
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
CRISPR Génération / cas9 Mediated monoallélique Suppressions pour étudier la fonction Enhancer dans souris cellules souches embryonnaires
Dans cet article
Résumé
Experimental validation of enhancer activity is best approached by loss-of-function analysis. Presented here is an efficient protocol that uses CRISPR/Cas9 mediated deletion to study allele-specific regulation of gene transcription in F1 ES cells which contain a hybrid genome (Mus musculus129 x Mus castaneus).
Résumé
Enhancers control cell identity by regulating tissue-specific gene expression in a position and orientation independent manner. These enhancers are often located distally from the regulated gene in intergenic regions or even within the body of another gene. The position independent nature of enhancer activity makes it difficult to match enhancers with the genes they regulate. Deletion of an enhancer region provides direct evidence for enhancer activity and is the gold standard to reveal an enhancer's role in endogenous gene transcription. Conventional homologous recombination based deletion methods have been surpassed by recent advances in genome editing technology which enable rapid and precisely located changes to the genomes of numerous model organisms. CRISPR/Cas9 mediated genome editing can be used to manipulate the genome in many cell types and organisms rapidly and cost effectively, due to the ease with which Cas9 can be targeted to the genome by a guide RNA from a bespoke expression plasmid. Homozygous deletion of essential gene regulatory elements might lead to lethality or alter cellular phenotype whereas monoallelic deletion of transcriptional enhancers allows for the study of cis-regulation of gene expression without this confounding issue. Presented here is a protocol for CRISPR/Cas9 mediated deletion in F1 mouse embryonic stem (ES) cells (Mus musculus129 x Mus castaneus). Monoallelic deletion, screening and expression analysis is facilitated by single nucleotide polymorphisms (SNP) between the two alleles which occur on average every 125 bp in these cells.
Introduction
Des éléments de régulation transcriptionnelle sont critiques pour réglage fin spatio-temporelle de l' expression des gènes au cours du développement 1 et la modification de ces éléments peut entraîner une maladie due à l' expression génique aberrante 2. De nombreuses régions associées à des maladies identifiées par le génome des études d'association sont larges dans les régions non codantes et présentent des caractéristiques d'amplificateurs de transcription 3-4. L' identification des amplificateurs et les harmoniser avec les gènes qu'ils régulent est compliqué car ils sont souvent situés à plusieurs kilobases à l' écart des gènes qu'ils régulent et peuvent être activées d'une manière spécifique au tissu 5-6. Prévisions Enhancer sont généralement basées sur des marques de modification des histones, des complexes de médiateur-cohesin et la liaison de la cellule transcription spécifique de type facteurs 7-10. La validation des amplificateurs de prédits est le plus souvent réalisé par un dosage à base de vecteur dans lequel l'activateur active l' expression d'un gène rapporteur 11-12. Ces données fournissent vinformations ressources précieuses sur le potentiel de régulation des séquences amplificatrices putatives, mais ne révèlent pas leur fonction dans leur contexte génomique endogène ou d'identifier les gènes qu'ils régulent. édition du génome est un outil puissant pour étudier la fonction des éléments de régulation transcriptionnelle dans leur contexte endogène par l'analyse de perte de fonction.
Les progrès récents dans l'édition du génome, à savoir le / cas9 système de montage du génome CRISPR, faciliter l'enquête de la fonction du génome. Le système / cas9 CRISPR est facile à utiliser et adaptable à de nombreux systèmes biologiques. La protéine cas9 est ciblé à un site spécifique par une ARN de guidage (ARNg) 13 dans le génome. Le complexe SpCas9 / gARN analyse du génome pour sa séquence génomique cible qui doit être 5 'à une séquence protospacer motif adjacent (PAM), NGG 14-15. Appariement de bases de l'ARNg à sa cible, un 20 nucleotides (nt) la séquence complémentaire de l'ARNg, active une activité nuclease SpCas9 résultant en un doublpause e brin (DSB) de 3 pb en amont de la séquence PAM. La spécificité est obtenue par appariement complet de la base dans la région de la graine gARN, le 12.06 nt adjacent à l'APM; à l' inverse, mal assortit 5 'de la graine sont généralement tolérés 16-17. L'ORD a introduit peut être réparé soit par l'extrémité non homologue de jonction (NHEJ) réparation de l'ADN ou d'homologie de réparation dirigée (HDR) mechanisms.NHEJ réparation de l'ADN crée souvent insertion / délétion (indels) de quelques pb au niveau du site cible qui peut perturber le cadre ouvert de lecture (ORF) d'un gène. Pour générer de plus grandes deletions dans le génome de deux ARNg, qui flanquent la région d'intérêt, peuvent être utilisés 18 à 19. Cette approche est particulièrement utile pour l'étude des amplificateurs de transcription regroupés en régions de contrôle de locus ou super-amplificateurs qui sont plus grands que des amplificateurs conventionnels 9,18,20-22.
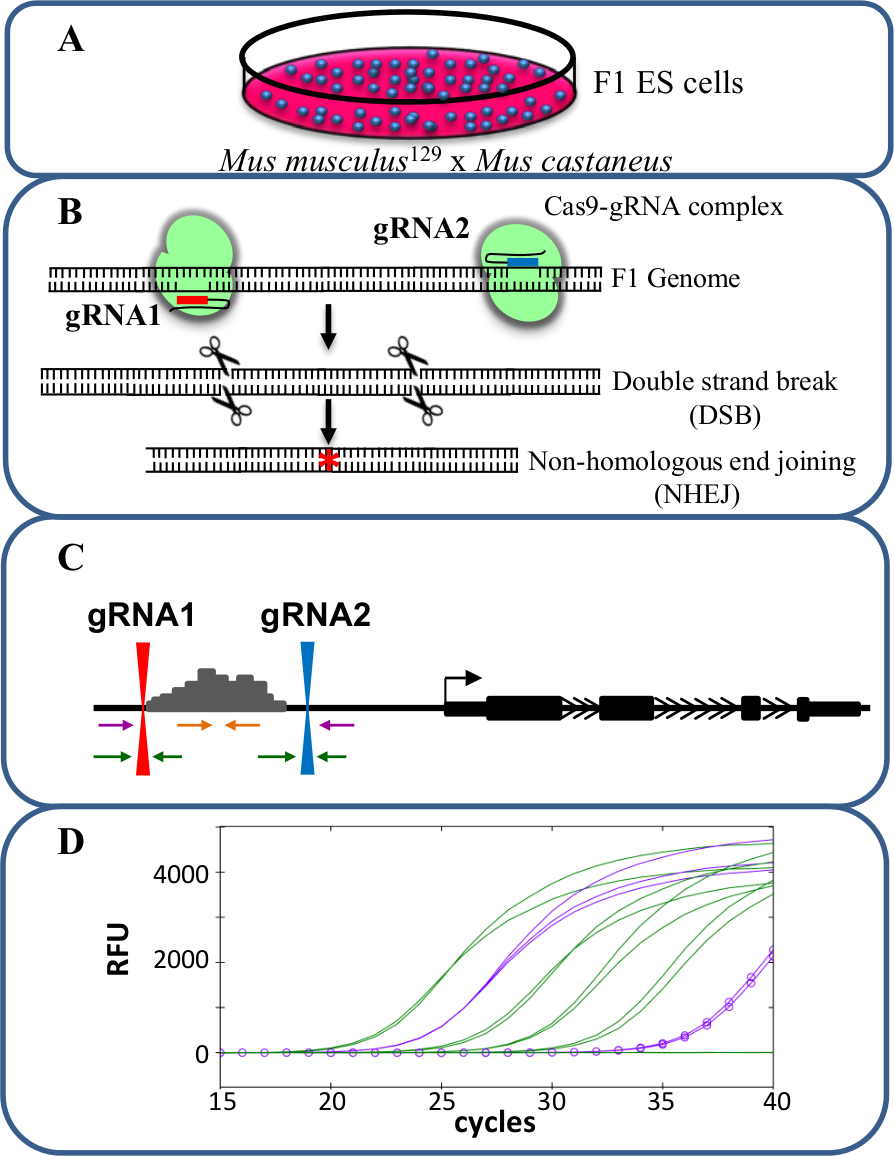
Suppressions monoallélique sont un modèle précieux pour l' étude cis - régulation de la transcription. Le chang observée au niveau de la transcription après la suppression monoallélique d'un activateur est en corrélation avec le rôle de cet activateur dans la régulation des gènes sans les effets de confusion qui peuvent se produire lorsque la transcription des deux allèles est affecté potentiellement influencer remise en forme cellulaire. L'évaluation de l'expression réduite est difficile sans toutefois la capacité de distinguer la suppression de l'allèle de type sauvage. En outre, le génotypage délétions à chaque allèle sans la capacité de distinguer les deux allèles est difficile, surtout pour les grandes suppressions de> 10 kb à 1 Mb 23 , dans lequel il est difficile d'amplifier toute la région de type sauvage par PCR. L'utilisation de cellules ES F1 générées par croisement avec 129 Mus musculus Mus castaneus permet aux deux allèles d'être différenciés par PCR spécifique d'allèle 18,24. Le génome hybride dans ces cellules facilite le dépistage de la suppression spécifique de l'allèle et l'analyse d'expression. En moyenne, il y a un SNP tous les 125 pb entre ces deux génomes, Offrant une flexibilité dans la conception de l'amorce d'expression et de génotypage analyse. La présence d'un SNP peut influer sur la température d' amorce de fusion (Tm) et la spécificité de cible en temps réel par PCR quantitative (qPCR) d' amplification permettant une discrimination des deux allèles 25. En outre , un mésappariement à l'intérieur de l' extrémité 3 'de l'amorce influence grandement la capacité de l' ADN polymérase pour s'étendre à partir de l'amorce empêchant l' amplification de l'allèle cible indésirable 26. Décrite dans le protocole suivant est l'utilisation de cellules ES F1 pour allèle spécifique enhancer suppressions de plus de 1 kb et analyse de l' expression suivante en utilisant le / cas9 système de montage du génome CRISPR (figure 1).

Figure 1. suppression Enhancer utilisant CRISPR / cas9 pour étudier cis -regulation de l' expression génique. (A) Les cellules ES F1 générées par un croisement entre Mus musculus 129 et Mus castaneus sont utilisés pour permettre la suppression allèle spécifique. (B) Deux ARN de guidage (ARNg) sont utilisés pour induire une grande deletion à médiation par cas9 de la région amplificatrice. (C) Les amorces sont utilisées pour identifier les grandes mono- et délétions bi-alléliques. Les amorces orange sont les amorces à l'intérieur, les amorces violet sont les amorces externes et les amorces vertes sont les amorces flanquant gARN. (D) Les changements dans l' expression des gènes sont surveillés à l' aide spécifique d'allèle qPCR. RFU désigne des unités de fluorescence relative. S'il vous plaît cliquez ici pour voir une version plus grande de cette figure.
{kind=link}
Protocole
1. Concevoir et construire l'gARN
- Pour supprimer les régions activatrices transcriptionnelles utilisent deux ARNg, une 5 'et une 3' de la région d'intérêt. Utilisez la piste de UCSC de la souris du navigateur du génome généré par le laboratoire Zhang pour identifier des séquences uniques gARN (http://www.genome-engineering.org 15). Vérifiez ensuite ces ARNg et leur PAM adjacent pour les SNP et indels en utilisant des outils en ligne fournis par l'Institut Sanger (www.sanger.ac.uk/sanger/Mouse_SnpViewer/rel-1211) 27-28. Pour cibler les deux allèles avec une efficacité égale, éviter gARN séquences / PAM qui contiennent un SNP ou indel.
- Tout en choisissant le gARN, vérifier la faisabilité de concevoir des amorces spécifiques d'allèles pour le génotypage la suppression. Reportez-vous à la section 5 pour la conception d'amorce allèle spécifique.
- Assembler les deux plasmides gARN basés sur le protocole décrit au Mali et al. 2013 15. Incorporer le 20 pb séquence cible int uniques sélectionnéeo les oligonucléotides 61mer comme indiqué dans le tableau 7 ( les séquences sont affichés dans le sens 5 'à 3', et les bases sont en gras la séquence cible de 20 pb , qui sont les compléments inverses de l'autre).
- Mélanger 10 ul de 10 uM gARN Primer_F et 10 pl de 10 pM Primer_R complémentaire dans un tube.
- Anneler les amorces par incubation du mélange d'amorce à 100 ° C pendant 5 min, puis refroidir à 1 ° C / s à 25 ° C. Pour cette étape, utiliser une machine PCR ou placer le tube dans l'eau bouillante et laisser refroidir à température ambiante.
- Pour le mélange amorce hybridée, ajouter le mélange réactionnel suivant et incuber à 72 ° C pendant 30 min pour étendre chaque amorce: 18,5 pi d'eau, 10 pl de tampon 5x HF, 1 pl de mM dNTP 10 mélanger et 0,5 pi de haut polymérase fidélité ADN.
- Exécuter 10 ul de fragment cible sur un gel d'agarose à 2% pour confirmer les fragments de 100 pb de guidage ont été produites.
- Linéariser le vecteur gARN (un cadeau de GeoEglise rge; Addgene plasmide # 41824) 15 avec Afl II en utilisant la réaction suivante : mise en place: 5 pi de gARN squelette du vecteur (2-4 pg), 5 pi de tampon 10x, 3 pi de Afl II (20 unités / ul) et 32 ul de l'eau. Incuber le mélange réactionnel pendant 3 heures à 37 ° C.
- Exécuter le produit de digestion sur un gel d'agarose à 1% et on purifie la bande d'ADN correspondant aux 3,5 kb gARN vecteur linéarisé en utilisant un kit d'extraction de gel en suivant les instructions du fabricant.
- Mettre en place des réactions d'assemblage Gibson 29-30 linéarisé en utilisant vecteur gARN et le fragment de l' étape 1.2.3 cible comme suit: 1 pl de vecteur linéaire gARN (50 ng / pl), 1 pl de fragment cible, 10 pl de 2x Gibson ensemble master mix et 8 pi d'eau. Incuber réactions à 50 ° C pendant 60 min.
- Transformation de cellules de E. coli avec assemblé gARN Vector.
- Mélanger 1 pl du vecteur gARN assemblé à partir de 1.2.7et 50 ul de DH5a (souche E. coli) des cellules dans un tube. Transformer les cellules DH5a par la méthode du choc thermique en exposant les cellules à 42 ° C pendant 45 secondes.
- Snap refroidir les tubes sur de la glace pendant 5 min; puis ajouter 400 ul de milieu SOC et incuber à 37 ° C pendant 45 min dans un incubateur à agitation.
- Étaler 100 pi des cellules DH5a sur un LB-kanamycine (50 pg / ml) plaque pour la sélection positive des cellules transformées et incuber O / N à 37 ° C.
- Dépistage positif E. coli Colonies pour gARN Insérer.
- Choisir une colonie résistante à la kanamycine et à remettre en suspension dans 3 ml de LB contenant 50 ug / ml de kanamycine. Répétez la même pour 6-8 colonies et incuber tous les tubes à 37 ° CO / N dans un incubateur à agitation.
- Extraire des plasmides de l'O / N adulte culture utilisant le plasmide mini kit de préparation en suivant le manuel du fabricant.
- Préparer un mélange de réaction de digestion EcoR I pour vérifier la séquence insert gARNdans le plasmide. Pour chaque échantillon, préparer le mélange de réaction comme suit: 2 ul de tampon enzymatique, 1 pl d'EcoR I, 15 ul d'eau. Aliquoter le mélange réactionnel dans 1,5 ml tubes et ajouter 2 pi de plasmide. Incuber les tubes à 37 ° C pendant 2 heures.
- Exécuter le produit de digestion sur un gel d'agarose à 1,5%.
Remarque: Les échantillons avec insert affiche une taille de la bande de 475 pb qui est de 100 pb plus élevé que les clones sans inserts.
Note: Alternativement, les clones positifs peuvent être criblés par une colonie PCR utilisant SP6 (avant) et T7 (inverse) amorces (tableau 7) qui se lient à la séquence de vecteur pour donner un fragment de taille 642 pb en présence d'un gARN insert. L'approche PCR de colonie est avantageux quand il y a un site EcoR I de restriction dans la séquence gARN.
- Confirmer la séquence de l'gARN Insertion par séquençage de l'ADN en utilisant l'amorce T7.
2. transfection
Remarque:L'électroporation est une méthode efficace de transfection des plasmides dans des cellules ES. Le procédé décrit ici utilise la technologie microporateur de transfection.
- Cultiver des cellules ES F1 dans une capsule de gélatine enrobées 10 cm contenant 10 ml de milieu cellulaire ES (tableau 1) à 37 ° C / 5% de CO 2. Lorsque les cellules atteignent 85% de confluence supprimer les médias et ajouter 2 ml de trypsine. Incuber à 37 ° C dans l'incubateur à CO 2 pendant 5 min.
Remarque: les cellules ES F1 ont été obtenues à partir de Barbara Panning 24 et sont disponibles sur demande. - Neutraliser la trypsine en ajoutant 10 ml de milieu par centrifugation (tableau 2). à plusieurs reprises Pipette pour détacher les cellules complètement.
- Collecter toutes les cellules dans un tube de 15 ml et un essorage à 300 xg pendant 5 min. Remettre en suspension dans 3 ml de PBS et compte les cellules en utilisant un hémocytomètre ou compteur de cellules automatisé.
- Sédimenter 1 x 10 6 cellules ES dans un tube de 1,5 ml par centrifugation à 300 g pendant 5 minutes et remise en suspension dans 100 pi de R (resuspension) tampon fourni par le fabricant du kit.
- Ajouter 5 ug chacun de pCas9_GFP (un cadeau de Kiran Musunuru; Addgene plasmide # 44719) 31, 5 'et 3' plasmides gARN pour la suppression de la région cible et mélanger doucement avec une pipette pour éviter l'introduction de bulles.
- Utilisez la pointe de la pipette électronique pour aspirer 100 ul du mélange d'électroporation, en prenant soin d'éviter une bulle dans la pointe.
- Programmer le volts, la largeur et des impulsions pour l'électroporation. Pour les cellules ES F1, utilisez 1.400 V, 10 msec pour 3 impulsions.
- Alors que le électroporation est en cours d'exécution d'observer la pointe pour voir des éventuelles étincelles dans la solution. Une étincelle indique la présence d'une bulle d'air et va interférer avec la transfection.
- Éjecte les cellules ES transfectées dans une capsule en gélatine revêtues 10 cm contenant 10 ml de milieu cellulaire ES (tableau 1) et incuber à 37 ° C / 5% de CO 2.
3. FACS de tri des cellules transfectées
- Au bout de 48 h, détacher les cellules par addition de 2 ml de trypsine et on incube à 37 ° C dans l'incubateur à CO 2 pendant 5 min.
- Neutraliser la plaque par addition de 10 ml d' un tampon de collecte (tableau 3). Recueillir les cellules dans un tube de 15 ml et un essorage à 300 xg pendant 5 min.
- Jeter le surnageant et remettre en suspension les cellules dans 1 ml de tampon de tri (tableau 4). Compter les cellules et dilué en fonction de la plate-forme de tri. Diluer les cellules à 0,5-1 x 10 6 cellules / ml pour le tri en tubes de 15 ml et pour trier des cellules individuelles directement dans des plaques à 96 puits, diluer les cellules à 2-5 x 10 6 cellules / ml.
- Trier les cellules cas9-GFP + ES en utilisant un flux FACS cytomètre 32. Recueillir les cellules en vrac dans des tubes avec 2 ml de milieu de récupération (tableau 5) et la plaque comme décrit dans 3.5 pour la cueillette des colonies, ou des cellules de tri individuels directement dans des plaques à 96 puits revêtues de gélatine contenant 100 pi de milieu de cellules ES / puits ( Tableau 1).
- Semences de 1 à 1,5 x 10 4 cellules GFP + cellules ES dans une capsule de gélatine enrobées 10 cm contenant 10 milieu de cellules ES ml (tableau 1). Placage à cette faible densité facilitera la cueillette des colonies de cellules ES individuelles.
4. Les clones en culture pour le génotypage, l'analyse d'expression et des stocks de cellules de congélation
- Au jour 4-5 après le tri, le score de chaque puits de plaques à 96 puits triés directement pour déterminer la présence de colonies de cellules ES.
- Dissocier colonies de cellules ES en supprimant les médias et en ajoutant 30 pi de trypsine. Incuber à 37 ° C pendant 5 min. Neutraliser la trypsine par addition de 170 ul de milieu cellulaire ES (tableau 1) et la pipette de haut en bas pour la dissociation complète de la colonie dans des cellules individuelles. Cultiver les cellules à 37 ° C / 5% de CO 2 jusqu'à ce que la plupart des puits sont plus de 70% de confluence (habituellement 2-3 jours).
- Vous pouvez également choisir des colonies de cellules ES individuels à partir de boîtes de 10 cm en utilisantun microscope inversé. Après aspiration de la colonie dans la pointe de pipette suivi l'étape 4.1.1 plaçant chaque colonie dans un puits d'une plaque à 96 puits, prétraitées avec de la gélatine et contenant 30 pi de trypsine.
Remarque: Les colonies peuvent siéger à la trypsine à TA pendant toute une rangée de colonies est capté. - Une fois que toutes les colonies ont été cueillies et dissociées dans les médias poussent les cellules à 37 ° C dans l'incubateur à CO 2 jusqu'à ce que la plupart des puits sont plus de 70% confluentes (généralement 2 jours).
- Lorsque les plaques à 96 puits sont prêts pour le fractionnement, retirez le support, ajouter 30 ul de trypsine et incuber à 37 ° C pendant 5 min. Neutraliser la trypsine en ajoutant 180 ul de milieu de cellules ES (tableau 1) à chaque puits et la pipette de haut en bas pour la dissociation complète en cellules individuelles.
- Du 210 pi résultant, ensemencer 70 pi en trois gélatine revêtu des plaques à 96 puits contenant chacun 130 pl d'ES médias de cellules / puits (tableau 1). Utilisez ces plaques pour genotyping, l'analyse d'expression et la congélation des stocks de cellules pour chaque clone comme décrit ci-dessous.
- Lorsque la plaque de génotypage atteint 70-85% de confluence, traiter la plaque comme décrit au chapitre 6 «génotypage la suppression".
- Lorsque la plaque d'analyse d'expression atteint 70-85% de confluence, retirez le support, sceller la plaque avec du ruban adhésif et un magasin d'étanchéité à -80 ° C jusqu'à ce que les clones ont été génotypés.
Remarque: La plaque d'analyse d'expression est utile pour analyser les changements dans l'expression génétique au début des passages des clones. Analyse de l'expression génique à partir de la plaque de 96 puits est possible, mais que le nombre de cellules est faible d'un kit de micro-extraction de l'ARN est recommandée. - Préparation du gel de 96 puits Plate Stock:
- Lorsque la plaque pour les stocks de cellules congelées (stock-1) atteint 70-85% de confluence, aspirez les médias, ajouter 30 ul de trypsine et incuber à 37 ° C pendant 5 min.
- Neutraliser la trypsine en ajoutant 100 ul de milieu de cellules ES ( voir le tableau 1) to chaque puits et la pipette de haut en bas pour la dissociation complète en cellules individuelles.
- Transfert de 15 pi de cellules en suspension dans chaque puits à deux plaques à 96 puits de gélatine revêtues, chacune contenant 185 pi d'ES milieu cellulaire (tableau 1) et permettre de croître à 37 ° C / 5% de CO 2.
Note: Ceci est pour le stock-2 et -3 plaques qui sont des clones de back-up en cas supplémentaires ranimant les cellules du stock-1 ne réussit pas.
- Pendant ce temps, les 100 pi des cellules restantes de la plaque de 96 puits (actions 1), ajouter 100 ul de 2x gel médias (tableau 6). Sceller la plaque avec une bande d'étanchéité et rapidement inverser la plaque 4-5 fois pour un bon mélange. Conservez la plaque à -80 ° C jusqu'à ce que les clones sont génotypés.
- Lorsque les plaques de stock-2 et de stock-3 sont prêts pour la congélation Aspirer les médias, ajouter 30 ul de trypsine et incuber à 37 ° C pendant 5 min. Neutraliser la trypsine en ajoutant 70 ul de milieu de cellules ES (Tmesure 1) à chaque puits et la pipette de haut en bas pour la dissociation complète en cellules individuelles.
- Ajouter 100 ul de 2x médias congélation, sceller la plaque avec du ruban d'étanchéité et inverser rapidement la plaque 4-5 fois pour un bon mélange. Conservez la plaque à -80 ° C jusqu'à ce que ces plaques sont nécessaires.
Conception Primer 5. allèle spécifique
- Conception 4 ensembles d'amorces (figure 1C) à l' écran des clones pour la suppression souhaitée: à l' intérieur des amorces, des amorces externes et gARN flanquant amorces (pour les 5 'et 3' des sites cibles gARN) comme décrit ci - dessous.
- Obtenir la piste SNP correspondant aux 129 et Cast génotypes à http://labs.csb.utoronto.ca/mitchell/crispr.html. La piste donnée montre des substitutions de base entre 129 et Cast à des coordonnées dans l'ensemble du génome de la souris mm9.
Remarque: Le lien sur le site ci-dessus va rediriger vers le navigateur de génome UCSC et ajouter une piste personnalisée contenant le SNP entre le 129 et Cast genomes. - Entrer les coordonnées de la région à supprimer. Zoom sur une région d'environ 500 pb dans le milieu de la suppression souhaitée contenant> 3 SNP.
- Allez à voir> ADN dans la barre d'options et cliquez sur obtenir l'ADN pour télécharger la séquence cible dans tous les formats majuscules.
- Créer deux séquences FASTA; un pour 129 et un pour Moulage par substitution de base à la position SNP. Marquez les SNPs par un boîtier inférieur.
- Aller à Primer3 plus (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi/) et collez le SNP substitué 129 séquence. Utilisez les paramètres par défaut pour concevoir des amorces.
- Pour concevoir les amorces spécifiques d'allèles intérieur sélectionner Primer_List dans le menu tâche de déposer et cliquez sur Choisir apprêts. Choisir une amorce avant ou arrière qui a un SNP soit en extrémité ou dans les 4 bases de l'extrémité 3 '3 et tombe à l'intérieur de la région à supprimer.
Note: Amorces qui ont un SNP à l'affichage de la spécificité de l'extrémité 3 'ont augmenté allèle qPCR. - Pour sélectionner le second retour d'amorce à la page principale, sélectionnez Détection dans le menu déroulant de la tâche et coller la première séquence d'amorce dans la case appropriée au bas de la page. Dans l'onglet Paramètres généraux modifier le réglage pour la gamme de taille de produit à 80-200 pb et cliquez sur Choisir apprêts. Choisissez un ensemble d'amorces des paires d'amorces énumérées; ce seront les 129 amorces spécifiques d'allèles à l'intérieur.
- Répétez les étapes 5.1.5 à 5.1.7 pour concevoir des amorces pour l'allèle Cast.
- Retour au navigateur de génome UCSC et entrez les coordonnées de la région à supprimer. Allez à voir> ADN dans la barre d'options, sous Options Sequence Retrieval Région ajouter 1000 pb en amont et en aval cliquez obtenir l'ADN pour télécharger la séquence cible.
- Marquer la séquence cible gARN entre parenthèses. Sauvegardez cette séquence entière avant de poursuivre.
- Pour concevoir les amorces externes éliminer la séquence entre les deux séquences cibles gARN. Répétez l'étape 5.1.4-5.1.8 pour concevoir l'extérieur amorce spécifique d'allèles mais changer la taille du produit 400-800 pb.
- Diviser la séquence obtenue à l'étape 05/01/10 en deux séquences, chacune ayant 500 paires de bases 5 'et 3' de la séquence cible gARN. Répétez les étapes 5.1.5 à 5.1.7 conception des amorces spécifiques non allèles pour les régions flanquant gARN mais changer la taille du produit 400-800 pb.
Remarque: Pour des amorces spécifiques non allèles, soit 129 ou la séquence Cast peut être utilisé et les amorces doivent être choisies qui ne contiennent pas un SNP. Il est conseillé de définir une taille de produit de 400-800 pb pour la conception de l'extérieur et gARN flanquant amorces. Cela permet une amplification, même si les petits indels sont présents.
- Obtenir la piste SNP correspondant aux 129 et Cast génotypes à http://labs.csb.utoronto.ca/mitchell/crispr.html. La piste donnée montre des substitutions de base entre 129 et Cast à des coordonnées dans l'ensemble du génome de la souris mm9.
- Testez les amorces à l'intérieur de la spécificité de l'allèle par qPCR utilisant de l'ADN génomique de la souche 129 et Cast pur à 2 ng / ul. Suivez l'étape 6,2-6,4 pour mettre en place la réaction qPCR.
Remarque: si le génotype 129 dans la région cible est identique à celle C57BL / 6J, l'ADN provenant C57BL / 6J peut être utilisé à la place de 129 l'ADN. amorces spécifiques d'allèles devraient afficher au moins 5 cyclesdifférence entre la valeur Ct (seuil de cycle) sur le bon par rapport au génotype incorrect. Les amorces externes peuvent être testés afin de s'assurer qu'ils amplifient la suppression à l'aide cas9 / ARNg transfecté les cellules ES et les commandes de l'ADN génomique de F1 respectivement positives et négatives. L'allèle-spécificité des amorces externes peuvent être testés une fois que les clones monoalléliques ont été identifiés.
6. génotypage la suppression
- Extraire l'ADN génomique à partir de l'analyse génotypique plaque à 96 puits en utilisant la plaque de l'étape 4.6 qui est généré après l'expansion de la colonie.
- Préparer la génomique extraction d'ADN mélange: 89 pi d'eau, 10 pi de 10x tampon et 1 pl de réactif d'extraction (fourni par le fabricant). Ajouter 100 pi de génomique extraction d'ADN mélange à chaque puits et sceller la plaque avec un ruban d'étanchéité.
- Incuber la plaque à 75 ° C pendant 5 minutes puis 95 ° C pendant 5 min.
- Laisser refroidir la plaque par incubation sur de la glace pendant quelques minutes und, puis centrifuger brièvement pour régler toute condensation au fond du puits. Cela sert de plaque d'ADN matrice pour le criblage de suppression.
- Mettre en place les réactions qPCR en double pour chaque clone comme suit: 5 pi de 2x SYBR qPCR mélange, vers l'avant et l'amorce (3 uM) chaque 1 pl et 1 pl d'eau inverse. Utiliser une pipette multicanaux pour ajouter 2 ul d'ADN matrice, suivie de 8 pi de mélange réactionnel dans chaque puits d'une plaque à 384 puits.
- Sceller la plaque avec du ruban adhésif et de spin d'étanchéité à 600 g pendant 2 min pour mélanger le contenu. Placez la plaque de 384 puits tableau dans la vraie cycleur de temps.
- Programme réel cycleur de temps pour une PCR en deux étapes, suivie par une analyse de courbe de fusion avec la détection de la manière suivante: 1 cycle à 95 ° C pendant 10 min, 40 cycles de 95 ° C pendant 15 s, 62 ° C pendant 30 secondes avec la plaque de lecture et 95 ° C pendant 10 s, 65 ° C à 95 ° C avec un incrément de 5 ° C pendant 5 s + plaque de lecture.
Remarque: En plus de l'amorce conception, la qPCR miparamètres x et le cycle contribuent également à l'amorce de spécificité. Les paramètres décrits ci-dessus et des réactifs mentionnés dans les matériaux donnent plus souvent l'amplification allèle spécifique. - Analyse des résultats de qPCR
- Vérifiez chaque allèle pour l'amplification avec des amorces spécifiques d'allèles à l'intérieur. Aucune amplification d'un allèle ou des différences de valeur élevée Ct (> 5 cycles) entre allèles suggèrent que ces clones portent une délétion hétérozygote de l'allèle avec la valeur élevée / absent Ct. Aucune amplification des deux alleles suggère qu'ils portent une délétion homozygote.
- Vérifiez chaque allèle pour l'amplification avec l'extérieur amorces spécifiques d'allèles. Lorsque la suppression de la cible est supérieure à 1 kb amplification avec des amorces extérieures se produit uniquement quand une délétion est présente. Une valeur Ct de 22-28 confirme la suppression. Pour les suppressions de cibles plus petites que 1 kb, confirmer la taille de l'amplicon par électrophorèse.
Remarque: Si les amorces externes ne montrent que l' allèle spécificité modérée (voir Figure 2), les amplicons peuvent être obtenues avec les deux jeux d'amorces alléliques dans les clones monoalléliques en raison de l' amplification de cible des amorces extérieures. Dans ce cas, une différence de valeur Ct entre les deux allèles d'au moins cinq cycles doit confirmer l'allèle correcte (valeur de Ct plus faible) est supprimée sur la base des résultats obtenus à partir des amorces internes. Si la cible par rapport à l'allèle hors cible Ct différence est inférieure à cinq cycles concevoir de nouveaux allèle amorces externes spécifiques. - Dans clones de deletion monoalléliques vérifier l'intégrité de l'allèle non-supprimé en utilisant le criblage secondaire, gARN flanquant amorces.
Remarque: indels de> 25 taille de pb autour du site cible gARN peuvent être identifiés par l'observation d'un décalage de la courbe de fusion dans la qPCR pour 400-800 amplicons pb. En variante, les amplicons des amorces flanquant gARN peuvent être séquencés pour détecter de petites indels de <25 pb.- Effectuer qPCR avec 2 ensembles de gARN amorces flanquant -à- dire, 5 'et 3' gARN utilisé dans la génération de CRISPR suppression. Aucune amplification avec ces ensembles d'amorces indique indels plus grande que l'amplicon qPCR sont présents au niveau du site cible gARN sur l'allèle non délété de clones de deletion monoalléliques. clones Jeter contenant ces grandes indels de une analyse plus approfondie que les résultats peuvent être difficiles à interpréter sans connaître l'étendue de la suppression.
- Purifier les amplicons obtenus à partir de la réaction qPCR de l'amorce externe en utilisant une PCR nettoyer kit suivant les instructions du fabricant.
- Confirmer la séquence de l'allèle supprimé par séquençage d'ADN du produit de PCR purifié à partir de l'étape précédente. Utilisez les amorces d'amplification qPCR pour l'avant et le séquençage inverse.
Remarque: A ce stade SNP au sein de l'acte de amplicon comme une confirmation secondaire du génotype de l'allèle supprimé.
7. Analyse Expression avec allèles Amorces spécifiques
- Décongeler le 96 puits stock de cellules plate stockés à -80 ° C (Stock-1 de l'étape 4.9) en le plaçant sur un bain de perles chaud. Lorsque plus de la moitié des puits de la plaque sont décongelées, rotation à 300 xg pendant 5 min.
- Avec précaution, retirer la bande d'étanchéité et transférer rapidement les cellules de la suppression des puits positifs dans, plaques à 12 puits revêtues de gélatine contenant 1 ml de milieu de cellules ES (tableau 1) et incuber à 37 ° C / 5% de CO 2.
- Lorsque la plaque atteint 70-85% de confluence, le passage des cellules et les diviser en trois puits d'une plaque à 6 puits revêtues de gélatine, contenant chacun 2 ml de milieu de cellules ES (tableau 1). Utiliser deux puits pour préparer les 2 flacons de stocks de cellules congelées pour le stockage à long terme dans de l'azote liquide (décrit dans l'étape 8) et le troisième puits pour l'extraction de l'ARN.
- L'ARN extrait en utilisant un kit d'extraction d'ARN.
- Convertir 100-500 ng d'ARN en ADNc par transcription inverse (RT) de l'ARN en utilisant le kit de synthèse d'ADNc en suivant le protocole du fabricant. Inclure une RT negative réaction pour chaque échantillon d'ARN pour surveiller la quantité d'ADN contaminant dans les échantillons d'ARN.
- Diluer l'ADNc avant qPCR dans un rapport compris entre 1: 2 et 1: 4; en fonction du niveau du gène cible dans des cellules ES de l'expression.
- Réglez le qPCR comme décrit ci-dessus, y compris F1 ADN génomique comme courbe standard (5 dilutions de pliage de 250 à 0,08 ng / ul) pour la quantification absolue des niveaux de transcription. Comparer l'expression d'un allèle du gène d'intérêt dans chaque clone a confirmé supprimé à un gène de contrôle approprié, par exemple Gapdh (amorces énumérées dans le tableau 7).
Nota: Les amorces de gène de contrôle ne doivent pas nécessairement être allèle spécifique. la conception d'amorce allèle-spécifique est le même pour les amorces de RT-qPCR comme décrit pour les amorces de génotypage à l'exception de la région cible pour l'amplification. La séquence du gène doit être utilisé; si l'on utilise des amorces pour un seul exon ou une limite exon-intron (pour contrôler la transcription primaire) F1 ADN génomique peut être utilisé pourla courbe standard. Pour plus de détails sur RT-qPCR s'il vous plaît se référer à Forlenza et al. 2012 33.
8. Gel Stock Préparation pour le stockage à long terme de cellules ES
- Ajouter 300 pi de trypsine à chaque puits 6 (étape 7.3) et on incube pendant 5 minutes à 37 ° C. Ajouter 2 ml de milieu de spin (tableau 2) pour neutraliser la trypsine et de la pipette vers le haut et vers le bas plusieurs fois pour se dissocier dans des cellules individuelles.
- Transférer les cellules dans un tube de 15 ml et un essorage à 300 xg pendant 5 min.
- Aspirer le surnageant et ajouter 500 ul de milieu de cellules ES (tableau 1). Pipeter vers le haut et vers le bas pour remettre en suspension les cellules.
- Transférer le contenu dans un tube cryofiole 1,5 ml et ajouter 500 pl de 2x congélation de cellules ES supports (tableau 3). Bien mélanger en inversant le tube et placer le tube dans un récipient de congélation de cellules sans alcool. Placez cette cellule de congélation des récipients à -80 ° C pendant au moins 12 heures avant la transferrineg dans un liquide de cuve de stockage d'azote.
Résultats
Le protocole décrit ici utilise des cellules ES F1 pour étudier cis - régulation de l' expression génique dans les cellules amplificatrices supprimé monoalléliques générés en utilisant CRISPR / cas9 édition du génome (Figure 1). Le gARN et la conception de l'amorce spécifique d'allèle pour le génotypage et l'expression génique sont les facteurs clés de cette approche. Chaque ensemble d'amorces spécifiques de l'allèle doi...
Discussion
CRISPR / cas9 technologie d'édition du génome médiation fournit une méthode simple, rapide et peu coûteuse pour la modification du génome. La méthode détaillée ici pour générer et analyser monoallélique suppression d'activateur pour la caractérisation fonctionnelle enhancer tire profit de SNP dans les cellules de souris F1. Les avantages de ce type d'approche sont: 1) les suppressions d'activateurs monoalléliques ne produisent pas des effets de confusion qui se produisent quand un activateu...
Déclarations de divulgation
Les auteurs ont lu les politiques de JoVE sur les conflits d'intérêts et ne pas avoir à divulguer les conflits.
Remerciements
We would like to thank all the members of the Mitchell lab for helpful discussions. This work was supported by the Canadian Institutes of Health Research, the Canada Foundation for Innovation and the Ontario Ministry of Research and Innovation (operating and infrastructure grants held by JAM).
matériels
| Name | Company | Catalog Number | Comments |
| Phusion High-Fidelity DNA Polymerase | NEB | M0530S | high fidelity DNA polymerase used in gRNA assembly |
| Gibson Assembly Master Mix | NEB | E2611L | |
| gRNA_Cloning Vector | Addgene | 41824 | A target sequence is cloned into this vector to create the gRNA plasmid |
| pCas9_GFP | Addgene | 44719 | Codon-optimized SpCas9 and EGFP co-expression plasmid |
| AflII | NEB | R0520S | |
| EcoRI | NEB | R3101S | |
| Neon Transfection System 100 µL Kit | Life Technologies | MPK10096 | Microporator transfection technology |
| prepGEM | ZyGEM | PT10500 | genomic DNA extraction reagent |
| Nucleo Spin Gel & PCR Clean-up | Macherey-Nagel | 740609.5 | |
| High-Speed Plasmid Mini Kit | Geneaid | PD300 | |
| Maxi Plasmid Kit Endotoxin Free | Geneaid | PME25 | |
| SYBR select mix for CFX | Life Technologies | 4472942 | qPCR reagent |
| iScript cDNA synthesis kit | Bio-rad | 170-8891 | Reverse transcription reagent |
| 0.25% Trypsin with EDTA | Life Technologies | 25200072 | |
| PBS without Ca/Mg2+ | Sigma | D8537 | |
| 0.5 M EDTA | Bioshop | EDT111.500 | |
| HBSS | Life Technologies | 14175095 | |
| 1 M HEPES | Life Technologies | 13630080 | |
| BSA fraction V (7.5%) | Life Technologies | 15260037 | |
| Max Efficiency DH5α competent cells | Invitrogen | 18258012 | |
| FBS | ES cell qualified | FBS is subjected to a prior testing in mouse ES cells for pluripotency | |
| DMSO | Sigma | D2650 | |
| Glutamax | Invitrogen | 35050 | |
| DMEM | Life Technologies | 11960069 | |
| Pencillin/Streptomycin | Invitrogen | 15140 | |
| Sodium pyruvate | Invitrogen | 11360 | |
| Non-essential aminoacid | Invitrogen | 11140 | |
| β-mercaptoethanol | Sigma | M7522 | |
| 96-well plate | Sarstedt | 83.3924 | |
| Sealing tape | Sarstedt | 95.1994 | |
| CoolCell LX | Biocision | BCS-405 | alcohol-free cell freezing container |
| CHIR99021 | Biovision | 1748-5 | Inhibitor for F1 ES cell culture |
| PD0325901 | Invivogen | inh-pd32 | Inhibitor for F1 ES cell culture |
| LIF | Chemicon | ESG1107 | Inhibitor for F1 ES cell culture |
Références
- Sagai, T., Hosoya, M., Mizushina, Y., Tamura, M., Shiroishi, T. Elimination of a long-range cis-regulatory module causes complete loss of limb-specific Shh expression and truncation of the mouse limb. Development. 132 (4), 797-803 (2005).
- Kleinjan, D. A., Lettice, L. A. Long-range gene control and genetic disease. Adv Genet. 61, 339-388 (2008).
- Visel, A., Rubin, E. M., Pennacchio, L. A. Genomic views of distant-acting enhancers. Nature. 461 (7261), 199-205 (2009).
- Maurano, M. T., et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 337 (6099), 1190-1195 (2012).
- Heintzman, N. D., et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 459 (7243), 108-112 (2009).
- Shen, Y., et al. A map of the cis-regulatory sequences in the mouse genome. Nature. 488 (7409), 116-120 (2012).
- Johnson, D. S., Mortazavi, A., Myers, R. M., Wold, B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 316 (5830), 1497-1502 (2007).
- Rhee, H. S., Pugh, B. F. Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell. 147 (6), 1408-1419 (2011).
- Whyte, W. A., et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 153 (2), 307-319 (2013).
- Chen, C. Y., Morris, Q., Mitchell, J. A. Enhancer identification in mouse embryonic stem cells using integrative modeling of chromatin and genomic features. BMC Genomics. 13 (1), 152 (2012).
- Patwardhan, R. P., et al. Massively parallel functional dissection of mammalian enhancers in vivo. Nat Biotechnol. 30 (3), 265-270 (2012).
- Melnikov, A., et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat Biotechnol. 30 (3), 271-277 (2012).
- Jinek, M., et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337 (6096), 816-821 (2012).
- Cong, L., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Mali, P., et al. RNA-guided human genome engineering via Cas9. Science. 339 (6121), 823-826 (2013).
- Hsu, P. D., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 31 (9), 827-832 (2013).
- Cho, S. W., et al. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 24 (1), 132-141 (2014).
- Zhou, H. Y., et al. A Sox2 distal enhancer cluster regulates embryonic stem cell differentiation potential. Genes Dev. 28 (24), 2699-2711 (2014).
- Fujii, W., Kawasaki, K., Sugiura, K., Naito, K. Efficient generation of large-scale genome-modified mice using gRNA and CAS9 endonuclease. Nucleic Acids Res. 41 (20), e187 (2013).
- Tuan, D. Y., Solomon, W. B., London, I. M., Lee, D. P. An erythroid-specific, developmental-stage-independent enhancer far upstream of the human 'beta-like globin' genes. Proc Natl Acad Sci U S A. 86 (8), 2554-2558 (1989).
- Amano, T., et al. Chromosomal dynamics at the Shh locus: limb bud-specific differential regulation of competence and active transcription. Dev Cell. 16 (1), 47-57 (2009).
- Li, Y., et al. CRISPR reveals a distal super-enhancer required for Sox2 expression in mouse embryonic stem cells. PLoS One. 9 (12), e114485 (2014).
- Canver, M. C., et al. Characterization of genomic deletion efficiency mediated by clustered regularly interspaced palindromic repeats (CRISPR)/Cas9 nuclease system in mammalian cells. J Biol Chem. 289 (31), 21312-21324 (2014).
- Mlynarczyk-Evans, S., et al. X chromosomes alternate between two states prior to random X-inactivation. PLoS Biol. 4 (6), e159 (2006).
- Lefever, S., Pattyn, F., Hellemans, J., Vandesompele, J. Single-nucleotide polymorphisms and other mismatches reduce performance of quantitative PCR assays. Clin Chem. 59 (10), 1470-1480 (2013).
- Huang, M. M., Arnheim, N., Goodman, M. F. Extension of base mispairs by Taq DNA polymerase: implications for single nucleotide discrimination in PCR. Nucleic Acids Res. 20 (17), 4567-4573 (1992).
- Keane, T. M., et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 477 (7364), 289-294 (2011).
- Yalcin, B., et al. Sequence-based characterization of structural variation in the mouse genome. Nature. 477 (7364), 326-329 (2011).
- Gibson, D. G., et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 6 (5), 343-345 (2009).
- Gibson, D. G., Smith, H. O., Hutchison, C. A., Venter, J. C., Merryman, C. Chemical synthesis of the mouse mitochondrial genome. Nat Methods. 7 (11), 901-903 (2010).
- Ding, Q., et al. Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell. 12 (4), 393-394 (2013).
- Basu, S., Campbell, H. M., Dittel, B. N., Ray, A. Purification of specific cell population by fluorescence activated cell sorting (FACS). J Vis Exp. (41), (2010).
- Forlenza, M., Kaiser, T., Savelkoul, H. F., Wiegertjes, G. F. The use of real-time quantitative PCR for the analysis of cytokine mRNA levels. Methods Mol Biol. 820, 7-23 (2012).
- Wu, J. H., Hong, P. Y., Liu, W. T. Quantitative effects of position and type of single mismatch on single base primer extension. J Microbiol Methods. 77 (3), 267-275 (2009).
- Sanyal, A., Lajoie, B. R., Jain, G., Dekker, J. The long-range interaction landscape of gene promoters. Nature. 489 (7414), 109-113 (2012).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.