和修改这些元素的过程中的基因表达的时空微调可导致疾病的关键。通过全基因组关联研究发现了许多疾病相关的区域是在非编码区,并有转录增强子3-4的功能。识别增强剂并将它们与它们调节是复杂的,因为它们通常位于从它们调节基因几千个碱基远并且可以以组织特异性方式5-6被激活的基因匹配。增强剂的预测通常是基于组蛋白修饰的标记,介体黏着复合物和细胞类型特异性转录结合因子7-10。预测增强剂的验证是最经常通过一个基于矢量的测定,其中所述增强剂激活报告基因11-12的表达进行。这些数据提供了v有关推测的增强子序列的调节潜力aluable信息,但不透露自己的功能其内生基因组范围内或识别它们调节的基因。基因组编辑充当一个强有力的工具来研究由失功能分析在它们的内源上下文转录调控元件的功能。
在基因组编辑,即CRISPR / Cas9基因组编辑系统的最新进展,有利于基因功能的研究。的CRISPR / Cas9系统易于使用和适应性对于许多生物系统。所述Cas9蛋白靶向于由导的RNA(gRNA)13中的基因组中的特定位点。所述SpCas9 / gRNA复杂扫描对其靶基因组序列的基因组中它必须是5'到protospacer相邻基序(PAM)的序列,NGG 14-15。的gRNA到其目标,一个20个核苷酸(nt)的序列与gRNA互补的碱基配对,激活导致域金字塔之戒SpCas9核酸酶活Ë链断裂(DSB)3碱基的序列PAM的上游。特异性是通过在gRNA种子区域完全碱基配对来实现,所述6-12 nt下邻近于PAM;相反地,不匹配5'种子的通常耐受16-17。引入的DSB可以修复或者由非同源末端连接(NHEJ)的DNA修复或同源性定向修复(HDR)mechanisms.NHEJ DNA修复往往造成在目标部位的几个碱基对,可以破坏的插入/缺失(插入缺失)的基因的开放阅读框(ORF)。以产生在基因组2 gRNAs,侧翼感兴趣的区域大的缺失,可以使用18-19。这种方法是对聚成基因座控制区或超增强剂它比常规增强剂9,18,20-22较大转录增强子的研究中特别有用的。
单等位基因缺失是研究转录顺 -regulation一个有价值的模型。观察到昌E在转录水平的增强子的单等位基因缺失之后关联到在基因调控该增强剂的不当两个等位基因的转录可能受影响的影响蜂窝健身时可能出现的混杂影响的作用。评估减少的表达是困难的但不区分野生型等位基因的删除的能力。此外,基因分型在每个等位基因缺失而不区分两个等位基因的能力是具有挑战性的,尤其是对大缺失> 10kb的至1兆23,其中它是难以通过PCR扩增整个野生型区域。使用通过杂交小家鼠 129与小家鼠castaneus生成的F1 ES细胞的允许两个等位基因通过等位基因特异性PCR 18,24区别开来。在这些细胞中的基因组杂交便于等位基因特异缺失筛选和表达分析。上平均有一个SNP位这两个基因组之间的每一个125 bp的为表达和基因分型提供在引物设计的灵活性的分析。一种SNP的存在可以影响引物的熔化温度(T M)与靶实时定量PCR(qPCR的)扩增特异性允许两个等位基因25的歧视。此外,引物的3'末端中的一个错配极大地影响DNA聚合酶从引物防止不期望的等位基因靶26的扩增延伸的能力。描述在下面的协议是使用CRISPR / Cas9基因组编辑系统( 图1)大于1 kb的等位基因特异性增强缺失和随后的表达分析使用F1 ES细胞。

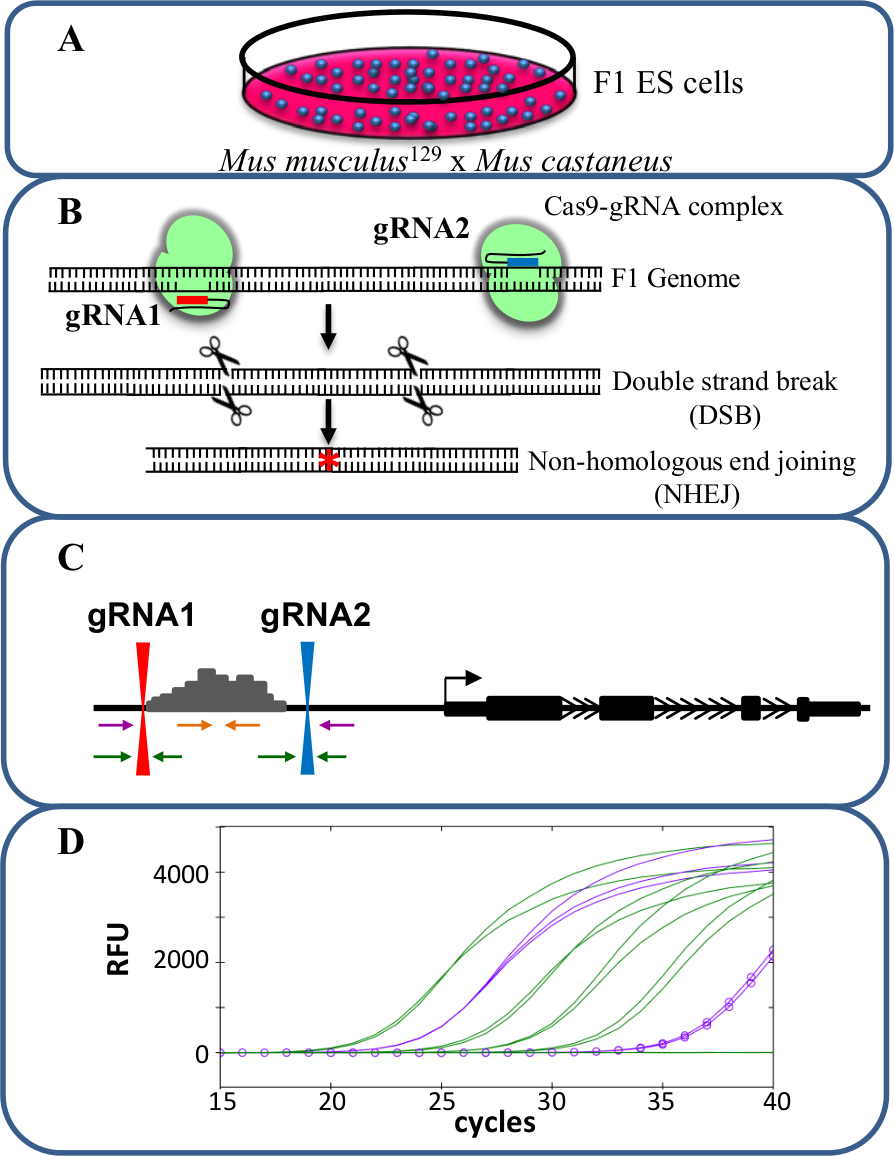
图1.增强删除使用CRISPR / Cas9研究 顺 -reg基因表达的ulation。(A)中由小家鼠 129和小家鼠castaneus之间的交叉产生的F1 ES细胞用于允许等位基因特异性缺失。 (B)中的两个导向的RNA(gRNA)用于诱导增强子区的一个大Cas9介导的缺失。 (C)的引物组被用于识别大的单-和双等位基因缺失。橙色引物是内引物,紫色引物外侧的引物和绿色的引物的gRNA侧翼引物。 (D)基因表达的变化是使用等位基因特异性qPCR的监控。俄罗斯足协表示相对荧光单位。 请点击此处查看该图的放大版本。
{kind=link}
研究方案
1.设计和建造的gRNA
- 删除转录增强子区域使用两个gRNAs一5'和一个3'的感兴趣区域的。使用由张实验室产生的鼠标UCSC基因组浏览器的轨道,以确定独特的gRNA序列(http://www.genome-engineering.org 15)。接下来检查这些gRNAs和他们的使用由桑格研究所(www.sanger.ac.uk/sanger/Mouse_SnpViewer/rel-1211)27-28提供在线工具SNP和插入缺失相邻的PAM。要定位具有同等效率两个等位基因,避免含有SNP或插入缺失gRNA / PAM序列。
- 在选择gRNA,检查设计等位基因特异性引物进行基因分型缺失的可行性。参见第5节等位基因特异性引物设计。
- 组装基于在马里等人2013 15描述的协议两个gRNA质粒。包括所选择的独特的20 bp的靶序列INTO作为在表7所示的61mer寡核苷酸(序列显示在5'至3'方向,且粗体的碱是是彼此反向互补的20碱基对的靶序列)。
- 混合10μM的gRNA Primer_F的10微升和10微升10μM的互补Primer_R中的管。
- 通过温育在100℃下的引物混合5分钟退火的引物,然后冷却1℃/秒至25℃。对于此步骤,使用PCR仪或放置在沸水中的试管,并且允许其冷却至室温。
- 到退火的引物混合物中,添加以下反应混合物孵育在72℃进行30分钟至延伸每个引物:18.5微升水,10微升5×HF缓冲液中,1微升10毫的dNTP混合和0.5微升高的高保真DNA聚合酶。
- 运行在2%琼脂糖凝胶上10微升靶片段的确认100bp的指南片段已经产生。
- 线性化gRNA向量(来自地球的礼物RGE教堂; Addgene质粒#41824)15用的Afl II通过使用以下的反应设置:5微升gRNA载体骨架(2-4微克),5微升10X缓冲液,3微升的Afl II的(20单位/微升)和32微升的水。孵育3小时将反应混合物在37℃。
- 运行在1%琼脂糖凝胶的消化产物和纯化对应于3.5kb的该DNA带用凝胶提取试剂盒按照制造商的说明书线性化gRNA载体。
- 1μl的线性gRNA载体(50纳克/微升),1μl的靶片段,10微升2×吉布森组件主混合物的组成:用线性gRNA矢量和目标从步骤1.2.3片段如下设置吉布森装配反应29-30和8微升的水。在50℃下孵育反应60分钟。
- 大肠杆菌细胞,组装gRNA载体转化。
- 混合1微升组装gRNA矢量从1.2.7和50μl的DH5α( 大肠杆菌菌株)细胞在管的。通过将细胞暴露于42℃45秒变换DH5α细胞通过热休克法。
- 卡扣冷却冰上5分钟管;然后加入400μl的SOC培养基和在振荡培养箱中于37℃孵育45分钟。
- 散布在LB卡那霉素(50微克/毫升)板转化细胞的阳性选择100微升DH5α细胞中,并在37℃孵育O / N。
- 筛查呈阳性大肠杆菌菌落gRNA插入。
- 挑在3ml LB卡那霉素抗性菌落重悬含有50μg/ ml卡那霉素。重复同样的6-8的殖民地和在37°CO / N所有的管子在摇床。
- 通过按照制造商的说明书提取使用质粒微型制备试剂盒从O / N生长培养质粒。
- 准备一个EcoRI位 I酶切反应混合物来检查gRNA序列插入在质粒。 2微升酶缓冲液,1微升EcoRI位我的,15微升的水:对于每个样品,如下制备反应混合物。分装反应混合物成1.5毫升管中,加入2微升质粒。孵育管在37℃下2小时。
- 运行上的1.5%琼脂糖凝胶的消化产物。
注:将样品将显示一个475 bp的条带大小比无插件的克隆高100个基点。
注意:可替换地,阳性克隆可通过菌落PCR,使用SP6启动(正向)和T7(反向)引物( 表7),该结合到载体序列,得到一个gRNA的存在下插入一个642 bp的大小片段进行筛选。菌落PCR方法中是有利的,当在gRNA序列内一个EcoRI位 I限制位点。
- 确认gRNA插入的DNA测序采用T7引物序列。
2.转
注意:电穿孔是转染质粒导入ES细胞的一种有效的方法。这里所描述的方法使用微孔器转染技术。
- 生长在含有10ml的ES细胞培养基( 表1)的10cm的明胶包被培养皿F1 ES细胞在37℃/ 5%CO 2。当细胞达到85%汇合删除媒体及加2ml胰蛋白酶。中的CO 2培养箱中5分钟,在37℃。
注:芭芭拉平移24获得F1 ES细胞,并根据要求提供。 - 通过加入10ml自旋介质( 表2)的中和胰蛋白酶。移液器反复彻底分离细胞。
- 收集在一个15毫升管和旋转所有的细胞在300×g离心5分钟。在3ml PBS中重悬,并计算使用血球或自动细胞计数器细胞。
- 沉淀1×10 6 ES细胞中通过离心1.5ml的管中,在300×g离心于100μl的R 5分钟,重悬(再悬浮)缓冲器如由试剂盒制造商提供。
- 添加每个pCas9_GFP 5微克(从吉兰Musunuru的礼物; Addgene质粒#44719)31,5'和3'gRNA质粒对靶区域的缺失并用移液管轻轻混匀,以避免引入气泡。
- 使用电子枪头吸100微升的电组合,小心以避免在尖端泡沫。
- 程序的伏,宽度和电穿孔脉冲。对于F1胚胎干细胞,用1400伏,10毫秒3个脉冲。
- 虽然电运行观察尖端观看解决方案中的任何火花。的火花表示气泡的存在,并且将与转染干扰。
- 弹出转染的ES细胞入含有10ml的ES细胞培养基( 表1)在10cm明胶涂层培养皿并在37℃/ 5%CO 2孵育。
3.流式细胞分选转染细胞
- 48小时后,通过加入2 ml胰蛋白酶的分离的细胞,并在CO 2培养箱中于37℃孵育5分钟。
- 通过加入10 ml收集缓冲液( 表3)中和该板。收集在一个15毫升管和自旋的细胞以300×g离心5分钟。
- 弃上清,重悬细胞于1ml分拣缓冲液( 表4)。算基于排序的平台上的细胞,并稀释。稀释细胞以0.5-1×10 6细胞/ ml的对分类到15ml试管和用于单个细胞直接分选入96孔板,稀释细胞以2-5×10 6个细胞/ ml。
- 排序Cas9-GFP + ES细胞使用FACS流式细胞仪32。作为用于菌落采摘,或排序的单个细胞在3.5中描述直接到含有100微升的ES细胞培养基/孔明胶包被的96孔板收集细胞散装在用2ml恢复介质( 表5)和板管( 表1)。
- 种子1-1.5×10 4个 GFP + ES细胞在含有10毫升的ES细胞培养基( 表1)在10cm明胶包被培养皿中。在这样低的密度电镀有利于选择单独的ES细胞集落。
4.培养克隆基因分型,表达分析和冷冻细胞股票
- 关于排序后4-5天,得分直接排序96孔板的每个孔用于ES细胞集落的存在。
- 通过除去培养基并加入30微升的胰蛋白酶解离ES细胞集落。孵育在37℃下5分钟。通过加入170微升的ES细胞培养基( 表1)的中和胰蛋白酶,和吸管上下的集落的完整解离成单细胞。生长的细胞在37℃/ 5%CO 2,直到大部分井超过70%汇合(通常2-3天)。
- 使用或者挑选个人ES细胞集落10 cm的培养皿倒置显微镜。吸取菌落成枪头后续步骤4.1.1将每个集落于96孔平板的一个孔中,用明胶预处理和含有30微升胰蛋白酶之后。
注:菌落胰蛋白酶在室温下坐而殖民地之一整行拾取。 - 一旦所有的菌落都被拾取和离解成培养基上生长的细胞在37℃的CO 2培养箱中 ,直到大部分井超过70%汇合(通常为2天)。
- 当96孔板准备好拆分,取出介质,添加30微升的胰蛋白酶,并在37℃下孵育5分钟。通过加入180微升的ES细胞培养基( 表1),以每孔和吸管上下完全解离成单个细胞中和胰蛋白酶。
- 从所得210微升,种子70微升为三个明胶包被的96孔各自含有130微升的ES细胞培养基/孔( 表1)的平板上。使用这些板块为GEnotyping,表达分析和如下所述的冷冻细胞种群的每个克隆。
- 当基因分型板达到70-85%汇合,在第6章"基因分型删除"中所述治疗板。
- 当表达分析板达到70-85%汇合,除去介质,具有直到克隆已基因型在-80℃的密封胶带和存储密封板。
注意:表达分析板是有用的克隆的早期传代来分析基因表达的变化。从96孔板的基因表达分析是可能的,但作为细胞数是低建议一个RNA微提取试剂盒。 - 96孔板冻结股票的制备:
- 当板对细胞冻存物(股票-1)达到70-85%汇合时,吸出介质中,添加30微升的胰蛋白酶,并在37℃下孵育5分钟。
- 通过加入100μl的ES细胞培养基中和胰蛋白酶( 表1)TØ每孔吸管向上和向下的完全分离成单个细胞。
- 转印悬浮的细胞的15微升每孔两个明胶包被的96孔板,每片含185微升的ES细胞培养基( 表1),并允许在37℃/ 5%CO 2生长。
注:这是股票-2和-3板,它们分别时提供额外的备份克隆现货-1重振细胞不成功。
- 同时,在96孔板(股票-1)的100微升剩余的细胞,加入100μl的2×的冷冻培养基( 表6)。用密封胶带密封该板,并迅速翻转板4-5次进行适当的混合。在-80℃保存,直到盘克隆基因分型。
- 当股票-2和库存-3-板是准备用于冷冻抽吸介质中,添加30微升的胰蛋白酶,并在37℃下孵育5分钟。通过加入70微升的ES细胞培养基中和胰蛋白酶(T能1)至每孔并移液器上下完全解离成单细胞。
- 加入100微升2个冷冻介质,具有密封胶带密封板,迅速反转板适当混合4-5倍。直到需要这些板块在-80℃保存板。
5.等位基因特异性引物设计
- 设计4组引物( 图1C)以筛选所需的删除克隆:内部引物,外引物,以及gRNA侧翼引物(对于5'和3'gRNA目标位点),如下所述。
- 在获取相应http://labs.csb.utoronto.ca/mitchell/crispr.html的129和演员基因型SNP轨道。给定的轨迹显示在该MM9小鼠基因组组装的坐标129和演员之间的碱基替换。
注:在上述网站的链接重定向到UCSC基因组浏览器,并添加含有129之间的SNP定制的跟踪和GE铸造nomes。 - 进入该区域的坐标被删除。放大在含有> 3个SNP所需的删除的中间约500bp的区域。
- 去查看> DNA在选项栏,点击获取DNA下载所有大写格式靶序列。
- 创建两个FASTA序列;一个用于129和一个用于在SNP位置饰演通过碱基取代。由小写的标记单核苷酸多态性。
- 去引物3加号(http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi/)并粘贴SNP取代129序列。使用默认的设置来设计引物。
- 为了设计里面等位基因特异性引物,在任务下拉菜单中选择Primer_List并单击选择引物。选择正向或反向引物,有一个SNP无论是在3'末端或内从3 4基"以及落入内的区域被删除。
注意:必须在3'末端显示一个SNP引物的qPCR增加等位基因特异性。 - 选择第二个底返回到主页面,在任务下拉菜单中选择检测,并在页面的底部粘贴在相应的框中的第一个引物序列。在常规设置选项卡更改产品尺寸范围设置为80-200基点,并单击选择引物。选择了底漆从上市引物对设置;这些将是129内等位基因特异性引物。
- 重复步骤5.1.5至5.1.7设计引物的演员等位基因。
- 返回到UCSC基因组浏览器,并输入该区域的坐标被删除。去查看> DNA在选项栏,在顺序检索区选项上游增加1000 bp和下游点击获取DNA下载靶序列。
- 马克括号中的gRNA靶序列。继续之前保存这个整个序列。
- 为了设计外引物拆除两个gRNA靶序列之间的序列。重复步骤5.1.4-5.1.8设计外等位基因特异性引物小号但改变产品尺寸400-800基点。
- 拆分在步骤5.1.10得到的序列分成两个序列,每500 bp的5'和3'的gRNA靶序列。重复步骤gRNA侧翼区5.1.5到5.1.7设计非等位基因特异性引物,但改变产品尺寸400-800基点。
注意:对于非等位基因特异性引物,无论是129或可用于铸造序列和引物的选择应不包含一个SNP。明智的做法是设置400-800 bp的产品尺寸设计之外,gRNA侧翼引物。这允许放大即使小插入缺失存在。
- 在获取相应http://labs.csb.utoronto.ca/mitchell/crispr.html的129和演员基因型SNP轨道。给定的轨迹显示在该MM9小鼠基因组组装的坐标129和演员之间的碱基替换。
- 使用纯129和演员株的基因组DNA在2毫微克/微升通过qPCR测试等位基因特异性引物内。按照步骤6.2-6.4建立定量PCR反应。
注意:如果129基因型在目标区域是相同的C57BL / 6J,从C57BL / 6J DNA可在129 DNA的地方使用。等位基因特异性引物应显示至少5个循环在正确的Ct(循环阈值)值与不正确的基因型差异。外面的引物进行测试,以确保它们扩增使用Cas9 / gRNA转染的ES细胞的缺失,与F1基因组DNA分别作为阳性和阴性对照。外引物的等位基因特异性可以一旦单等位基因的克隆已经鉴定测试。
6.基因分型的缺失
- 提取使用来自步骤4.6所菌落膨胀后所产生的板的基因分型的96孔板的基因组DNA。
- 制备的基因组DNA的提取混合物:89微升水,10微升10X缓冲液和1μl的提取试剂(由生产商提供)。加入100μl的基因组DNA的提取混合物的向每个孔和密封用密封胶带的板。
- 孵育所述板在75℃下进行5分钟,随后95℃5分钟。
- 允许该板在冰上温育几个min的冷却ð然后短暂离心解决任何冷凝井的底部。此作为模板DNA板为删除筛选。
- 设置一式两份的qPCR反应对每个克隆如下:5微升2×SYBR qPCR的混合物,正向和反向引物(3μM)各1微升和1微升的水。使用多道移液器到2微升模板DNA,接着8微升反应混合物添加至各孔384孔板的。
- 密封带600 XG密封带和旋转2分钟以混合内容物的板。放置在实时PCR仪的384孔板阵列。
- 程序进行熔融曲线分析,检出了两步PCR的实时PCR仪如下:在95℃下1个循环10分钟,15秒95℃40个循环,62℃,用读板30秒和95℃10秒,65℃至95℃与5℃增量为5秒+读板。
注意:除了引物设计中,定量PCR英里x和循环参数也有助于引物的特异性。和在材料中列出上述参数试剂更频繁地产生等位基因特异性扩增。 - 定量PCR分析结果
- 检查每个等位基因与内等位基因特异性引物的扩增。一个等位基因的不扩增或等位基因之间高Ct值差异(> 5周期)表明这些克隆携带的高/缺席Ct值的等位基因的杂合缺失。两个等位基因的无扩增表明它们携带纯合缺失。
- 检查每个等位基因与外部等位基因特异性引物的扩增。当目标缺失比与外部引物1 KB的扩增较大时才会发生缺失存在。 22-28 Ct值确认删除。对于大于1 KB的目标缺失,确认通过电泳扩增子大小。
注意:如果外引物仅显示适度的等位基因特异性(见Figu重2),扩增子可以与由于外部的引物脱靶扩增两个等位基因的引物组在单等位基因的克隆而获得。在这种情况下,至少五个循环的两个等位基因之间的Ct值差异应该确认正确等位基因(低级Ct值),是基于从内侧引物获得的结果被删除。如果目标与脱靶等位基因的Ct差小于五个周期设计出新的等位基因特异性引物外。 - 在单等位基因缺失克隆通过使用二次筛选,gRNA侧翼引物检查未删除等位基因的完整性。
注:gRNA目标部位周围> 25基点大小的插入缺失,可以通过观察400-800 bp的扩增子在qPCR的熔体曲线的移动来识别。或者,从gRNA侧翼引物的扩增子进行测序,以检测<25个碱基对小插入缺失。- 用2台gRNA侧翼引物,即 ,5'和3'G执行的qPCR生成CRISPR删除使用RNA。与这些组引物无扩增指示插入缺失比的qPCR扩增较大存在于上单等位基因缺失克隆的未删除等位基因的gRNA目标部位。含有从进一步分析的结果,这些大插入缺失丢弃克隆可能难以在不知道删除的范围来解释。
- 净化从使用PCR清理试剂盒依照制造商的指令外引物的qPCR反应中得到的扩增子。
- 确认删除的等位基因的通过DNA测序来自前面步骤的纯化的PCR产物的序列。使用定量PCR扩增的引物为正向和反向测序。
注意:在扩增子充当删除等位基因的基因型的二次确认内这一阶段的SNP。
7.等位基因特异性引物表达分析
- 解冻96孔细胞库存复吃了被放置在一个温暖的澡珠储存在-80°C(股票1步骤4.9)。当超过半数在板孔中解冻,旋在300×g离心5分钟。
- 小心,除去密封带,并迅速从删除阳性孔转移细胞进入含有1ml ES细胞培养基( 表1)的明胶包被的12孔板,并在37℃/ 5%CO 2孵育。
- 当板达到70-85%汇合时,通道中的细胞,并分成三个孔中明胶包被,6孔板,每个含2ml ES细胞培养基( 表1)。使用两个孔以制备2瓶细胞冻存物的为在液氮长期贮存(步骤8中描述的)和用于RNA提取的第三阱。
- 使用RNA提取试剂盒提取RNA。
- 通过逆转录(RT)使用按照制造商的协议中的cDNA合成试剂盒的RNA转变100-500毫微克的RNA成cDNA。包括RTñ对于每个RNA样品egative反应监测RNA样本中污染DNA的量。
- 稀释的qPCR之前的cDNA中的1之间的比值:2和1:4;取决于在ES细胞中靶基因的表达水平。
- 上述包括F1基因组DNA作为转录水平的绝对定量标准曲线(从250 5倍稀释至0.08纳克/微升)的说明设置的qPCR。比较,在每个确认删除克隆到一个适当的控制基因感兴趣的基因的各等位基因的表达,例如GAPDH( 表7中所列的引物)。
注意:控制基因的引物不需要是等位基因特异性。等位基因特异性引物的设计是作为与目标区域的异常扩增用于基因分型的引物描述用于RT-qPCR的引物是相同的。基因序列应该使用;如果使用引物对单个外显子或外显子 - 内含子边界(监测初级转录物)的F1基因组DNA,可用于标准曲线。有关RT-qPCR的进一步详情,请参阅Forlenza 等 。 2012 33。
8.冻结股票准备ES细胞的长期储存
- 添加300μl的胰蛋白酶的每个6孔(来自步骤7.3)中并在37℃孵育5分钟。加2ml自旋介质( 见表2),以中和胰蛋白酶和吸管上下数次以解离成单细胞。
- 转移细胞进入一个15ml试管和自旋在300×g离心5分钟。
- 吸出上清液,并加入500μl的ES细胞培养基( 表1)的。吸管上下悬浮细胞。
- 内容转移到1.5 ml离心管管加入500微升2个胚胎干细胞冷冻介质( 表3)。颠倒离心管拌匀管放入无酒精的细胞冷冻集装箱。把这个细胞冷冻容器在-80℃下转前至少12小时克到液氮贮罐。
结果
这里所描述的协议使用的F1 ES细胞,研究基因表达的顺式 -regulation使用CRISPR / Cas9基因组编辑( 图1)产生的单等位基因增强剂删除的细胞。用于基因分型和基因表达的gRNA和等位基因特异性引物的设计是在该方法的关键因素。每个等位基因特异性引物组必须通过qPCR进行验证,以确认等位基因特异性。等位基因特异性引物仅扩增各自的基因组DNA靶标是理想?...
讨论
CRISPR / Cas9介导的基因组编辑技术为基因改造一个简单,快捷,廉价的方法。这里详细生成和分析单等位基因缺失的增强功能性增强特性的方法发生在F1小鼠细胞单核苷酸多态性的优势的。这种类型的方法的优点是:1)单等位基因增强剂缺失不产生时的临界增强剂是从两个等位基因缺失, 即 ,在规定的基因导致细胞杀伤力的蛋白水平大大降低的或改变的发生的混杂影响表型; 2)如果单等位?...
披露声明
笔者看了朱庇特的关于利益冲突的政策,并没有冲突披露。
致谢
We would like to thank all the members of the Mitchell lab for helpful discussions. This work was supported by the Canadian Institutes of Health Research, the Canada Foundation for Innovation and the Ontario Ministry of Research and Innovation (operating and infrastructure grants held by JAM).
材料
| Name | Company | Catalog Number | Comments |
| Phusion High-Fidelity DNA Polymerase | NEB | M0530S | high fidelity DNA polymerase used in gRNA assembly |
| Gibson Assembly Master Mix | NEB | E2611L | |
| gRNA_Cloning Vector | Addgene | 41824 | A target sequence is cloned into this vector to create the gRNA plasmid |
| pCas9_GFP | Addgene | 44719 | Codon-optimized SpCas9 and EGFP co-expression plasmid |
| AflII | NEB | R0520S | |
| EcoRI | NEB | R3101S | |
| Neon Transfection System 100 µL Kit | Life Technologies | MPK10096 | Microporator transfection technology |
| prepGEM | ZyGEM | PT10500 | genomic DNA extraction reagent |
| Nucleo Spin Gel & PCR Clean-up | Macherey-Nagel | 740609.5 | |
| High-Speed Plasmid Mini Kit | Geneaid | PD300 | |
| Maxi Plasmid Kit Endotoxin Free | Geneaid | PME25 | |
| SYBR select mix for CFX | Life Technologies | 4472942 | qPCR reagent |
| iScript cDNA synthesis kit | Bio-rad | 170-8891 | Reverse transcription reagent |
| 0.25% Trypsin with EDTA | Life Technologies | 25200072 | |
| PBS without Ca/Mg2+ | Sigma | D8537 | |
| 0.5 M EDTA | Bioshop | EDT111.500 | |
| HBSS | Life Technologies | 14175095 | |
| 1 M HEPES | Life Technologies | 13630080 | |
| BSA fraction V (7.5%) | Life Technologies | 15260037 | |
| Max Efficiency DH5α competent cells | Invitrogen | 18258012 | |
| FBS | ES cell qualified | FBS is subjected to a prior testing in mouse ES cells for pluripotency | |
| DMSO | Sigma | D2650 | |
| Glutamax | Invitrogen | 35050 | |
| DMEM | Life Technologies | 11960069 | |
| Pencillin/Streptomycin | Invitrogen | 15140 | |
| Sodium pyruvate | Invitrogen | 11360 | |
| Non-essential aminoacid | Invitrogen | 11140 | |
| β-mercaptoethanol | Sigma | M7522 | |
| 96-well plate | Sarstedt | 83.3924 | |
| Sealing tape | Sarstedt | 95.1994 | |
| CoolCell LX | Biocision | BCS-405 | alcohol-free cell freezing container |
| CHIR99021 | Biovision | 1748-5 | Inhibitor for F1 ES cell culture |
| PD0325901 | Invivogen | inh-pd32 | Inhibitor for F1 ES cell culture |
| LIF | Chemicon | ESG1107 | Inhibitor for F1 ES cell culture |
参考文献
- Sagai, T., Hosoya, M., Mizushina, Y., Tamura, M., Shiroishi, T. Elimination of a long-range cis-regulatory module causes complete loss of limb-specific Shh expression and truncation of the mouse limb. Development. 132 (4), 797-803 (2005).
- Kleinjan, D. A., Lettice, L. A. Long-range gene control and genetic disease. Adv Genet. 61, 339-388 (2008).
- Visel, A., Rubin, E. M., Pennacchio, L. A. Genomic views of distant-acting enhancers. Nature. 461 (7261), 199-205 (2009).
- Maurano, M. T., et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 337 (6099), 1190-1195 (2012).
- Heintzman, N. D., et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 459 (7243), 108-112 (2009).
- Shen, Y., et al. A map of the cis-regulatory sequences in the mouse genome. Nature. 488 (7409), 116-120 (2012).
- Johnson, D. S., Mortazavi, A., Myers, R. M., Wold, B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 316 (5830), 1497-1502 (2007).
- Rhee, H. S., Pugh, B. F. Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell. 147 (6), 1408-1419 (2011).
- Whyte, W. A., et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 153 (2), 307-319 (2013).
- Chen, C. Y., Morris, Q., Mitchell, J. A. Enhancer identification in mouse embryonic stem cells using integrative modeling of chromatin and genomic features. BMC Genomics. 13 (1), 152 (2012).
- Patwardhan, R. P., et al. Massively parallel functional dissection of mammalian enhancers in vivo. Nat Biotechnol. 30 (3), 265-270 (2012).
- Melnikov, A., et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat Biotechnol. 30 (3), 271-277 (2012).
- Jinek, M., et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337 (6096), 816-821 (2012).
- Cong, L., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Mali, P., et al. RNA-guided human genome engineering via Cas9. Science. 339 (6121), 823-826 (2013).
- Hsu, P. D., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 31 (9), 827-832 (2013).
- Cho, S. W., et al. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 24 (1), 132-141 (2014).
- Zhou, H. Y., et al. A Sox2 distal enhancer cluster regulates embryonic stem cell differentiation potential. Genes Dev. 28 (24), 2699-2711 (2014).
- Fujii, W., Kawasaki, K., Sugiura, K., Naito, K. Efficient generation of large-scale genome-modified mice using gRNA and CAS9 endonuclease. Nucleic Acids Res. 41 (20), e187 (2013).
- Tuan, D. Y., Solomon, W. B., London, I. M., Lee, D. P. An erythroid-specific, developmental-stage-independent enhancer far upstream of the human 'beta-like globin' genes. Proc Natl Acad Sci U S A. 86 (8), 2554-2558 (1989).
- Amano, T., et al. Chromosomal dynamics at the Shh locus: limb bud-specific differential regulation of competence and active transcription. Dev Cell. 16 (1), 47-57 (2009).
- Li, Y., et al. CRISPR reveals a distal super-enhancer required for Sox2 expression in mouse embryonic stem cells. PLoS One. 9 (12), e114485 (2014).
- Canver, M. C., et al. Characterization of genomic deletion efficiency mediated by clustered regularly interspaced palindromic repeats (CRISPR)/Cas9 nuclease system in mammalian cells. J Biol Chem. 289 (31), 21312-21324 (2014).
- Mlynarczyk-Evans, S., et al. X chromosomes alternate between two states prior to random X-inactivation. PLoS Biol. 4 (6), e159 (2006).
- Lefever, S., Pattyn, F., Hellemans, J., Vandesompele, J. Single-nucleotide polymorphisms and other mismatches reduce performance of quantitative PCR assays. Clin Chem. 59 (10), 1470-1480 (2013).
- Huang, M. M., Arnheim, N., Goodman, M. F. Extension of base mispairs by Taq DNA polymerase: implications for single nucleotide discrimination in PCR. Nucleic Acids Res. 20 (17), 4567-4573 (1992).
- Keane, T. M., et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 477 (7364), 289-294 (2011).
- Yalcin, B., et al. Sequence-based characterization of structural variation in the mouse genome. Nature. 477 (7364), 326-329 (2011).
- Gibson, D. G., et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 6 (5), 343-345 (2009).
- Gibson, D. G., Smith, H. O., Hutchison, C. A., Venter, J. C., Merryman, C. Chemical synthesis of the mouse mitochondrial genome. Nat Methods. 7 (11), 901-903 (2010).
- Ding, Q., et al. Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell. 12 (4), 393-394 (2013).
- Basu, S., Campbell, H. M., Dittel, B. N., Ray, A. Purification of specific cell population by fluorescence activated cell sorting (FACS). J Vis Exp. (41), (2010).
- Forlenza, M., Kaiser, T., Savelkoul, H. F., Wiegertjes, G. F. The use of real-time quantitative PCR for the analysis of cytokine mRNA levels. Methods Mol Biol. 820, 7-23 (2012).
- Wu, J. H., Hong, P. Y., Liu, W. T. Quantitative effects of position and type of single mismatch on single base primer extension. J Microbiol Methods. 77 (3), 267-275 (2009).
- Sanyal, A., Lajoie, B. R., Jain, G., Dekker, J. The long-range interaction landscape of gene promoters. Nature. 489 (7414), 109-113 (2012).
转载和许可
请求许可使用此 JoVE 文章的文本或图形
请求许可探索更多文章
This article has been published
Video Coming Soon
版权所属 © 2025 MyJoVE 公司版权所有,本公司不涉及任何医疗业务和医疗服务。