
È necessario avere un abbonamento a JoVE per visualizzare questo. Accedi o inizia la tua prova gratuita.
Method Article
Generazione CRISPR / Cas9 Mediated monoallelic eliminazioni per studiare la funzione Enhancer nel topo cellule staminali embrionali
In questo articolo
Riepilogo
Experimental validation of enhancer activity is best approached by loss-of-function analysis. Presented here is an efficient protocol that uses CRISPR/Cas9 mediated deletion to study allele-specific regulation of gene transcription in F1 ES cells which contain a hybrid genome (Mus musculus129 x Mus castaneus).
Abstract
Enhancers control cell identity by regulating tissue-specific gene expression in a position and orientation independent manner. These enhancers are often located distally from the regulated gene in intergenic regions or even within the body of another gene. The position independent nature of enhancer activity makes it difficult to match enhancers with the genes they regulate. Deletion of an enhancer region provides direct evidence for enhancer activity and is the gold standard to reveal an enhancer's role in endogenous gene transcription. Conventional homologous recombination based deletion methods have been surpassed by recent advances in genome editing technology which enable rapid and precisely located changes to the genomes of numerous model organisms. CRISPR/Cas9 mediated genome editing can be used to manipulate the genome in many cell types and organisms rapidly and cost effectively, due to the ease with which Cas9 can be targeted to the genome by a guide RNA from a bespoke expression plasmid. Homozygous deletion of essential gene regulatory elements might lead to lethality or alter cellular phenotype whereas monoallelic deletion of transcriptional enhancers allows for the study of cis-regulation of gene expression without this confounding issue. Presented here is a protocol for CRISPR/Cas9 mediated deletion in F1 mouse embryonic stem (ES) cells (Mus musculus129 x Mus castaneus). Monoallelic deletion, screening and expression analysis is facilitated by single nucleotide polymorphisms (SNP) between the two alleles which occur on average every 125 bp in these cells.
Introduzione
Elementi regolatori trascrizionali sono fondamentali per la messa a punto spazio-temporale dell'espressione genica durante lo sviluppo 1 e la modifica di questi elementi può portare a malattia a causa di espressione genica aberranti 2. Molte regioni associate alla malattia identificati da studi di associazione genome wide sono nelle regioni non codificanti e hanno caratteristiche di esaltatori trascrizionale 3-4. Identificare esaltatori e abbinandoli con i geni che regolano è complicato in quanto sono spesso trovano diversi kilobases lontano dai geni che regolano e possono essere attivati in maniera tessuto-specifica 5-6. Previsioni Enhancer sono comunemente basate su segni modificazione degli istoni, complessi mediatore-cohesin e vincolante di trascrizione specifici per tipo di cellula fattori 7-10. Convalida di esaltatori previsti è più spesso fatto attraverso un saggio basato vettore in cui l'enhancer attiva espressione di un gene reporter 11-12. Questi dati forniscono vinformazioni aluable circa il potenziale normativo di sequenze enhancer putativi, ma non rivelano la loro funzione nel loro contesto genomico endogena o identificare i geni che regolano. la modifica del genoma serve come un potente strumento per studiare la funzione di elementi regolatori trascrizionali nel loro contesto endogena di analisi perdita-di-funzione.
I recenti progressi nella modifica del genoma, cioè il / Cas9 sistema di editing genoma CRISPR, facilitano la ricerca della funzione del genoma. Il sistema / Cas9 CRISPR è facile da usare e adattabile per molti sistemi biologici. La proteina Cas9 si rivolge ad un sito specifico nel genoma da un RNA guida (gRNA) 13. Il complesso SpCas9 / gRNA esegue la scansione del genoma per il suo obiettivo di sequenza genomica che deve essere 5 'ad una sequenza protospacer motivo adiacente (PAM), NGG 14-15. appaiamento delle basi del gRNA al suo obiettivo, a 20 nucleotidi (nt) sequenza complementare al gRNA, attiva SpCas9 attività nucleasi con un conseguente Doublpausa e filamento (DSB) 3 bp a monte della sequenza PAM. Specificità è ottenuto attraverso l'accoppiamento base completa nella regione seme gRNA, il 6-12 nt adiacente al PAM; Al contrario, disallineamenti 5 'del seme di solito sono tollerati 16-17. La DSB introdotta può essere riparato entro la fine non omologa unirsi (NHEJ) riparazione del DNA o di omologia riparazione diretta (HDR) mechanisms.NHEJ riparazione del DNA spesso crea inserzione / delezione (indels) di pochi bp al sito di destinazione che possono disturbare l'open reading frame (ORF) di un gene. Per generare delezioni grandi nel genoma due gRNAs, che fiancheggiano la regione di interesse, può essere utilizzato 18-19. Questo approccio è particolarmente utile per lo studio di esaltatori di trascrizione raggruppati in regioni di controllo locus o super-stimolatori che sono più grandi di esaltatori convenzionali 9,18,20-22.
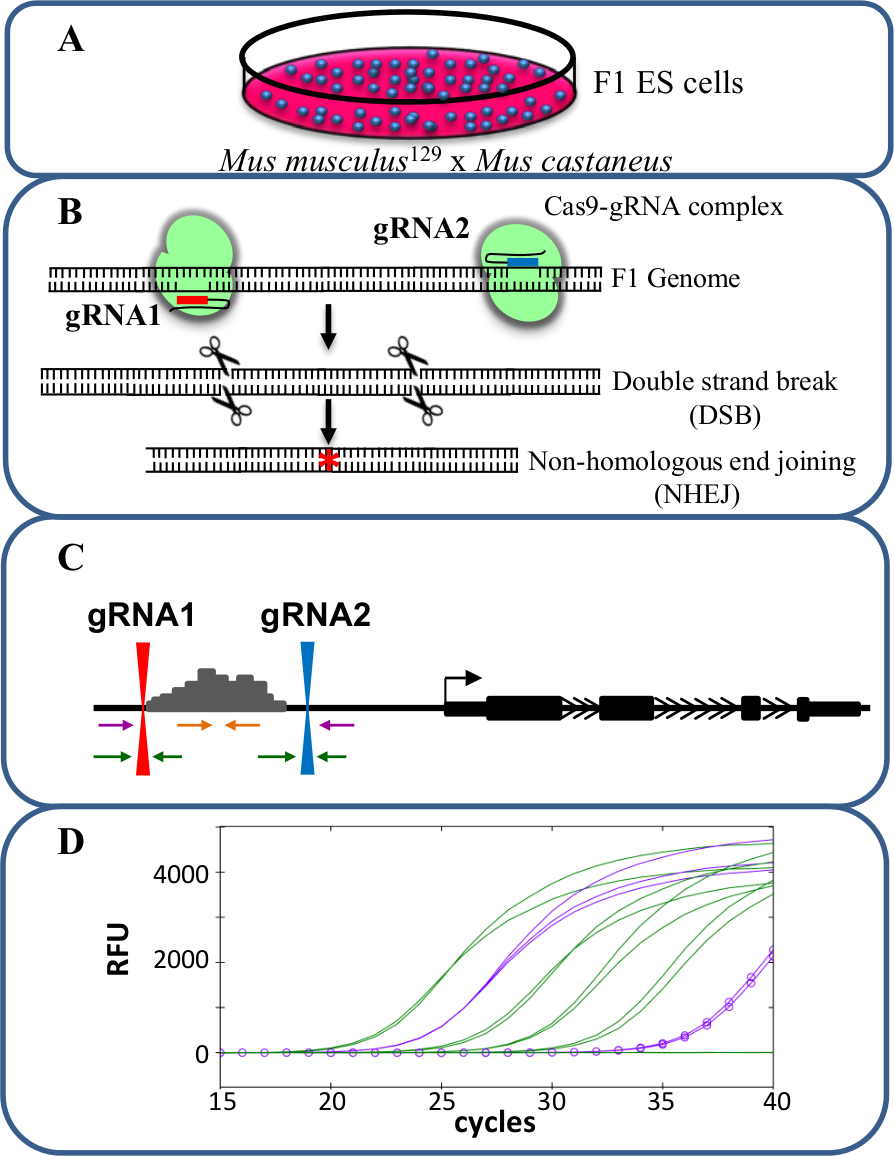
Eliminazioni monoallelic sono un modello valido per lo studio -Regolamento cis di trascrizione. Il chang osservatae nel livello di trascrizione dopo la cancellazione monoallelic di un potenziatore correlato al ruolo di detta enhancer nella regolazione genica senza gli effetti confondenti che possono verificarsi quando la trascrizione di entrambi gli alleli è influenzata potenzialmente influenzare il fitness cellulare. Valutare ridotta espressione è difficile ma senza la capacità di distinguere la eliminato dal tipo di allele selvatico. Inoltre, genotipizzazione delezioni in ogni allele senza la capacità di distinguere i due alleli è impegnativo, soprattutto per le grandi delezioni di> 10 kb a 1 Mb 23 in cui è difficile per amplificare l'intera regione di tipo selvatico mediante PCR. L'uso di cellule ES F1 generati dal passaggio a Mus musculus 129 con Mus castaneus permette ai due alleli per essere differenziati mediante PCR allele-specifica 18,24. Il genoma ibrido in queste cellule facilita allele specifico di screening cancellazione e analisi di espressione. In media c'è un SNP ogni 125 bp tra questi due genomi, Fornendo flessibilità nella progettazione di primer per l'espressione e la genotipizzazione analisi. La presenza di uno SNP può influenzare la temperatura di innesco di fusione (T m) e destinazione specificità amplificazione real-time PCR quantitativa (qPCR) consentendo la discriminazione dei due alleli 25. Inoltre un disadattamento all'interno all'estremità 3 'del primer influenza notevolmente la capacità della DNA polimerasi di estendere dal primer impedendo amplificazione del bersaglio indesiderato allele 26. Descritto nella seguente protocollo è l'uso di cellule ES F1 per l'allele specifico delezioni enhancer di maggiore di 1 KB e la successiva analisi di espressione utilizzando il / Cas9 sistema di editing genoma CRISPR (Figura 1).

Figura 1. eliminazione Enhancer usando CRISPR / Cas9 per studiare cis -REGlamento di espressione genica. cellule (A) F1 ES generati da un incrocio tra Mus musculus 129 e Mus castaneus sono utilizzati per consentire l'eliminazione allele specifico. (B) Due RNA guida (gRNA) vengono utilizzati per indurre una grande delezione Cas9-mediata della regione enhancer. (C) Set di primer sono utilizzati per identificare grande mono e bi-delezioni alleliche. I primer di colore arancione sono i primer all'interno, i primer viola sono i primer esterni ei primer verde sono i primer fiancheggianti gRNA. (D) Le variazioni di espressione genica sono monitorati utilizzando allele-specifica qPCR. RFU denota unità di fluorescenza relativa. Clicca qui per vedere una versione più grande di questa figura.
{kind=link}
Protocollo
1. Progettare e Costruire il gRNA
- Per eliminare regioni enhancer utilizzano due gRNAs, una 5 'e uno 3' della regione di interesse. Utilizzare il mouse UCSC pista del browser genoma generato dal laboratorio Zhang per identificare uniche sequenze gRNA (http://www.genome-engineering.org 15). Successivo controllare questi gRNAs e la loro PAM adiacente per SNP e indels utilizzando strumenti online forniti dal Sanger Institute (www.sanger.ac.uk/sanger/Mouse_SnpViewer/rel-1211) 27-28. Per indirizzare entrambi gli alleli con uguale efficienza, evitare di sequenze / PAM gRNA che contengono un SNP o indel.
- Mentre la scelta del gRNA, verificare la fattibilità di progettazione di primer allele-specifici per la genotipizzazione l'eliminazione. Riferirsi alla sezione 5 per la progettazione di primer allele-specifica.
- Montare i due plasmidi gRNA in base al protocollo descritto in Mali et al. 2013 15. Incorporare l'unico 20 bp sequenza bersaglio selezionato into gli oligonucleotidi 61mer come indicato nella tabella 7 (le sequenze vengono visualizzati nella 5 'a 3' orientamento, e le basi in grassetto sono la sequenza bersaglio 20 bp che sono complementi d'inversione di ogni altro).
- Mescolare 10 ml di 10 mM gRNA Primer_F e 10 ml di 10 micron Primer_R complementare in un tubo.
- Ricuocere i primers incubando la miscela di primer a 100 ° C per 5 minuti e poi raffreddare 1 ° C / sec a 25 ° C. Per questo passaggio, utilizzare una macchina PCR o posizionare il tubo in acqua bollente e lasciare raffreddare a temperatura ambiente.
- Per la miscela di primer ricotta, aggiungere il seguente mix di reazione e incubare a 72 ° C per 30 minuti per estendere ogni primer: 18,5 ml di acqua, 10 ml di tampone 5x HF, 1 ml di 10 mM dNTP Mix e 0,5 l di alta polimerasi fedeltà DNA.
- Eseguire 10 ml di frammento bersaglio su un gel di agarosio al 2% per confermare sono stati prodotti 100 frammenti guida bp.
- Linearizzare il vettore gRNA (un dono di GeoRGE Chiesa; Addgene plasmide # 41824) 15 con Afl II utilizzando la seguente reazione costituita: 5 ml di gRNA vettore backbone (2-4 mg), 5 ml di tampone 10x, 3 microlitri di Afl II (20 unità / microlitri) e 32 microlitri d'acqua. Incubare la miscela di reazione per 3 ore a 37 ° C.
- Eseguire il prodotto digerito su un 1% gel e purificare la band DNA corrispondente alle 3,5 kb linearizzato gRNA vettore utilizzando un kit di estrazione del gel seguendo le istruzioni del produttore.
- Impostare reazioni di assemblaggio Gibson 29-30 usando linearizzato vettore gRNA e indirizzare frammento dal punto 1.2.3 come segue: 1 ml di lineare gRNA vettore (50 ng / ml), 1 ml di frammento bersaglio, 10 ml di 2x Gibson montaggio Master Mix e 8 ml di acqua. Incubare le reazioni a 50 ° C per 60 min.
- Trasformazione di cellule di E.coli con Assemblato gRNA Vector.
- Mescolare 1 ml di vettore gRNA assemblato da 1.2.7e 50 ml di DH5α (ceppo E.coli), le cellule in una provetta. Trasformare le cellule DH5α con il metodo di shock termico esponendo le cellule a 42 ° C per 45 sec.
- Snap raffreddare le provette in ghiaccio per 5 min; poi aggiungere 400 ml di mezzo SOC e incubare a 37 ° C per 45 minuti in un incubatore agitazione.
- Distribuire 100 ml di cellule DH5α su LB-kanamicina (50 ug / ml) Piastra per selezione positiva di cellule trasformate e incubare O / N a 37 ° C.
- Screening positivo E.coli Colonie per gRNA Inserisci.
- Scegliere una colonia resistente kanamicina e risospendere in 3 ml di LB contenente 50 mg / ml di kanamicina. Ripetere la stessa per 6-8 colonie e incubare tutte le provette a 37 ° CO / N in un incubatore agitazione.
- Estrarre plasmidi dal / N cultura adulta O utilizzando plasmide mini kit di preparazione seguendo il manuale del produttore.
- Preparare una miscela di reazione di digestione EcoR I per verificare la sequenza di inserimento gRNAnel plasmide. Per ogni campione, preparare la miscela di reazione come segue: 2 microlitri di tampone enzimatico, 1 ml di EcoR I, 15 ml di acqua. Aliquota la miscela di reazione in provette da 1,5 ml e aggiungere 2 ml di plasmide. Incubare le provette a 37 ° C per 2 ore.
- Eseguire il prodotto digerita su un gel di agarosio 1,5%.
Nota: I campioni con inserto in mostreranno una dimensione band 475 bp che è al 100 bp superiore ai cloni senza inserti.
Nota: In alternativa, i cloni positivi possono essere vagliate da una colonia PCR usando Sp6 (in avanti) e primer T7 (reverse) (Tabella 7) che si legano alla sequenza di vettore per dare un frammento di dimensioni 642 bp in presenza di un gRNA inserto. L'approccio colonia PCR è vantaggiosa quando vi è un sito di restrizione EcoR I all'interno della sequenza gRNA.
- Confermare la sequenza delle gRNA Inserisci per sequenziamento del DNA Utilizzando il T7 Primer.
2. trasfezione
Nota:L'elettroporazione è un metodo efficace di trasfezione plasmidi in cellule ES. Il metodo descritto qui utilizza la tecnologia microporator trasfezione.
- Crescere le cellule ES F1 in un piatto di gelatina-rivestite da 10 cm contenente 10 ml di supporti cellule ES (Tabella 1) a 37 ° C / 5% CO 2. Quando le cellule raggiungono l'85% di confluenza rimuovere i media e aggiungere 2 ml di tripsina. Incubare a 37 ° C in incubatore CO 2 per 5 min.
Nota: le cellule ES F1 sono stati ottenuti da Barbara Panoramica 24 e sono disponibili su richiesta. - Neutralizzare la tripsina aggiungendo 10 ml di mezzi di rotazione (Tabella 2). Pipetta ripetutamente per staccare completamente le cellule.
- Raccogliere tutte le cellule in una provetta da 15 ml e far girare a 300 xg per 5 min. Risospendere in 3 ml di PBS e contare le cellule utilizzando un emocitometro o contatore di cellule automatizzato.
- Pellet 1 x 10 6 cellule ES in una provetta da 1,5 ml per centrifugazione a 300 xg per 5 minuti e risospendere in 100 ml di R (risospensione) tampone come fornito dal produttore del kit.
- Aggiungere 5 mg ciascuno di pCas9_GFP (un dono di Kiran Musunuru; Addgene plasmide # 44719) 31, 5 'e 3' plasmidi gRNA per la cancellazione della regione di destinazione e mescolare delicatamente con una pipetta per evitare l'introduzione di bolle.
- Usare la punta della pipetta elettronica per aspirare 100 ml di mix elettroporazione, facendo attenzione ad evitare una bolla nella punta.
- Programmare il volt, larghezza e impulsi per elettroporazione. Per le cellule ES F1, utilizzare 1.400 V, 10 msec per 3 impulsi.
- Mentre l'elettroporazione è in funzione osservare la punta di guardare per eventuali scintille nella soluzione. Una scintilla indica la presenza di una bolla d'aria e interferisce con la trasfezione.
- Espellere le cellule ES transfettate in un piatto di gelatina rivestite 10 centimetri contenente 10 ml ES supporto cellulare (Tabella 1) e incubare a 37 ° C / 5% di CO 2.
3. FACS Ordinamento cellule transfettate
- Dopo 48 ore, staccare le cellule aggiungendo 2 ml di tripsina e incubare a 37 ° C in incubatore CO 2 per 5 min.
- Neutralizzare il piatto aggiungendo 10 ml di tampone di raccolta (Tabella 3). Raccogliere le cellule in una provetta da 15 ml e far girare a 300 xg per 5 min.
- Eliminare il surnatante e risospendere le cellule in 1 ml di tampone di smistamento (Tabella 4). Contare le celle e diluito basati sulla piattaforma di smistamento. Diluire le cellule a 0,5-1 x 10 6 cellule / ml per l'ordinamento in 15 ml provette e per classificare singole celle direttamente in piastre a 96 pozzetti, diluire le cellule di 2-5 x 10 6 cellule / ml.
- Ordina cellule Cas9-GFP + ES utilizzando un flusso FACS citometro 32. Raccogliere le cellule alla rinfusa in tubi con 2 ml di supporti di ripristino (Tabella 5) e la piastra come descritto in 3.5 per colonia picking, o cellule sorta singoli direttamente in piastre da 96 pozzetti rivestiti di gelatina contenenti 100 microlitri mezzi cellule ES / pozzetto ( Tabella 1).
- Seme 1-1.5 x 10 4 GFP + cellule ES in un piatto di gelatina rivestita 10 cm contenente 10 mL supporti cellule ES (Tabella 1). Placcatura in questa bassa densità faciliterà scegliere i singoli colonie di cellule ES.
4. cloni la coltura per la genotipizzazione, analisi di espressione e degli stock cella di congelamento
- Il giorno 4-5 dopo la cernita, segnare ciascun pozzetto di piastre filtrate diretti 96 pozzetti per la presenza di colonie di cellule ES.
- Dissociarsi colonie di cellule ES rimuovendo il materiale e l'aggiunta di 30 ml di tripsina. Incubare a 37 ° C per 5 min. Neutralizzare la tripsina aggiungendo 170 ml di mezzi di cellule ES (Tabella 1), e pipetta su e giù per una completa dissociazione della colonia in singole cellule. Crescere le cellule a 37 ° C / 5% di CO 2 fino maggior pozzi sono più di 70% confluenti (solitamente 2-3 giorni).
- In alternativa scegliere singole colonie di cellule ES da 10 piatti cm utilizzandoun microscopio invertito. Dopo aspirare la colonia nella punta della pipetta seguito passo 4.1.1 mettendo ogni colonia in un pozzetto di una piastra a 96 pozzetti, pretrattati con gelatina e contenente 30 ml di tripsina.
Nota: Le colonie possono sedersi in tripsina a temperatura ambiente, mentre una intera fila di colonie viene prelevato. - Una volta che tutte le colonie sono state raccolte e dissociato in supporti crescono le cellule a 37 ° C in incubatore CO 2 fino maggior pozzi sono oltre il 70% confluenti (solitamente 2 giorni).
- Quando le piastre a 96 pozzetti sono pronti per la scissione, rimuovere il supporto, aggiungere 30 ml di tripsina e incubare a 37 ° C per 5 min. Neutralizzare la tripsina con l'aggiunta di 180 ml di mezzi di cellule ES (Tabella 1) per ciascun bene e pipetta su e giù per una completa dissociazione in singole cellule.
- Dalla risultante 210 ml, semi di 70 ml in tre rivestito di gelatina piastre a 96 pozzetti contenenti ciascuno 130 ml di ES supporti delle cellule / pozzetto (Tabella 1). Utilizzare questi piatti per genotyping, analisi di espressione e il congelamento delle scorte cellulari per ogni clone come descritto di seguito.
- Quando la piastra di genotipizzazione raggiunge 70-85% di confluenza, trattare la piastra come descritto nel capitolo 6 "Genotipizzazione la cancellazione".
- Quando la piastra analisi di espressione raggiunge 70-85% di confluenza, rimuovere il supporto, sigillare la piastra con nastro di tenuta e conservare a -80 ° C fino a che i cloni sono stati genotipizzati.
Nota: La piastra di analisi di espressione è utile per analizzare i cambiamenti nell'espressione genica nelle prime passaggi dei cloni. analisi di espressione genica dalla piastra a 96 pozzetti è possibile, ma come i numeri di cellulare sono bassi si raccomanda un RNA kit di micro estrazione. - Preparazione di Fermo a 96 pozzetti zolla Stock:
- Quando la piastra per gli stock di cellule congelate (stock-1) raggiunge il 70-85% di confluenza, aspirare i media, aggiungere 30 ml di tripsina e incubare a 37 ° C per 5 minuti.
- Neutralizzare la tripsina aggiungendo 100 ml di mezzi di cellule ES (Tabella 1) to ogni bene e pipetta su e giù per una completa dissociazione in singole cellule.
- Trasferimento 15 ml di cellule in sospensione da ogni pozzetto a due rivestito di gelatina piastre a 96 pozzetti, ciascuno contenente 185 ml di mezzi di cellule ES (Tabella 1) e consentono di crescere a 37 ° C / 5% di CO 2.
Nota: Questo è per magazzino-2 e -3 piatti che sono complementari cloni di back-up nel caso in cui far rivivere le cellule a magazzino-1 non è riuscita.
- Nel frattempo, per i 100 ml di cellule rimanenti nella piastra a 96 pozzetti (magazzino-1), aggiungere 100 ml di 2x congelamento dei media (Tabella 6). Sigillare la piastra con un nastro sigillante e rapidamente capovolgere la piastra 4-5 volte per una corretta miscelazione. Conservare la piastra a -80 ° C fino cloni vengono genotipizzati.
- Quando le piastre magazzino-2 e magazzino-3 sono pronti per il congelamento aspirare i media, aggiungere 30 ml di tripsina e incubare a 37 ° C per 5 minuti. Neutralizzare la tripsina con l'aggiunta di 70 ml di mezzi di cellule ES (Tgrado 1) in ciascun pozzetto e pipetta su e giù per una completa dissociazione in singole cellule.
- Aggiungere 100 ml di 2x supporti di congelamento, sigillare il piatto con del nastro di tenuta e invertire rapidamente la piastra 4-5 volte per una corretta miscelazione. Conservare la piastra a -80 ° C fino servono questi piatti.
5. allele-specifica progettazione Primer
- Progettazione 4 set di primer (Figura 1C) per schermo i cloni per l'eliminazione desiderato: all'interno di primer, primer al di fuori, e gRNA fiancheggiamento primer (per entrambi 5 'e 3' siti di destinazione gRNA) come descritto di seguito.
- Ottenere la pista SNP corrispondente alle 129 e gettato genotipi a http://labs.csb.utoronto.ca/mitchell/crispr.html. Il dato mostra pista sostituzioni di basi tra i 129 ei Fusioni alle coordinate in assemblea genoma MM9 mouse.
Nota: il collegamento al sito di cui sopra reindirizzerà al browser genoma UCSC e aggiungere una traccia personalizzato contenente l'SNP tra il 129 e il cast genomes. - Inserire le coordinate della regione da eliminare. Zoom su una regione di circa 500 bp nel mezzo della eliminazione desiderato contenente> 3 SNP.
- Vai a visualizzare> DNA nella barra delle opzioni e fare clic su ottenere il DNA per scaricare la sequenza bersaglio in tutti i formati maiuscolo.
- Creare due sequenze FASTA; uno per il 129 e una per Fusioni per sostituzione di base nella posizione SNP. Segnare i SNPs da un minuscolo.
- Vai Primer3 più (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi/) e incollare il SNP sostituito 129 sequenza. Utilizzare le impostazioni predefinite per la progettazione di primer.
- Per progettare i primer allele-specifici all'interno selezionare Primer_List nel menu a discesa compito e fare clic su Seleziona Primer. Scegli un primer in avanti o retromarcia che ha un SNP sia in 'fine o all'interno di 4 basi dal 3' del 3 e cade all'interno della regione da eliminare.
Nota: I primer che hanno un SNP sul display 3'end aumentato allele specificità qPCR. - Per selezionare il secondo ritorno fondo alla pagina principale, selezionare Rilevamento nel menu delle applicazioni e incollare la prima sequenza di innesco nella casella appropriata nella parte inferiore della pagina. Nella scheda Impostazioni generali modificare l'impostazione per la gamma di dimensioni del prodotto per 80-200 bp e fare clic su Seleziona Primer. Ha scelto un set di primer dalle coppie di primer quotate; questi saranno i 129 all'interno di primer allele-specifici.
- Ripetere i punti 5.1.5 a 5.1.7 per la progettazione di primer per l'allele Cast.
- Tornare al browser genoma UCSC e inserire le coordinate della regione da eliminare. Vai a visualizzare> DNA nella barra delle opzioni, in Opzioni Sequenza Recupero Regione aggiungere 1.000 bp a monte ea valle cliccare ottenere DNA per scaricare la sequenza bersaglio.
- Segnare la sequenza bersaglio gRNA tra parentesi. Salva questo intera sequenza prima di procedere.
- Per progettare i primer fuori togliere la sequenza tra le due sequenze bersaglio gRNA. Ripetere il passaggio 5.1.4-5.1.8 per progettare l'esterno di primer allele-specificas ma cambiare la dimensione del prodotto 400-800 bp.
- Dividere la sequenza ottenuta nel passaggio 5.1.10 in due sequenze, ciascuna con 500 bp 5 'e 3' della sequenza bersaglio gRNA. Ripetere i punti 5.1.5 a 5.1.7 progettazione di primer specifici non allele per gRNA regioni fiancheggianti, ma cambiare la dimensione del prodotto 400-800 bp.
Nota: Per i primer specifici non allele, o 129 o la sequenza Fusioni può essere utilizzato e primer dovrebbe essere scelto che non contengono un SNP. Si consiglia di impostare una dimensione del prodotto di 400-800 bp per la progettazione di fuori e gRNA di accompagnamento primer. Questo permette di amplificazione, anche se di piccole dimensioni indels sono presenti.
- Ottenere la pista SNP corrispondente alle 129 e gettato genotipi a http://labs.csb.utoronto.ca/mitchell/crispr.html. Il dato mostra pista sostituzioni di basi tra i 129 ei Fusioni alle coordinate in assemblea genoma MM9 mouse.
- Testare i primer interne per allele specificità da qPCR utilizzando puro 129 e Fusioni ceppo DNA genomico a 2 ng / ml. Seguire passo 6,2-6,4 per impostare la reazione qPCR.
Nota: Se il genotipo 129 nella regione di destinazione è la stessa C57BL / 6J, DNA da C57BL / 6J può essere utilizzato in luogo di 129 DNA. primer allele-specifici dovrebbero visualizzare almeno 5 ciclidifferenza tra il valore di Ct (ciclo soglia) la corretta vs. genotipo corretto. I primer esterni possono essere testati per assicurare che amplificano la cancellazione usando Cas9 / gRNA transfettate cellule ES, e controlli F1 DNA genomico rispettivamente positivi e negativi. L'allele-specificità del primer al di fuori può essere testato una volta che i cloni monoallelic sono stati identificati.
6. Genotyping la Cancellazione
- Estrarre il DNA genomico dalla genotipizzazione piastra a 96 pozzetti con la piastra dal punto 4.6 che si genera dopo l'espansione delle colonie.
- Preparare la miscela di estrazione del DNA genomico: 89 ml di acqua, 10 ml di 10x tampone e 1 ml di reagente di estrazione (fornito dal produttore). Aggiungere 100 ml di genomica mix estrazione del DNA di ogni bene e sigillare il piatto con un nastro sigillante.
- Incubare la piastra a 75 ° C per 5 minuti seguiti da 95 ° C per 5 min.
- Lasciare raffreddare la piastra incubando in ghiaccio per alcuni minuti und quindi centrifugare brevemente per risolvere qualsiasi condensazione al fondo del pozzo. Questo serve come il piatto DNA stampo per l'eliminazione di screening.
- Impostare le reazioni qPCR in duplicato per ogni clone come segue: 5 ml di 2x SYBR qPCR mix, in avanti e Primer (3 micron) ogni 1 ml e 1 ml di acqua inversa. Utilizzare una pipetta multicanale per aggiungere 2 ml di DNA stampo seguiti da 8 ml di mix di reazione per ciascun pozzetto di una piastra da 384 pozzetti.
- Sigillare la piastra con nastro di tenuta e far girare a 600 xg per 2 minuti per mescolare il contenuto. Posizionare la matrice piastra a 384 pozzetti nel vero cycler tempo.
- Programma reale cycler tempo per un 2-step PCR seguita da analisi della curva di fusione con rilevamento come segue: 1 ciclo a 95 ° C per 10 minuti, 40 cicli di 95 ° C per 15 s, 62 ° C per 30 sec con piastra lettura e 95 ° C per 10 s, 65 ° C a 95 ° C con incrementi di 5 ° C per 5 sec + piastra lettura.
Nota: oltre a primer progettazione, la qPCR miparametri x e il ciclo contribuiscono anche a primer specificità. I parametri sopra descritti e reagenti elencati nei materiali resa più frequentemente amplificazione allele specifica. - Analizzando qPCR Risultati
- Controllare ogni allele per l'amplificazione con primer allele-specifici all'interno. Nessuna amplificazione di un allele o le differenze di alto valore Ct (> 5 cicli) tra gli alleli suggeriscono questi cloni portano una delezione eterozigote dell'allele con l'assenza alto valore / Ct. Nessuna amplificazione di entrambi gli alleli suggerisce che portano una delezione omozigote.
- Controllare ogni allele per l'amplificazione con l'esterno primer allele-specifici. Quando l'eliminazione di destinazione è maggiore di 1 kb di amplificazione con primer esterni si verifica solo quando una cancellazione è presente. Un valore Ct di 22-28 conferma l'eliminazione. Per cancellazioni bersaglio più piccolo di 1 KB, confermare il formato amplicone mediante elettroforesi.
Nota: Se i primer esterni mostrano solo moderata allele-specificità (vedi FiguRE 2), ampliconi possono essere ottenuti con entrambi i set di primer alleliche in cloni monoallelic a causa di amplificazione del target fuori dei primer esterni. In questo caso la differenza del valore Ct tra i due alleli di almeno cinque cicli dovrebbe confermare l'allele corretta (minor valore Ct) è soppresso in base ai risultati ottenuti con i primer interni. Se il bersaglio contro il fuori allele differenza Ct è inferiore a cinque cicli di progettazione di nuovi allele specifici primer esterni. - In monoallelic eliminazione cloni verificare l'integrità del allele non eliminati utilizzando lo screening secondario, gRNA fiancheggiamento primer.
Nota: indels di> 25 bp dimensioni intorno al sito di destinazione gRNA possono essere identificati osservando uno spostamento della curva di fusione nella qPCR per 400-800 ampliconi bp. In alternativa, gli ampliconi dei primer fiancheggianti gRNA possono essere sequenziato per rilevare piccole indels di <25 bp.- Eseguire qPCR con 2 set di gRNA primer fiancheggianti cioè, 5 'e 3' gRNA usato nella generazione CRISPR eliminazione. Nessuna amplificazione con questi set di primer indica indels più grande della amplicone qPCR sono presenti presso il sito di destinazione gRNA sul allele non cancellato del monoallelic cancellazione cloni. Scartare i cloni che contengono queste grandi indels da ulteriori analisi, come i risultati possono essere difficili da interpretare senza conoscere l'entità della cancellazione.
- Purificare gli ampliconi ottenuti dall'esterno innesco reazione qPCR utilizzando una PCR clean up kit seguendo le istruzioni del produttore.
- Confermare la sequenza dell'allele cancellato dallo DNA sequenziamento del prodotto purificato PCR dal passaggio precedente. Utilizzare i primer di amplificazione qPCR per la marcia avanti e la sequenza inversa.
Nota: A questo SNP fase nell'atto amplicone come una conferma secondaria del genotipo dell'allele cancellato.
7. Espressione Analizzare con alleliche primer specifici
- Scongelare il 96 pozzetti delle cellule stock plate conservati a -80 ° C (magazzino-1 dal punto 4.9) ponendolo in un bagno caldo tallone. Quando più di metà dei pozzetti della piastra viene scongelato, centrifugare a 300 xg per 5 min.
- Cautela, rimuovere il nastro sigillante e trasferire rapidamente le cellule dai pozzetti positivi soppressione in, 12 pozzetti rivestiti di gelatina contenenti 1 ml di mezzi di cellule ES (Tabella 1) e incubare a 37 ° C / 5% CO 2.
- Quando la piastra raggiunge 70-85% di confluenza, le cellule passaggio e suddiviso in tre pozzetti di una piastra da 6 pozzetti rivestiti di gelatina, ciascuno contenente 2 ml di media cellule ES (Tabella 1). Utilizzare due pozzi per preparare 2 fiale di scorte di cellule congelate per la conservazione a lungo termine in azoto liquido (descritto al punto 8) e il terzo pozzo per l'estrazione di RNA.
- Estrarre l'RNA utilizzando un kit di estrazione di RNA.
- Convertire 100-500 ng di RNA a cDNA da trascrizione inversa (RT) l'RNA utilizzando il kit di sintesi di cDNA seguendo il protocollo del produttore. Includere un RT nreazione egative per ciascun campione di RNA per monitorare la quantità di DNA contaminante nei campioni di RNA.
- Diluire il cDNA prima qPCR in un rapporto compreso tra 1: 2 e 1: 4; a seconda del livello di espressione del gene bersaglio in cellule ES.
- Impostare la qPCR come descritto in precedenza compreso F1 DNA genomico come curva standard (5 diluizioni da 250 a 0,08 ng / ml) per la quantificazione assoluta dei livelli di trascrizione. Confrontare l'espressione di ogni allele del gene di interesse in ogni clone soppresso confermato un gene di controllo adatto, per esempio GAPDH (primer elencati nella Tabella 7).
Nota: primer gene di controllo non devono essere allele specifico. progettazione di primer allele-specifica è la stessa per primer RT-qPCR come descritto per primer genotipizzazione con l'eccezione della regione bersaglio per l'amplificazione. La sequenza genica deve essere utilizzato; se utilizzando primer per un singolo esone o un confine esone-introne (per monitorare trascritto primario) DNA genomico F1 può essere utilizzato perla curva standard. Per ulteriori dettagli su RT-qPCR consultare Forlenza et al. 2012 33.
8. gelata Stock Preparazione per la conservazione a lungo termine delle cellule staminali embrionali
- Aggiungere 300 ml di tripsina per ogni 6-bene (dal punto 7.3) e incubare per 5 minuti a 37 ° C. Aggiungere 2 ml di mezzi di rotazione (Tabella 2) per neutralizzare tripsina e pipetta su e giù diverse volte a dissociarsi in singole cellule.
- Trasferire le cellule in una provetta da 15 ml e far girare a 300 xg per 5 min.
- Aspirare il surnatante e aggiungere 500 ml di mezzi di cellule ES (Tabella 1). Pipetta su e giù per risospendere le cellule.
- Trasferire il contenuto da un tubo esageratamente 1,5 ml e aggiungere 500 ml di cella di congelamento supporti 2x ES (Tabella 3). Mescolare bene invertendo il tubo e posizionare il tubo in un contenitore di congelamento delle cellule senza alcool. Posizionare questa cella contenitori di congelamento a -80 ° C per almeno 12 ore prima della transferrinag in un serbatoio di stoccaggio di azoto liquido.
Risultati
Il protocollo qui descritto utilizza cellule ES F1 per studiare -Regolamento cis dell'espressione genica in cellule enhancer cancellato monoallelic generati utilizzando CRISPR / modifica del genoma Cas9 (Figura 1). Il gRNA e la progettazione di primer allele-specifica per la genotipizzazione e dell'espressione genica sono i fattori chiave di questo approccio. Ogni set di primer allele-specifica deve essere convalidato da qPCR per confermare allele specif...
Discussione
tecnologia di editing genoma mediata CRISPR / Cas9 fornisce un metodo semplice, veloce e poco costoso per la modifica del genoma. Il metodo descritto qui per generare e analizzare l'eliminazione enhancer monoallelic per la caratterizzazione funzionale potenziatore sfrutta SNPs nelle cellule di topo F1. I vantaggi di questo tipo di approccio sono: 1) le eliminazioni enhancer monoallelic non producono effetti confondenti che si verificano quando un esaltatore di critica viene eliminato da entrambi gli alleli, cio?...
Divulgazioni
Gli autori hanno letto le politiche di Giove sul conflitto di interessi e non hanno conflitti di rivelare.
Riconoscimenti
We would like to thank all the members of the Mitchell lab for helpful discussions. This work was supported by the Canadian Institutes of Health Research, the Canada Foundation for Innovation and the Ontario Ministry of Research and Innovation (operating and infrastructure grants held by JAM).
Materiali
| Name | Company | Catalog Number | Comments |
| Phusion High-Fidelity DNA Polymerase | NEB | M0530S | high fidelity DNA polymerase used in gRNA assembly |
| Gibson Assembly Master Mix | NEB | E2611L | |
| gRNA_Cloning Vector | Addgene | 41824 | A target sequence is cloned into this vector to create the gRNA plasmid |
| pCas9_GFP | Addgene | 44719 | Codon-optimized SpCas9 and EGFP co-expression plasmid |
| AflII | NEB | R0520S | |
| EcoRI | NEB | R3101S | |
| Neon Transfection System 100 µL Kit | Life Technologies | MPK10096 | Microporator transfection technology |
| prepGEM | ZyGEM | PT10500 | genomic DNA extraction reagent |
| Nucleo Spin Gel & PCR Clean-up | Macherey-Nagel | 740609.5 | |
| High-Speed Plasmid Mini Kit | Geneaid | PD300 | |
| Maxi Plasmid Kit Endotoxin Free | Geneaid | PME25 | |
| SYBR select mix for CFX | Life Technologies | 4472942 | qPCR reagent |
| iScript cDNA synthesis kit | Bio-rad | 170-8891 | Reverse transcription reagent |
| 0.25% Trypsin with EDTA | Life Technologies | 25200072 | |
| PBS without Ca/Mg2+ | Sigma | D8537 | |
| 0.5 M EDTA | Bioshop | EDT111.500 | |
| HBSS | Life Technologies | 14175095 | |
| 1 M HEPES | Life Technologies | 13630080 | |
| BSA fraction V (7.5%) | Life Technologies | 15260037 | |
| Max Efficiency DH5α competent cells | Invitrogen | 18258012 | |
| FBS | ES cell qualified | FBS is subjected to a prior testing in mouse ES cells for pluripotency | |
| DMSO | Sigma | D2650 | |
| Glutamax | Invitrogen | 35050 | |
| DMEM | Life Technologies | 11960069 | |
| Pencillin/Streptomycin | Invitrogen | 15140 | |
| Sodium pyruvate | Invitrogen | 11360 | |
| Non-essential aminoacid | Invitrogen | 11140 | |
| β-mercaptoethanol | Sigma | M7522 | |
| 96-well plate | Sarstedt | 83.3924 | |
| Sealing tape | Sarstedt | 95.1994 | |
| CoolCell LX | Biocision | BCS-405 | alcohol-free cell freezing container |
| CHIR99021 | Biovision | 1748-5 | Inhibitor for F1 ES cell culture |
| PD0325901 | Invivogen | inh-pd32 | Inhibitor for F1 ES cell culture |
| LIF | Chemicon | ESG1107 | Inhibitor for F1 ES cell culture |
Riferimenti
- Sagai, T., Hosoya, M., Mizushina, Y., Tamura, M., Shiroishi, T. Elimination of a long-range cis-regulatory module causes complete loss of limb-specific Shh expression and truncation of the mouse limb. Development. 132 (4), 797-803 (2005).
- Kleinjan, D. A., Lettice, L. A. Long-range gene control and genetic disease. Adv Genet. 61, 339-388 (2008).
- Visel, A., Rubin, E. M., Pennacchio, L. A. Genomic views of distant-acting enhancers. Nature. 461 (7261), 199-205 (2009).
- Maurano, M. T., et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 337 (6099), 1190-1195 (2012).
- Heintzman, N. D., et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 459 (7243), 108-112 (2009).
- Shen, Y., et al. A map of the cis-regulatory sequences in the mouse genome. Nature. 488 (7409), 116-120 (2012).
- Johnson, D. S., Mortazavi, A., Myers, R. M., Wold, B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 316 (5830), 1497-1502 (2007).
- Rhee, H. S., Pugh, B. F. Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell. 147 (6), 1408-1419 (2011).
- Whyte, W. A., et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 153 (2), 307-319 (2013).
- Chen, C. Y., Morris, Q., Mitchell, J. A. Enhancer identification in mouse embryonic stem cells using integrative modeling of chromatin and genomic features. BMC Genomics. 13 (1), 152 (2012).
- Patwardhan, R. P., et al. Massively parallel functional dissection of mammalian enhancers in vivo. Nat Biotechnol. 30 (3), 265-270 (2012).
- Melnikov, A., et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat Biotechnol. 30 (3), 271-277 (2012).
- Jinek, M., et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337 (6096), 816-821 (2012).
- Cong, L., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Mali, P., et al. RNA-guided human genome engineering via Cas9. Science. 339 (6121), 823-826 (2013).
- Hsu, P. D., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 31 (9), 827-832 (2013).
- Cho, S. W., et al. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 24 (1), 132-141 (2014).
- Zhou, H. Y., et al. A Sox2 distal enhancer cluster regulates embryonic stem cell differentiation potential. Genes Dev. 28 (24), 2699-2711 (2014).
- Fujii, W., Kawasaki, K., Sugiura, K., Naito, K. Efficient generation of large-scale genome-modified mice using gRNA and CAS9 endonuclease. Nucleic Acids Res. 41 (20), e187 (2013).
- Tuan, D. Y., Solomon, W. B., London, I. M., Lee, D. P. An erythroid-specific, developmental-stage-independent enhancer far upstream of the human 'beta-like globin' genes. Proc Natl Acad Sci U S A. 86 (8), 2554-2558 (1989).
- Amano, T., et al. Chromosomal dynamics at the Shh locus: limb bud-specific differential regulation of competence and active transcription. Dev Cell. 16 (1), 47-57 (2009).
- Li, Y., et al. CRISPR reveals a distal super-enhancer required for Sox2 expression in mouse embryonic stem cells. PLoS One. 9 (12), e114485 (2014).
- Canver, M. C., et al. Characterization of genomic deletion efficiency mediated by clustered regularly interspaced palindromic repeats (CRISPR)/Cas9 nuclease system in mammalian cells. J Biol Chem. 289 (31), 21312-21324 (2014).
- Mlynarczyk-Evans, S., et al. X chromosomes alternate between two states prior to random X-inactivation. PLoS Biol. 4 (6), e159 (2006).
- Lefever, S., Pattyn, F., Hellemans, J., Vandesompele, J. Single-nucleotide polymorphisms and other mismatches reduce performance of quantitative PCR assays. Clin Chem. 59 (10), 1470-1480 (2013).
- Huang, M. M., Arnheim, N., Goodman, M. F. Extension of base mispairs by Taq DNA polymerase: implications for single nucleotide discrimination in PCR. Nucleic Acids Res. 20 (17), 4567-4573 (1992).
- Keane, T. M., et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 477 (7364), 289-294 (2011).
- Yalcin, B., et al. Sequence-based characterization of structural variation in the mouse genome. Nature. 477 (7364), 326-329 (2011).
- Gibson, D. G., et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 6 (5), 343-345 (2009).
- Gibson, D. G., Smith, H. O., Hutchison, C. A., Venter, J. C., Merryman, C. Chemical synthesis of the mouse mitochondrial genome. Nat Methods. 7 (11), 901-903 (2010).
- Ding, Q., et al. Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell. 12 (4), 393-394 (2013).
- Basu, S., Campbell, H. M., Dittel, B. N., Ray, A. Purification of specific cell population by fluorescence activated cell sorting (FACS). J Vis Exp. (41), (2010).
- Forlenza, M., Kaiser, T., Savelkoul, H. F., Wiegertjes, G. F. The use of real-time quantitative PCR for the analysis of cytokine mRNA levels. Methods Mol Biol. 820, 7-23 (2012).
- Wu, J. H., Hong, P. Y., Liu, W. T. Quantitative effects of position and type of single mismatch on single base primer extension. J Microbiol Methods. 77 (3), 267-275 (2009).
- Sanyal, A., Lajoie, B. R., Jain, G., Dekker, J. The long-range interaction landscape of gene promoters. Nature. 489 (7414), 109-113 (2012).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati