
Se requiere una suscripción a JoVE para ver este contenido. Inicie sesión o comience su prueba gratuita.
Method Article
La generación de CRISPR / Cas9 mediada monoallelic deleciones para estudiar la función del reforzador en las células madre embrionarias de ratón
En este artículo
Resumen
Experimental validation of enhancer activity is best approached by loss-of-function analysis. Presented here is an efficient protocol that uses CRISPR/Cas9 mediated deletion to study allele-specific regulation of gene transcription in F1 ES cells which contain a hybrid genome (Mus musculus129 x Mus castaneus).
Resumen
Enhancers control cell identity by regulating tissue-specific gene expression in a position and orientation independent manner. These enhancers are often located distally from the regulated gene in intergenic regions or even within the body of another gene. The position independent nature of enhancer activity makes it difficult to match enhancers with the genes they regulate. Deletion of an enhancer region provides direct evidence for enhancer activity and is the gold standard to reveal an enhancer's role in endogenous gene transcription. Conventional homologous recombination based deletion methods have been surpassed by recent advances in genome editing technology which enable rapid and precisely located changes to the genomes of numerous model organisms. CRISPR/Cas9 mediated genome editing can be used to manipulate the genome in many cell types and organisms rapidly and cost effectively, due to the ease with which Cas9 can be targeted to the genome by a guide RNA from a bespoke expression plasmid. Homozygous deletion of essential gene regulatory elements might lead to lethality or alter cellular phenotype whereas monoallelic deletion of transcriptional enhancers allows for the study of cis-regulation of gene expression without this confounding issue. Presented here is a protocol for CRISPR/Cas9 mediated deletion in F1 mouse embryonic stem (ES) cells (Mus musculus129 x Mus castaneus). Monoallelic deletion, screening and expression analysis is facilitated by single nucleotide polymorphisms (SNP) between the two alleles which occur on average every 125 bp in these cells.
Introducción
Elementos reguladores de la transcripción son críticos para la puesta a punto espacio-temporal de la expresión génica durante el desarrollo 1 y la modificación de estos elementos puede dar lugar a la enfermedad debido a la expresión aberrante de genes 2. Muchas regiones asociadas a la enfermedad identificados por el genoma estudios de asociación de todo se encuentran en las regiones no codificantes y tienen características de potenciadores de la transcripción 3-4. La identificación de los potenciadores y emparejarlos con los genes que regulan se complica ya que a menudo se encuentran varias kilobases lejos de los genes que regulan y pueden ser activados de una manera específica de tejido 5-6. Predicciones Enhancer se basan comúnmente en marcas de modificación de las histonas, complejos de cohesinas-mediador y la unión de células de transcripción de tipo específico factores de 7-10. Validación de potenciadores predichos a menudo se realiza a través de un ensayo basado en vector en el que el potenciador activa la expresión de un gen indicador 11-12. Estos datos proporcionan valuable información sobre el potencial regulador de las secuencias potenciadoras putativos, pero no revelan su función en su contexto genómico endógeno o identificar los genes que regulan. edición genoma sirve como una poderosa herramienta para estudiar la función de los elementos reguladores de la transcripción en su contexto endógena mediante análisis de la pérdida de función.
Los avances recientes en la edición del genoma, es decir, el sistema de edición de genoma / Cas9 CRISPR, facilitar la investigación de la función del genoma. El sistema / Cas9 CRISPR es fácil de usar y adaptable para muchos sistemas biológicos. La proteína Cas9 está dirigida a un sitio específico en el genoma por un ARN de guía (gRNA) 13. El complejo SpCas9 / gRNA escanea el genoma de su secuencia genómica diana que debe ser de 5 'a una secuencia protospacer motivo adyacente (PAM), NGG 14-15. el apareamiento de bases de la gRNA a su diana, un (nt) secuencia de 20 nucleótidos complementaria a la gRNA, activa la actividad nucleasa SpCas9 que resulta en una doubldescanso e hebra (DSB) 3 pb aguas arriba de la secuencia de PAM. La especificidad se logra a través de apareamiento de bases completa en la región de la semilla gRNA, el 6-12 nt adyacente a la PAM; por el contrario, no corresponde a 5 'de la semilla son generalmente tolerada 16-17. El OSD introducido se puede reparar, ya sea por el extremo no homóloga (NHEJ) de reparación del ADN o reparación dirigida homología de reparación del ADN (HDR) mechanisms.NHEJ menudo crea inserción / deleción (indeles) de unos pocos pares de bases en el sitio diana que puede interrumpir el marco de lectura abierto (ORF) de un gen. Para generar deleciones de mayor tamaño en el genoma de dos gRNAs, que flanquean la región de interés, se puede utilizar 18-19. Este enfoque es particularmente útil para el estudio de los potenciadores de la transcripción agrupadas en regiones de control de locus o super-potenciadores que son más grandes que los potenciadores convencionales 9,18,20-22.
Supresiones monoallelic son un modelo valioso para el estudio de -Reglamento cis de la transcripción. El chang observadoe en el nivel de transcripción después de la eliminación monoallelic de un potenciador se correlaciona con el papel de promotor de que en la regulación de genes, sin los efectos de confusión que pueden ocurrir cuando la transcripción de ambos alelos se ve afectada potencialmente influir en la aptitud celular. Evaluación de la reducción de expresión es difícil, sin embargo, sin la capacidad de distinguir el borrado de el alelo de tipo salvaje. Además, la genotipificación de deleciones en cada alelo sin la capacidad de distinguir los dos alelos es un reto, especialmente para las grandes deleciones de> 10 kb a 1 Mb 23 en el que es difícil para amplificar toda la región de tipo salvaje por PCR. El uso de células ES F1 generados por musculus cruce Mus 129 con Mus castaneus permite que los dos alelos que se diferencian por alelo-específico PCR 18,24. El genoma híbrido en estas células facilita alelo cribado deleción específica y análisis de la expresión. En promedio hay un SNP cada 125 pb entre estos dos genomas, Proporcionando flexibilidad en el diseño de cebadores para la expresión y genotipado analiza. La presencia de un SNP puede influir en la temperatura de fusión del cebador (T m) y apuntar a la especificidad en la amplificación cuantitativa en tiempo real PCR (qPCR) lo que permite la discriminación de los dos alelos 25. Además, una falta de coincidencia dentro de el extremo 3 'del cebador influye en gran medida la capacidad de la ADN polimerasa para extender el cebador de la prevención de la amplificación de la diana alelo no deseado 26. Describe en el siguiente protocolo es el uso de células madre embrionarias para la F1 deleciones específicas de alelo potenciador de más de 1 kb y el análisis posterior expresión que utilizan el sistema de CRISPR / Cas9 edición genoma (Figura 1).

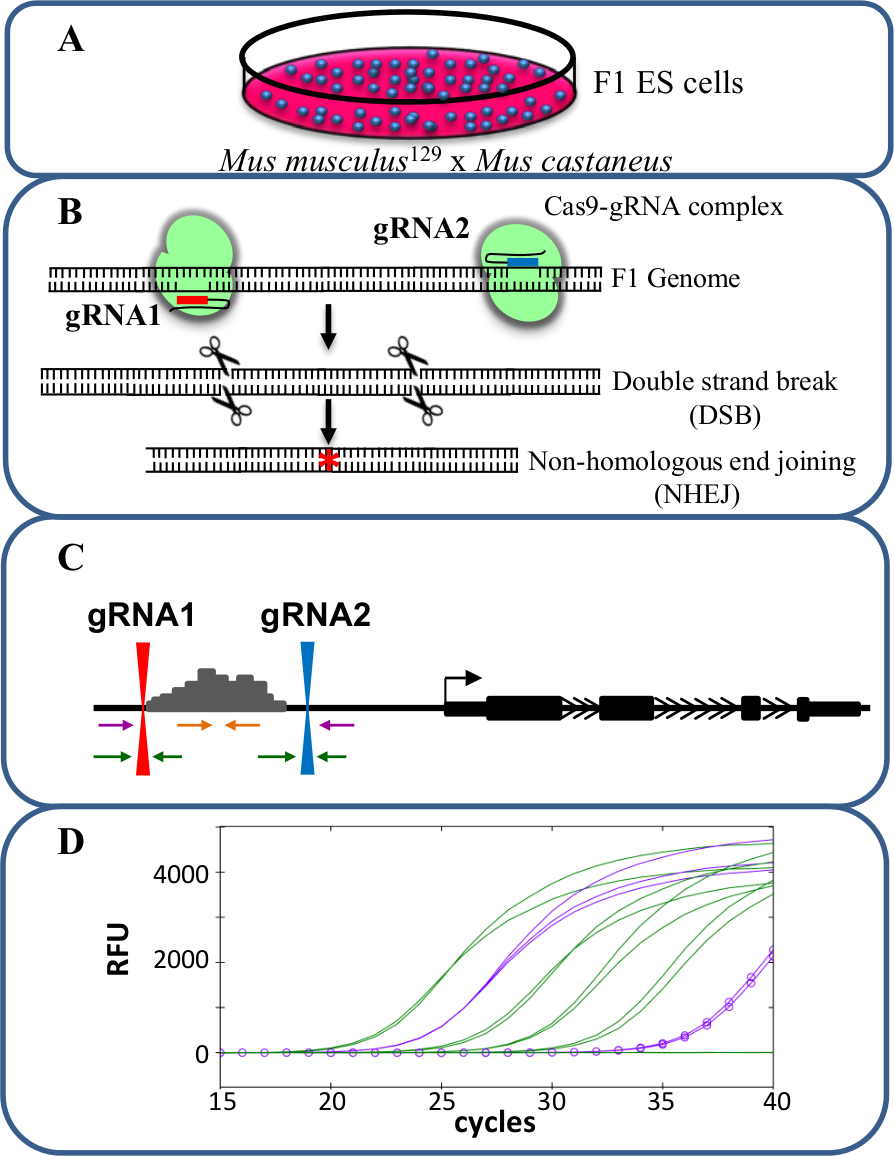
Figura 1. eliminación reforzador usando CRISPR / Cas9 para estudiar cis -regulación de la expresión génica. células F1 ES (A) generados por un cruce entre Mus musculus 129 y castaneus Mus se utilizan para permitir la supresión alelo específico. (B) dos ARN de guía (gRNA) se utilizan para inducir una gran deleción mediada por Cas9 de la región potenciadora. (C) Los conjuntos de cebadores se utilizan para identificar gran mono- y bi-deleciones alélicas. Los cebadores de color naranja son los iniciadores en el interior, los cebadores de color púrpura son los cebadores externos y los cebadores verdes son los cebadores que flanquean gRNA. (D) Los cambios en la expresión génica son monitoreados mediante qPCR específica de alelo. RFU denota unidades relativas de fluorescencia. Por favor, haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Protocolo
1. Diseño y construcción de la gRNA
- Para eliminar regiones potenciadoras transcripcionales utilizan dos, uno gRNAs 5 'y un 3' de la región de interés. Utilice la pista navegador UCSC genoma del ratón generados por el laboratorio de Zhang para identificar secuencias únicas gRNA (http://www.genome-engineering.org 15). A continuación comprobar estos gRNAs PAM y su adyacente para SNPs y indeles utilizando herramientas en línea proporcionados por el Instituto Sanger (www.sanger.ac.uk/sanger/Mouse_SnpViewer/rel-1211) 27-28. Para hacer referencia a los dos alelos con la misma eficacia, evitar secuencias / PAM gRNA que contienen un SNP o indel.
- Si bien la elección del gRNA, comprobar la viabilidad de diseñar cebadores específicos de alelo para el genotipado de la eliminación. Consulte la sección 5 para el diseño del cebador específico de alelo.
- Montar los dos plásmidos gRNA basado en el protocolo descrito en Mali et al. 2013 15. Incorporar el único de 20 pb secuencia diana seleccionada into los oligonucleótidos 61mer como se muestra en la Tabla 7 (secuencias se muestran en la 5 'a 3', y las bases en negrita son la secuencia diana 20 pb que son complementos inversos el uno del otro).
- Mezclar 10 l de 10 mM gRNA Primer_F y 10 l de 10 mM Primer_R complementaria en un tubo.
- Recocido de los cebadores mediante la incubación de la mezcla de cebadores a 100 ° C durante 5 min y luego enfriar 1 ° C / seg a 25 ° C. Para este paso, utilizar una máquina de PCR o colocar el tubo en agua hirviendo y dejar que se enfríe a temperatura ambiente.
- A la mezcla de cebadores recocidos, agregue la siguiente mezcla de reacción y se incuba a 72 ° C durante 30 minutos para extender cada cebador: 18,5 l de agua, 10 l de 5x tampón HF, 1 l de dNTP 10 mM mezclar y 0,5 l de alto ADN fidelidad de la polimerasa.
- Ejecutar 10 l de fragmento diana en un gel de agarosa al 2% para confirmar los 100 fragmentos de guía pb se han producido.
- Linealizar el vector gRNA (un regalo de GeoIglesia RGE; Addgene plásmido # 41824) 15 con Afl II mediante el uso de la siguiente reacción creó: 5 l de gRNA cadena principal del vector (2-4 g), 5 l de 10x tampón, 3 l de Afl II (20 unidades / l) y 32 l de agua. Incubar la mezcla de reacción durante 3 horas a 37 ° C.
- Ejecutar el producto digerido en un gel de agarosa al 1% y purificar la banda de ADN correspondiente a los 3,5 kb linealizado gRNA vector usando un kit de extracción de gel, siguiendo las instrucciones del fabricante.
- Configurar Gibson reacciones de montaje 29 a 30 usando linealizado vector gRNA y orientar fragmento de la etapa 1.2.3 como sigue: 1 l de vector gRNA lineal (50 ng / l), 1 l de fragmento diana, 10 l de montaje 2x Gibson mezcla maestra y 8 l de agua. Se incuban las reacciones a 50 ° C durante 60 minutos.
- La transformación de células de E. coli con Montado gRNA Vector.
- Mezclar 1 l del vector gRNA montado a partir de 1.2.7y 50 l de células en un tubo de DH5a (cepa de E. coli). Transformar las células DH5a por el método de choque de calor mediante la exposición de las células a 42 ° C durante 45 seg.
- Snap enfriar los tubos en hielo durante 5 min; a continuación, añadir 400 l de medio SOC y se incuba a 37 ° C durante 45 minutos en un incubador con agitación.
- Extender 100 l de las células DH5a en una LB-kanamicina (50 mg / ml) de placa para la selección positiva de células transformadas y se incuba O / N a 37 ° C.
- Las colonias de E. coli de detección positiva para gRNA Insertar.
- Elige una colonia resistente a la kanamicina y resuspender en 3 ml de LB que contiene 50 g / ml de kanamicina. Repetir lo mismo para 6-8 colonias e incubar los tubos a 37 ° CO / N en una incubadora con agitación.
- Extraer los plásmidos de la O / N cultivo crecido utilizando plásmido mini kit de preparación siguiendo el manual del fabricante.
- Preparar una mezcla de reacción de digestión EcoRI para verificar si hay gRNA secuencia del insertoen el plásmido. Para cada muestra, preparar la mezcla de reacción como sigue: 2 l de tampón de enzima, 1 l de EcoR I, 15 l de agua. Alícuota de la mezcla de reacción en tubos de 1,5 ml y añadir 2 l de plásmido. Incubar los tubos a 37 ° C durante 2 hr.
- Ejecutar el producto digerido en un gel de agarosa al 1,5%.
Nota: Las muestras con inserto mostrará un tamaño de banda de 475 pb que es 100 pb por encima de los clones sin insertos.
Nota: Como alternativa, los clones positivos se pueden cribar por una colonia PCR utilizando SP6 (hacia adelante) y T7 (inverso) primers (Tabla 7) que se unen a la secuencia del vector para dar un fragmento tamaño de 642 pb en presencia de un gRNA inserto. La colonia enfoque PCR es ventajoso cuando hay un sitio de restricción EcoR I dentro de la secuencia gRNA.
- Confirmar la secuencia de la gRNA Insertar por secuenciación de ADN Uso de la T7 Primer.
2. La transfección
Nota:La electroporación es un método eficiente de transfección de plásmidos en células ES. El método descrito aquí utiliza la tecnología microporator transfección.
- Se cultivan las células ES F1 en un plato de 10 cm recubierto con gelatina que contiene 10 ml de medio de células ES (Tabla 1) a 37 ° C / 5% de CO 2. Cuando las células alcanzan el 85% de confluencia eliminar los medios de comunicación y añadir 2 ml de tripsina. Incubar a 37 ° C en la incubadora de CO 2 durante 5 min.
Nota: Se obtuvieron células madre embrionarias a partir de F1 Barbara toma panorámica 24 y están disponibles bajo petición. - Neutralizar la tripsina mediante la adición de 10 ml de medio giro (Tabla 2). Pipeta varias veces para separar completamente las células.
- Recoge todas las células en un tubo de 15 ml y centrifugado a 300 xg durante 5 min. Resuspender en 3 ml de PBS y el recuento de las células utilizando un hemocitómetro o un contador de células automatizado.
- Se precipitan 1 x 10 6 células ES en un tubo de 1,5 ml por centrifugación a 300 xg durante 5 min y se resuspenden en 100 l de R (resuspensión) tampón suministrado por el fabricante del kit.
- Añadir 5 g de cada uno de pCas9_GFP (un regalo de Kiran Musunuru; Addgene plásmido # 44719) 31, 5 'y 3' plásmidos gRNA para supresión de la región de destino y mezclar suavemente con una pipeta para evitar la introducción de burbujas.
- Utilice la punta de la pipeta electrónica para aspirar 100 l de la mezcla de electroporación, teniendo cuidado de evitar una burbuja en la punta.
- Programa de la voltios, anchura y pulsos de electroporación. Para las células madre embrionarias F1, utilice 1.400 V, 10 ms de 3 pulsos.
- Mientras que la electroporación se está ejecutando observar la punta para detectar cualquier chispas en la solución. Una chispa indica la presencia de una burbuja de aire y va a interferir con la transfección.
- Expulsar las células ES transfectadas en un plato de 10 cm recubierto con gelatina que contiene 10 ml de medio de células ES (Tabla 1) y se incuba a 37 ° C / 5% de CO 2.
3. clasificación FACS células transfectadas
- Después de 48 horas, separar las células mediante la adición de 2 ml de tripsina y se incuba a 37 ° C en el incubador de CO2 durante 5 min.
- Neutralizar la placa mediante la adición de 10 ml de tampón de colección (tabla 3). Recoger las células en un tubo de 15 ml y centrifugado a 300 xg durante 5 min.
- Eliminar el sobrenadante y resuspender las células en 1 ml de tampón de clasificación (Tabla 4). Contar las células y diluir basados en la plataforma de clasificación. Diluir las células a 0,5-1 x 10 6 células / ml para la clasificación en tubos de 15 ml y para la clasificación de las células individuales directamente en placas de 96 pocillos, diluir las células de 2-5 x 10 6 células / ml.
- Ordenar las células GFP + Cas9-ES utilizando un citómetro de flujo FACS 32. Recoger las células a granel en tubos con 2 ml de medio de recuperación (Tabla 5) y la placa como se describe en 3.5 para el picking de colonia, o células de tipo individuales directamente en placas de 96 pocillos recubiertos de gelatina que contienen 100 l medio de células ES / pocillo ( Tabla 1).
- Seed 1-1,5 x 10 4 GFP + ES células en un plato de 10 cm recubierto con gelatina que contiene 10 ml de medio de células ES (Tabla 1). Plating a esta baja densidad facilitará recoger colonias de células ES individuales.
4. Los clones de cultivar durante genotipificación, análisis de expresión y las poblaciones de congelación celular
- En los días 4-5 después de la clasificación, la puntuación de cada pocillo de placas de 96 pocillos según directos para la presencia de colonias de células ES.
- Disociar las colonias de células ES mediante la eliminación de los medios de comunicación y la adición de 30 l de tripsina. Incubar a 37 ° C durante 5 min. Neutralizar la tripsina mediante la adición de 170 l de medio de células ES (Tabla 1), y la pipeta hacia arriba y abajo para la disociación completa de la colonia en células individuales. Cultivar las células a 37 ° C / 5% de CO 2 hasta que la mayoría de los pozos son más de 70% de confluencia (generalmente 2-3 días).
- Alternativamente recoger colonias de células ES individuales de placas de 10 cm usandoun microscopio invertido. Después de aspirar la colonia en la etapa de seguimiento punta de la pipeta 4.1.1 colocando cada colonia en un pocillo de una placa de 96 pocillos, previamente tratado con gelatina y que contiene 30 l de tripsina.
Nota: Las colonias pueden sentarse en tripsina a temperatura ambiente mientras se recoge toda una hilera de colonias. - Una vez que todas las colonias han sido recogidos y disociado en medios crecen las células a 37 ° C en el incubador de CO2 hasta que la mayoría de los pozos son más de 70% confluentes (por lo general 2 días).
- Cuando las placas de 96 pocillos están listos para partir, retirar los medios de comunicación, añadir 30 l de tripsina y se incuba a 37 ° C durante 5 min. Neutralizar la tripsina mediante la adición de 180 l de medio de células ES (Tabla 1) a cada pocillo y la pipeta hacia arriba y abajo para la disociación completa en células individuales.
- De los 210 l resultante, sembrar 70 l en tres placas de 96 pocillos recubiertos con gelatina que contienen cada uno 130 l de medio de células ES / pocillo (Tabla 1). Utilice estas placas para genotyping, análisis de la expresión y la congelación de las poblaciones de células para cada clon como se describe a continuación.
- Cuando la placa de genotipificación alcanza el 70-85% de confluencia, el tratamiento de la placa como se describe en la sección 6 "Genotipificación la eliminación".
- Cuando la placa de análisis de expresión alcanza el 70-85% de confluencia, retirar los medios de comunicación, sellar la placa con cinta de sellado y almacenar a -80 ° C hasta que los clones se han determinado el genotipo.
Nota: La placa de análisis de la expresión es útil para analizar los cambios en la expresión génica en los primeros pasos de los clones. La expresión de genes de la placa de 96 pocillos es posible, pero como el número de células son bajos, se recomienda un kit de extracción de micro ARN. - Preparación de congelación de 96 pocillos de la placa:
- Cuando la placa de las poblaciones de células congeladas (stock-1) alcanza el 70-85% de confluencia, aspirar los medios de comunicación, añadir 30 l de tripsina y se incuba a 37 ° C durante 5 min.
- Neutralizar la tripsina mediante la adición de 100 l de medio de células ES (Tabla 1) tO cada pocillo y la pipeta hacia arriba y abajo para la disociación completa en células individuales.
- Transferencia de 15 l de células suspendidas de cada pocillo a dos placas de 96 pocillos recubiertos con gelatina, que contienen cada uno 185 l de medio de células ES (Tabla 1) y permitir que crezca a 37 ° C / 5% de CO 2.
Nota: Esto es para la acción-2 y -3 placas que son clones de copia de seguridad adicionales en caso de que la reactivación de las células de la acción-1 no tiene éxito.
- Mientras tanto, para los 100 l de las células restantes en la placa de 96 pocillos (Stock-1), añadir 100 l de 2x medio de congelación (Tabla 6). Sellar la placa con una cinta de sellado y rápidamente se invierta la placa 4-5 veces por una mezcla adecuada. Guarde la placa a -80 ° C hasta que los clones son genotipo.
- Cuando el caldo-2 y social-3 placas están listas para la congelación de aspirado de los medios de comunicación, añadir 30 l de tripsina y se incuba a 37 ° C durante 5 min. Neutralizar la tripsina mediante la adición de 70 l de medio de células ES (Tcapaz 1) a cada pocillo y la pipeta hacia arriba y abajo para la disociación completa en células individuales.
- Añadir 100 l de 2x medios de congelación, sellar la placa con cinta aislante e invertir rápidamente la placa de 4-5 veces para una mezcla adecuada. Guarde la placa a -80 ° C hasta que se necesitan estas placas.
Primer diseño 5. El alelo-específica
- Diseño 4 conjuntos de cebadores (Figura 1C) a la pantalla de clones para la deleción deseada: dentro de cebadores, cebadores externos, y gRNA cebadores flanqueantes (por tanto 5 'y 3' sitios diana gRNA) como se describe a continuación.
- Obtener la pista SNP correspondientes a los 129 genotipos y moldeada en http://labs.csb.utoronto.ca/mitchell/crispr.html. La pista dada muestra sustituciones de bases entre 129 y moldeada en las coordenadas en el conjunto del genoma del ratón mm.9.
Nota: El enlace en el sitio anterior se redirigirá a la UCSC genoma navegador y añadir una pista personalizada que contiene los SNPs entre el 129 y moldeada genomos. - Introduzca las coordenadas de la región que desea eliminar. Zoom en una región de aproximadamente 500 pb en el medio de la supresión deseado que contiene> 3 SNPs.
- Ir a ver> ADN en la barra de opciones y haga clic en el ADN llegar a descargar la secuencia diana en todos los formatos de mayúsculas.
- Crear dos secuencias FASTA; uno de 129 y otro para moldeada por sustitución de bases en la posición de SNP. Marcar los SNPs por una letra minúscula.
- Ir a Primer3 más (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi/) y pegue el SNP 129 sustituido secuencia. Estos valores son válidos para diseñar cebadores.
- Para diseñar los cebadores específicos de alelo dentro Primer_List seleccionar en el menú desplegable de tareas y haga clic en Seleccionar cebadores. Elija un cebador directo o inverso que tiene un SNP, ya sea en el extremo o dentro de las 4 bases desde el extremo 3 '3 y cae dentro de la región que desea eliminar.
Nota: Los cebadores que tienen un SNP en el extremo 3 'de visualización mayor especificidad de alelo en qPCR. - Para seleccionar la segunda vuelta de imprimación a la página principal, seleccione Detección en el menú desplegable de tareas y pegar la primera secuencia del cebador en la casilla correspondiente en la parte inferior de la página. En la ficha Configuración general cambiar el ajuste de rango de tamaño de producto a 80-200 pb y haga clic en Seleccionar cebadores. Eligió un conjunto de cebadores de los pares de cebadores enumerados; estos serán los 129 cebadores dentro específicos de alelo.
- Repita los pasos 5.1.5 a 5.1.7 para diseñar cebadores para el alelo moldeada.
- Regresan a la UCSC genoma navegador y entrar en las coordenadas de la región que desea eliminar. Ir a ver> ADN en la barra de opciones, en Opciones de secuencia de recuperación Región añadir 1.000 pb aguas arriba y aguas abajo haga clic en obtener ADN para descargar la secuencia diana.
- Marque la secuencia diana gRNA entre paréntesis. Guardar esta secuencia entera antes de proceder.
- Para diseñar los cebadores fuera de suprimir la secuencia entre las dos secuencias diana gRNA. Repita el paso 5.1.4-5.1.8 para diseñar el cebador específico de alelo fueras, pero cambiar el tamaño del producto 400-800 pb.
- Dividir la secuencia obtenida en el paso 5.1.10 en dos secuencias, cada una con 500 pares de bases 5 'y 3' de la secuencia diana gRNA. Repita los pasos 5.1.5 a 5.1.7 el diseño de cebadores específicos de alelo no para las regiones que flanquean gRNA pero cambiar el tamaño del producto 400-800 pb.
Nota: Para los cebadores específicos de alelo no, ya sea 129 o secuencia fundido se pueden utilizar y cebadores debe ser elegido que no contienen un SNP. Es aconsejable establecer un tamaño de producto de 400-800 pb para el diseño exterior y gRNA cebadores flanqueantes. Esto permite la amplificación aunque contenga pequeños indeles están presentes.
- Obtener la pista SNP correspondientes a los 129 genotipos y moldeada en http://labs.csb.utoronto.ca/mitchell/crispr.html. La pista dada muestra sustituciones de bases entre 129 y moldeada en las coordenadas en el conjunto del genoma del ratón mm.9.
- Prueba de los cebadores dentro de especificidad alelo por qPCR utilizando ADN genómico de la cepa 129 y moldeada puro a 2 ng / l. Siga el paso 6,2-6,4 para configurar la reacción de PCR cuantitativa.
Nota: Si el genotipo 129 en la región diana es el mismo que C57BL / 6J, el ADN de C57BL / 6J se puede utilizar en el lugar de 129 ADN. cebadores específicos de alelo deben mostrar al menos 5 ciclosdiferencia entre el valor Ct (ciclo umbral) en la relación correcta entre el genotipo incorrecta. Los cebadores externos pueden ser probados para asegurar que amplifican la supresión usando Cas9 / gRNA transfectaron las células ES, y los controles F1 ADN genómico, respectivamente, como positivos y negativos. El alelo-especificidad de los cebadores externos puede ser revisada una vez se han identificado los clones monoallelic.
6. El genotipado la Supresión
- Extraer el ADN genómico a partir de la placa de 96 pocillos de genotipificación utilizando la placa de paso 4.6 que se genera después de la expansión de colonias.
- Preparar la mezcla de extracción de ADN genómico: 89 l de agua, 10 l de tampón 10x y 1 l de reactivo de extracción (suministrado por el fabricante). Añadir 100 l de mezcla de extracción de ADN genómico a cada pocillo y sellar la placa con una cinta de sellado.
- Incubar la placa a 75 ° C durante 5 min seguido de 95 ° C durante 5 min.
- Dejar que la placa se enfríe por incubación en hielo durante unos minutos unad después centrifugar brevemente para resolver cualquier condensación en el fondo del pozo. Esto sirve como la placa de plantilla de ADN para la detección de la eliminación.
- Configurar las reacciones de qPCR por duplicado para cada clon de la siguiente manera: 5 l de mezcla 2x SYBR qPCR, hacia adelante y el cebador (3 M) cada 1 l y 1 l de agua inversa. Usar una pipeta multicanal para añadir 2 l de ADN molde seguido de 8 l de mezcla de reacción a cada pocillo de una placa de 384 pocillos.
- Sellar la placa con cinta de sellado y centrifugado a 600 xg durante 2 minutos para mezclar el contenido. Coloque el conjunto de placa de 384 pocillos en el termociclador en tiempo real.
- Programa de la verdadera ciclador tiempo para una PCR de 2 etapas seguido de análisis de la curva de fusión con la detección de la siguiente manera: 1 ciclo a 95 ° C durante 10 min, 40 ciclos de 95 ° C durante 15 s, 62 ° C durante 30 seg con lectura placa y 95 ° C durante 10 s, 65 ° C a 95 ° C con un incremento de 5 ° C durante 5 seg + placa de leer.
Nota: Además de la cartilla de diseño, la qPCR millasparámetros x y el ciclo también contribuyen al cebador especificidad. Los parámetros descritos anteriormente y los reactivos mencionados en los materiales más frecuentemente producen la amplificación específica de alelo. - El análisis de qPCR resultados
- Compruebe cada alelo para la amplificación con cebadores específicos de alelo en el interior. Sin amplificación de un alelo o diferencias de alto valor Ct (> 5 ciclos) entre los alelos sugieren estos clones llevan una deleción heterocigota del alelo con el valor alto / ausente Ct. No amplificación de ambos alelos sugiere que llevan una deleción homocigótica.
- Compruebe cada alelo para la amplificación con cebadores externos específicos de alelo. Cuando la supresión de destino es mayor que 1 kb de amplificación con los cebadores externos se produce sólo cuando una deleción está presente. Un valor de Ct de 22-28 confirma la eliminación. Para las supresiones objetivo de menos de 1 kb, confirmar el tamaño de amplificación por electroforesis.
Nota: Si los cebadores externos muestran sólo moderada alelo-especificidad (ver Figure 2), los amplicones se pueden obtener con las dos conjuntos de cebadores alélicas en clones monoallelic debido a la amplificación de la diana fuera de los cebadores externos. En este caso una diferencia de valor de Ct entre los dos alelos de al menos cinco ciclos debe confirmar el alelo correcto (menor valor Ct) se elimina la base de los resultados obtenidos a partir de los cebadores en el interior. Si el objetivo en frente de alelo para la diferencia de Ct es menos de cinco ciclos de diseño de nuevos alelos cebadores externos específicos. - En clones de deleción monoallelic comprobar la integridad del alelo no eliminada mediante el uso de la selección secundaria, gRNA cebadores flanqueantes.
Nota: Indeles de> 25 pb tamaño alrededor del sitio de destino gRNA se pueden identificar mediante la observación de un desplazamiento de la curva de fusión en el qPCR de 400-800 pb amplicones. Como alternativa, los amplicones de los cebadores que flanquean gRNA pueden ser secuenciados para detectar pequeños indeles de <25 pb.- Realizar qPCR con 2 juegos de cebadores que flanquean gRNA es decir, 5 'y 3' gRNA utilizado en la generación de deleción CRISPR. No amplificación con estos conjuntos de cebadores indica indeles más grandes que el amplicón qPCR están presentes en el sitio diana gRNA en el alelo no borrado de clones de deleción monoallelic. Descartar clones que contienen estos grandes indeles del análisis adicional como los resultados pueden ser difíciles de interpretar sin conocer la extensión de la eliminación.
- Purificar los amplicones obtenidos a partir de la reacción de PCR cuantitativa imprimación exterior mediante una PCR kit de limpieza siguiendo las instrucciones del fabricante.
- Confirmar la secuencia del alelo suprimido por ADN la secuenciación del producto de PCR purificado de la etapa anterior. Usa los cebadores de amplificación qPCR para avance y la secuencia inversa.
Nota: En esta etapa SNPs en el acto de amplificación como una confirmación secundaria del genotipo del alelo suprimido.
7. Analizar la expresión de alelos con cebadores específicos
- Descongelar el 96 pocillos celular stock plate almacenado a -80 ° C (Stock-1 desde el paso 4.9) colocándolo en un baño caliente del grano. Cuando más de la mitad de los pocillos en la placa se descongeló, centrifugado a 300 xg durante 5 min.
- Con cuidado, retire la cinta de sellado y rápidamente la transferencia de las células de los pocillos positivos de deleción en, placas revestidas de gelatina 12 pocillos que contenían 1 ml de medio de células ES (Tabla 1) y se incuba a 37 ° C / 5% de CO 2.
- Cuando la placa alcanza 70 a 85% de confluencia, el paso de las células y los divide en tres pocillos de una placa de 6 pocillos recubierta con gelatina, que contienen cada uno 2 ml de medio de células ES (Tabla 1). Use dos pozos para preparar 2 viales de poblaciones de células congeladas para el almacenamiento a largo plazo en nitrógeno líquido (descrito en la etapa 8) y la tercera bien para la extracción de RNA.
- Extracto de ARN usando un kit de extracción de ARN.
- Convertir 100-500 ng de ARN a ADNc mediante transcripción inversa (RT) del ARN utilizando el kit de síntesis de ADNc siguiendo el protocolo del fabricante. Incluir un RT nreacción egative para cada muestra de ARN para controlar la cantidad de ADN contaminante en las muestras de ARN.
- Diluir el ADNc antes de qPCR en una relación de entre 1: 2 y 1: 4; dependiendo del nivel de expresión del gen diana en células madre embrionarias.
- Ajuste el qPCR como se ha descrito anteriormente, incluyendo ADN genómico de la F1 como curva estándar (5 diluciones desde 250 a 0,08 ng / l) para la cuantificación absoluta de los niveles de transcripción. Comparación de la expresión de cada alelo del gen de interés en cada clon eliminado confirmado que un gen de control adecuado, por ejemplo Gapdh (cebadores listados en la Tabla 7).
Nota: cebadores de genes de control no necesitan ser específica de alelo. el diseño del cebador específico de alelo es el mismo para los cebadores de RT-qPCR como se describe para los cebadores de genotipado con la excepción de la región diana para la amplificación. La secuencia del gen se debe utilizar; Si el uso de cebadores para un solo exón o un límite exón-intrón (para monitorear transcrito primario) ADN genómico F1 se puede utilizar parala curva estándar. Para más detalles sobre la RT-qPCR consulte Forlenza et al. 2012 33.
8. Freeze de la Preparación para el almacenamiento a largo plazo de las células madre embrionarias
- Añadir 300 l de tripsina a cada 6 pocillos (de la etapa 7.3) y se incuba durante 5 min a 37 ° C. Añadir 2 ml de medio giro (Tabla 2) para neutralizar la tripsina y la pipeta arriba y abajo varias veces para disociarse en células individuales.
- Transferencia de las células en un tubo de 15 ml y centrifugado a 300 xg durante 5 min.
- Aspirar el sobrenadante y añadir 500 l de medio de células ES (Tabla 1). Pipetear arriba y hacia abajo para volver a suspender las células.
- Transferir el contenido a un tubo criovial de 1,5 ml y añadir 500 l de medio de congelación de células 2x ES (Tabla 3). Mezclar bien invirtiendo el tubo y colocar el tubo en un recipiente de congelación de células sin alcohol. Colocar esta célula recipientes de congelación a -80 ° C durante al menos 12 h antes de la transferrinag en un tanque de almacenamiento de nitrógeno líquido.
Resultados
El protocolo descrito aquí utiliza células F1 ES para estudiar -regulación cis de la expresión génica en las células potenciador eliminado monoallelic generados usando CRISPR / edición genoma Cas9 (Figura 1). El gRNA y el diseño del cebador específico de alelo para el genotipado y la expresión génica son los factores clave en este enfoque. Cada conjunto de cebadores alelo-específica debe ser validado por qPCR para confirmar la especificidad de alelo....
Discusión
CRISPR / Cas9 tecnología de edición genoma mediada proporciona un método sencillo, rápido y barato para la modificación del genoma. El método detallado aquí para generar y analizar eliminación potenciador monoallelic para la caracterización funcional potenciador se aprovecha de SNPs en células de ratón F1. Las ventajas de este tipo de enfoque son: 1) deleciones potenciador monoallelic no producen efectos de confusión que se producen cuando un promotor de crítico se elimina de ambos alelos, es decir,
Divulgaciones
Los autores han leído las políticas de Júpiter sobre el conflicto de intereses y no tienen conflictos a revelar.
Agradecimientos
We would like to thank all the members of the Mitchell lab for helpful discussions. This work was supported by the Canadian Institutes of Health Research, the Canada Foundation for Innovation and the Ontario Ministry of Research and Innovation (operating and infrastructure grants held by JAM).
Materiales
| Name | Company | Catalog Number | Comments |
| Phusion High-Fidelity DNA Polymerase | NEB | M0530S | high fidelity DNA polymerase used in gRNA assembly |
| Gibson Assembly Master Mix | NEB | E2611L | |
| gRNA_Cloning Vector | Addgene | 41824 | A target sequence is cloned into this vector to create the gRNA plasmid |
| pCas9_GFP | Addgene | 44719 | Codon-optimized SpCas9 and EGFP co-expression plasmid |
| AflII | NEB | R0520S | |
| EcoRI | NEB | R3101S | |
| Neon Transfection System 100 µL Kit | Life Technologies | MPK10096 | Microporator transfection technology |
| prepGEM | ZyGEM | PT10500 | genomic DNA extraction reagent |
| Nucleo Spin Gel & PCR Clean-up | Macherey-Nagel | 740609.5 | |
| High-Speed Plasmid Mini Kit | Geneaid | PD300 | |
| Maxi Plasmid Kit Endotoxin Free | Geneaid | PME25 | |
| SYBR select mix for CFX | Life Technologies | 4472942 | qPCR reagent |
| iScript cDNA synthesis kit | Bio-rad | 170-8891 | Reverse transcription reagent |
| 0.25% Trypsin with EDTA | Life Technologies | 25200072 | |
| PBS without Ca/Mg2+ | Sigma | D8537 | |
| 0.5 M EDTA | Bioshop | EDT111.500 | |
| HBSS | Life Technologies | 14175095 | |
| 1 M HEPES | Life Technologies | 13630080 | |
| BSA fraction V (7.5%) | Life Technologies | 15260037 | |
| Max Efficiency DH5α competent cells | Invitrogen | 18258012 | |
| FBS | ES cell qualified | FBS is subjected to a prior testing in mouse ES cells for pluripotency | |
| DMSO | Sigma | D2650 | |
| Glutamax | Invitrogen | 35050 | |
| DMEM | Life Technologies | 11960069 | |
| Pencillin/Streptomycin | Invitrogen | 15140 | |
| Sodium pyruvate | Invitrogen | 11360 | |
| Non-essential aminoacid | Invitrogen | 11140 | |
| β-mercaptoethanol | Sigma | M7522 | |
| 96-well plate | Sarstedt | 83.3924 | |
| Sealing tape | Sarstedt | 95.1994 | |
| CoolCell LX | Biocision | BCS-405 | alcohol-free cell freezing container |
| CHIR99021 | Biovision | 1748-5 | Inhibitor for F1 ES cell culture |
| PD0325901 | Invivogen | inh-pd32 | Inhibitor for F1 ES cell culture |
| LIF | Chemicon | ESG1107 | Inhibitor for F1 ES cell culture |
Referencias
- Sagai, T., Hosoya, M., Mizushina, Y., Tamura, M., Shiroishi, T. Elimination of a long-range cis-regulatory module causes complete loss of limb-specific Shh expression and truncation of the mouse limb. Development. 132 (4), 797-803 (2005).
- Kleinjan, D. A., Lettice, L. A. Long-range gene control and genetic disease. Adv Genet. 61, 339-388 (2008).
- Visel, A., Rubin, E. M., Pennacchio, L. A. Genomic views of distant-acting enhancers. Nature. 461 (7261), 199-205 (2009).
- Maurano, M. T., et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 337 (6099), 1190-1195 (2012).
- Heintzman, N. D., et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 459 (7243), 108-112 (2009).
- Shen, Y., et al. A map of the cis-regulatory sequences in the mouse genome. Nature. 488 (7409), 116-120 (2012).
- Johnson, D. S., Mortazavi, A., Myers, R. M., Wold, B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 316 (5830), 1497-1502 (2007).
- Rhee, H. S., Pugh, B. F. Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell. 147 (6), 1408-1419 (2011).
- Whyte, W. A., et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 153 (2), 307-319 (2013).
- Chen, C. Y., Morris, Q., Mitchell, J. A. Enhancer identification in mouse embryonic stem cells using integrative modeling of chromatin and genomic features. BMC Genomics. 13 (1), 152 (2012).
- Patwardhan, R. P., et al. Massively parallel functional dissection of mammalian enhancers in vivo. Nat Biotechnol. 30 (3), 265-270 (2012).
- Melnikov, A., et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat Biotechnol. 30 (3), 271-277 (2012).
- Jinek, M., et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337 (6096), 816-821 (2012).
- Cong, L., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Mali, P., et al. RNA-guided human genome engineering via Cas9. Science. 339 (6121), 823-826 (2013).
- Hsu, P. D., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 31 (9), 827-832 (2013).
- Cho, S. W., et al. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 24 (1), 132-141 (2014).
- Zhou, H. Y., et al. A Sox2 distal enhancer cluster regulates embryonic stem cell differentiation potential. Genes Dev. 28 (24), 2699-2711 (2014).
- Fujii, W., Kawasaki, K., Sugiura, K., Naito, K. Efficient generation of large-scale genome-modified mice using gRNA and CAS9 endonuclease. Nucleic Acids Res. 41 (20), e187 (2013).
- Tuan, D. Y., Solomon, W. B., London, I. M., Lee, D. P. An erythroid-specific, developmental-stage-independent enhancer far upstream of the human 'beta-like globin' genes. Proc Natl Acad Sci U S A. 86 (8), 2554-2558 (1989).
- Amano, T., et al. Chromosomal dynamics at the Shh locus: limb bud-specific differential regulation of competence and active transcription. Dev Cell. 16 (1), 47-57 (2009).
- Li, Y., et al. CRISPR reveals a distal super-enhancer required for Sox2 expression in mouse embryonic stem cells. PLoS One. 9 (12), e114485 (2014).
- Canver, M. C., et al. Characterization of genomic deletion efficiency mediated by clustered regularly interspaced palindromic repeats (CRISPR)/Cas9 nuclease system in mammalian cells. J Biol Chem. 289 (31), 21312-21324 (2014).
- Mlynarczyk-Evans, S., et al. X chromosomes alternate between two states prior to random X-inactivation. PLoS Biol. 4 (6), e159 (2006).
- Lefever, S., Pattyn, F., Hellemans, J., Vandesompele, J. Single-nucleotide polymorphisms and other mismatches reduce performance of quantitative PCR assays. Clin Chem. 59 (10), 1470-1480 (2013).
- Huang, M. M., Arnheim, N., Goodman, M. F. Extension of base mispairs by Taq DNA polymerase: implications for single nucleotide discrimination in PCR. Nucleic Acids Res. 20 (17), 4567-4573 (1992).
- Keane, T. M., et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 477 (7364), 289-294 (2011).
- Yalcin, B., et al. Sequence-based characterization of structural variation in the mouse genome. Nature. 477 (7364), 326-329 (2011).
- Gibson, D. G., et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 6 (5), 343-345 (2009).
- Gibson, D. G., Smith, H. O., Hutchison, C. A., Venter, J. C., Merryman, C. Chemical synthesis of the mouse mitochondrial genome. Nat Methods. 7 (11), 901-903 (2010).
- Ding, Q., et al. Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell. 12 (4), 393-394 (2013).
- Basu, S., Campbell, H. M., Dittel, B. N., Ray, A. Purification of specific cell population by fluorescence activated cell sorting (FACS). J Vis Exp. (41), (2010).
- Forlenza, M., Kaiser, T., Savelkoul, H. F., Wiegertjes, G. F. The use of real-time quantitative PCR for the analysis of cytokine mRNA levels. Methods Mol Biol. 820, 7-23 (2012).
- Wu, J. H., Hong, P. Y., Liu, W. T. Quantitative effects of position and type of single mismatch on single base primer extension. J Microbiol Methods. 77 (3), 267-275 (2009).
- Sanyal, A., Lajoie, B. R., Jain, G., Dekker, J. The long-range interaction landscape of gene promoters. Nature. 489 (7414), 109-113 (2012).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados