
需要订阅 JoVE 才能查看此. 登录或开始免费试用。
Method Article
目标
摘要
A strategy for generating mutations in histone genes at their endogenous location in Saccharomyces cerevisiae is presented.
摘要
We describe a PCR- and homologous recombination-based system for generating targeted mutations in histone genes in budding yeast cells. The resulting mutant alleles reside at their endogenous genomic sites and no exogenous DNA sequences are left in the genome following the procedure. Since in haploid yeast cells each of the four core histone proteins is encoded by two non-allelic genes with highly homologous open reading frames (ORFs), targeting mutagenesis specifically to one of two genes encoding a particular histone protein can be problematic. The strategy we describe here bypasses this problem by utilizing sequences outside, rather than within, the ORF of the target genes for the homologous recombination step. Another feature of this system is that the regions of DNA driving the homologous recombination steps can be made to be very extensive, thus increasing the likelihood of successful integration events. These features make this strategy particularly well-suited for histone gene mutagenesis, but can also be adapted for mutagenesis of other genes in the yeast genome.
引言
四个核心组蛋白H2A,H2B,H3和H4中的压实,组织,和真核染色体的功能发挥中心作用。两套各这些组蛋白的形成的组蛋白八聚体,即指示本身周围的DNA的〜147碱基对的环绕一个分子卷轴,最终导致核小体1的形成。核小体是在各种基于染色体过程,如基因转录的调节和常染色质的形成和异跨染色体积极参与者,因此已经深入研究在过去几十年的过程中的焦点。已经描述了许多机制由核小体可以在方法,可以方便特定进程的执行被操纵 - 这些机制包括组蛋白残基的翻译后修饰,依赖ATP的核小体重构,和ATP依赖性核重组和装配/拆卸2,3。
在芽殖酵母是组蛋白功能的真核生物的理解一个特别强大的模型生物。这在很大程度上归因于整个域真核生物高度组蛋白的进化保守的和酵母的各种遗传和生化实验的方法顺从4。在酵母反向遗传方法已被广泛用于研究染色质生物学的各个方面的具体组蛋白突变的影响。对于这些类型的实验,常常优选使用,其中所述突变体组蛋白从其天然基因组位点表达的细胞,如从自主质粒表达可导致组蛋白的异常细胞内水平(由于细胞不同质粒的数量),并染色质恩伴随变更vironments,它可以混淆最终结果的解释。
在这里,我们描述了一种基于PCR的技术,其允许在不需要在所需突变的生成而不在基因组中剩余的外源DNA序列的克隆步骤和结果其天然基因组的位置的组蛋白的基因的靶向诱变。这种技术利用了有效的同源重组系统的在酵母和具有几个共同的特征与其它组开发的其他类似的技术-尤其是在Delitto普菲 ,位点特异性基因组(SSG)诱变和克隆-自由基于PCR的等位基因替换方法5,6,7。然而,我们描述的技术存在,使得它特别适合于组蛋白基因的突变的一个方面。在单倍体酵母细胞中,每四个核心组蛋白是由两个非一个编码llelic和高度同源的基因:例如,组蛋白H3由HHT1和HHT2基因编码,而两个基因的开放阅读框(ORFs)在序列90%以上相同。这种高度的同源性可以变得复杂设计成特异性靶向诱变两个组蛋白编码基因中的一个实验。而上述的方法通常要求使用的靶基因的ORF内的至少一些序列来驱动同源重组,我们在这里描述的技术利用侧翼组蛋白基因的开放阅读框(共用少得多的序列同源性)序列的的重组步骤,从而增加至所需的轨迹诱变成功定位的可能性。此外,驱动重组同源区可以是非常广泛的,从而进一步提高效率的有针对性的同源重组。
研究方案
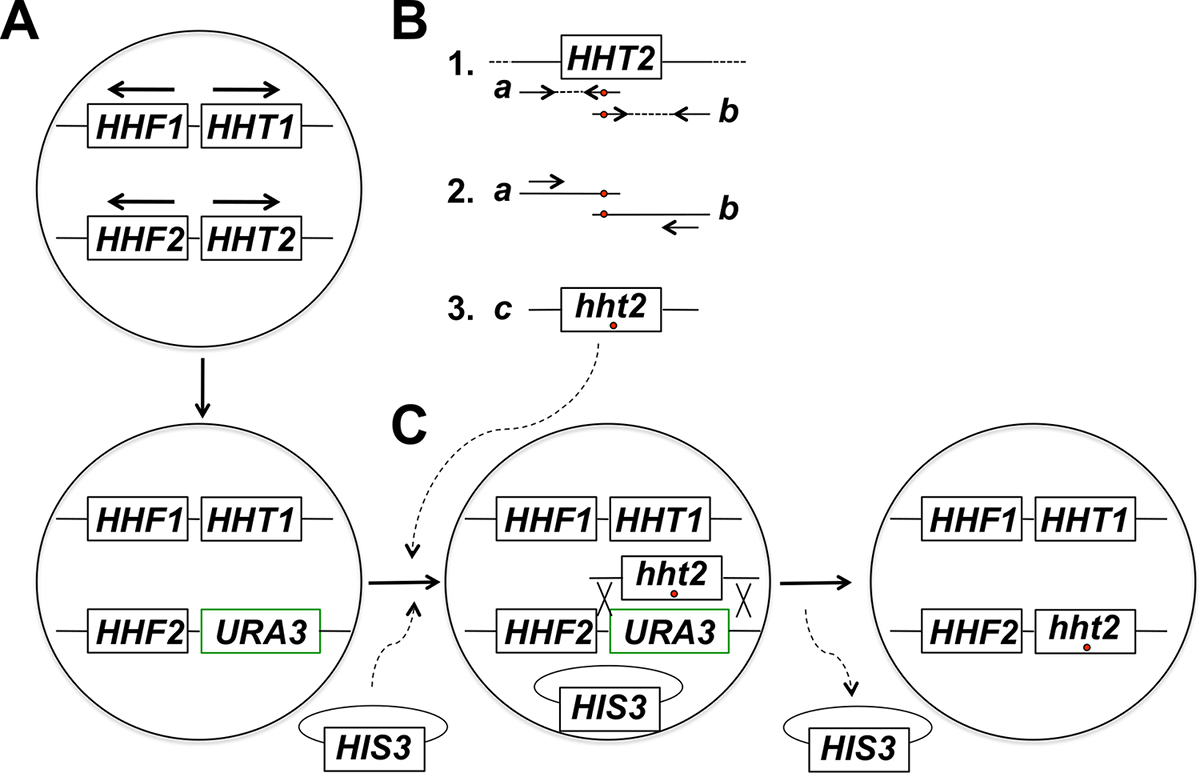
注:有针对性的原位组蛋白基因突变实验策略包括以下几个步骤( 图1中总结)。这些步骤包括:(1)与URA3基因的靶组蛋白基因的置换,(2)产生与对应于两个使用引物携带所需突变(s)时,目标组蛋白基因的部分重叠的片段的PCR产物的纯化(3 )这两个部分重叠的片段的融合的PCR,以获得充分的大小的PCR产物为一体,(4)的全尺寸的PCR产物和骨架质粒共转化,并选择对质粒的标记,(5)画面5-FOA抗性转化体,(6)5-FOA抗性菌落的纯化和骨架质粒的损失,和(7)的分子分析,以测定为突变等位基因的适当整合。

图1:目标在原地突变在芽殖酵母基因组蛋白的战略概述。在这个例子中,靶基因是HHT2的,但任何其它核心组蛋白基因也可使用这种策略诱变。 (A)的单倍体酵母细胞怀有布置在两个组蛋白H3编码基因(HHT1和HHT2)和两个组蛋白H4编码基因(HHF1和HHF2)如该图所示(该HHT1和HHF1基因位于第II染色体和HHT2和HHF2基因位于染色体XIV -在每种情况下,箭头指向在转录的方向上)。在该过程的第一步骤中,HHT2基因的ORF被替换为URA3基因,从而产生一种hht2Δ:: URA3菌株。 (B)在部分1中,从基因组DNA样本的HHT2基因的野生型拷贝用作模板两个PCR反应到属TE上基因的两个部分重叠的片段。用于第一反应中的反向引物包括一个或多个错配的核苷酸(用红色圆圈表示)对应于被引入到基因组中的期望的突变。用于第二反应的正向引物具有在反向互补构造(也用红色圆圈表示)相当于不匹配。在部分1中产生的两个PCR产物(产物a和b)随后被用作使用在部分2中示出退火在时尚产品a和b这导致全尺寸的PCR产生两种引物用于融合PCR的模板产品(产品C在第3部分)携带所需突变。 (C)中hht2Δ:: URA3应变然后共转化与全尺寸的PCR产物和骨架质粒(一个HIS3-标记在本实施例中的质粒),以及细胞被选择用于质粒的存在(在缺乏媒体H在本实施例istidine)。然后转化体进行筛选5-FOA抗性-抗性细胞是已经历同源重组事件导致URA3基因的PCR产物和切除的一体化,如图候选者。通过有丝分裂细胞分裂的骨架质粒的后续丧失导致最终所需组蛋白突变体菌株。我们已经找到了骨架质粒的该选择,然后在的正确整合事件的识别相比,对5-FOA平板直接选择,这主要是标识已获得自发URA3突变的细胞的高得多的频率筛选5-FOA抗性的结果。 (这个数字已经从14参考修改)。 请点击此处查看该图的放大版本。
{kind=link}
1.目标组蛋白基因的置换与URA3基因
- 执行标准PCR介导的一步法基因破坏与URA3基因8,9更换靶组蛋白基因的ORF。
注:建议使用携带ura3Δ0酵母细胞作为这种突变将删除整个内源性URA3的ORF,从而避免了PCR产物整合到URA3基因座8。可替代地, 乳酸克鲁维酵母URA3基因可有效地用于组蛋白替换的产生中的任何的ura3背景,因为它是在酿酒酵母中的功能,但具有与酿酒酵母URA3基因仅部分序列同源性。应变也应为至少一种化合物,这将允许在转化实验(见本协议的步骤4)的骨架质粒的选择是营养缺陷型。这一步是没有必要的,如果目标蛋白geneΔ:: URA3菌株已经可用。
2.生成和使用的引物携带所需突变对应于目标组蛋白基因的两个部分重叠的片段的PCR产物的纯化(S)
- 生成对应于该目标组蛋白基因的两个部分重叠的片段的PCR产物。
- 准备两个PCR反应如下:
- 产生对应于该基因( 图1B a)产物的前半部分,设置了下面的反应的PCR产物:1微升模板DNA,5μl10μM正向引物,5μl10μM反向引物,0.5微升(1.25 U)的热稳定DNA聚合酶,10微升5×聚合酶缓冲液,5微升dNTP混合液(2毫每个),和23.5微升的dh 2 O
注意:模板DNA可以是来自菌株的野生型目标组蛋白基因的基因组DNA使用标准亲分离cedures 10。考虑到在DNA浓度和在不同基因组制备杂质水平的变化,则建议通过使用未稀释的DNA或基因组制剂的不同稀释度,以优化反应( 例如,1:10和1:100)。正向引物应当退火到的区域中的靶基因的上游。反向引物应在长度的ORF内退火,是〜40个核苷酸,并含有所需的突变某处它的中间(参见图1B-1和代表结果为实施例部分)。使用高保真DNA聚合酶,以减少对PCR产物的合成过程中不希望的突变率,建议。 - 产生对应于该基因(产物B在图1B)的第二半的PCR产物,如在2.1.1.1但使用不同的引物表示设置了反应。
注:远期PRI聚体应在长度的ORF内退火,是〜40个核苷酸,并且包含在它的中间某处的所需突变。注意,在此引物的突变(s)是在步骤2.1.1.1在反向引物的突变的反向互补。反向引物应退火到靶基因的下游区域(参见实施例图1B-1和代表结果部分)。
- 产生对应于该基因( 图1B a)产物的前半部分,设置了下面的反应的PCR产物:1微升模板DNA,5μl10μM正向引物,5μl10μM反向引物,0.5微升(1.25 U)的热稳定DNA聚合酶,10微升5×聚合酶缓冲液,5微升dNTP混合液(2毫每个),和23.5微升的dh 2 O
- 放置在热循环仪反应使用以下设置:94℃30秒;的下列设置30个循环:98℃10秒,60℃5秒,72℃1.5分钟;和72℃10分钟。
注:可能需要特异性引物PCR组参数的优化和目标组蛋白基因。
- 准备两个PCR反应如下:
- 从在89毫摩尔Tris碱,89毫硼酸,2.5毫摩尔EDTA(TBE)缓冲液上0.9%的低熔点琼脂糖凝胶对PCR反应50微升材料 - 运行20。
- 含有PCR PR切下的琼脂糖凝胶切片使用干净的手术刀或刀片和从凝胶oducts每个转移到1.5ml微量离心管中。存储包含PCR产物的琼脂糖节,在-20℃,直到准备使用。
3.融合两个部分覆盖片段的PCR获取全尺寸PCR产品进行整合
- 准备PCR反应模板
- 通过在65℃放置离心管中的热块组5分钟熔融来自步骤2.3琼脂糖凝胶切片(或直至完全熔化)。涡流管每1 - 2分钟,以促进熔化过程。
- 从每个样品转移的熔融的设定量琼脂糖( 例如 ,每50微升,总共100微升)到单个的离心管,并通过涡旋混合。使用它作为在融合PCR反应的模板。发生在-20℃的管待用。
- 扩增全尺寸的PCR产物的量大(产品C在图1B)
- 设立六个PCR反应中,每个由以下部分组成:2微升模板DNA,10微升10μM的正向引物,10微升10μM反向引物,1微升(2.5 U)的耐热性DNA聚合酶,20微升5×聚合酶缓冲液,10微升的dNTP混合物(2mM的各)和47微升的dh 2 O.
注意:可以根据PCR效率来改变反应的数量。模板DNA(参见3.1.2)应该被加热到65℃直到熔化,通过涡旋混合,最后加入PCR反应混合物中。添加后,通过移液上下几次轻轻混匀,但彻底。考虑到在不同样品中的DNA浓度的变化,建议通过使用未稀释的模板或模板的不同稀释度的第一优化反应( 例如,1:10和1:100)。用于应退火到靶基因作为病的两个部分重叠的片段两个引物ustrated 图1B-2和被设计成使得最终PCR产物将具有在任一侧同源的区域侧翼的URA3 ORF将驱动同源重组步骤(参见实施例代表结果部分)的至少40个碱基对。使用高保真DNA聚合酶,以减少对PCR产物的合成过程中不希望的突变率,建议。 - 放置在热循环管具有以下设置:94℃30秒;的下列设置30个循环:98℃10秒,50℃15秒,72℃1.5分钟;和72℃10分钟。
注:可能需要特定的引物组和目标组蛋白基因的PCR参数的优化。
- 设立六个PCR反应中,每个由以下部分组成:2微升模板DNA,10微升10μM的正向引物,10微升10μM反向引物,1微升(2.5 U)的耐热性DNA聚合酶,20微升5×聚合酶缓冲液,10微升的dNTP混合物(2mM的各)和47微升的dh 2 O.
全尺寸PCR产品和骨架质粒4.共转化,与选择标记质粒
- 的PCR产物的浓度
- 池在S九PCR反应从步骤3.2.2成一个单一的离心管(600微升总),并通过涡旋混合。
- 拆分样品到离心管三块200微升等分。通过加入3M醋酸钠20微升(pH5.2)和550微升的100%乙醇沉淀在每个管中的DNA。充分搅拌溶解并在冰上放置至少15分钟。收集在〜14000 xg离心离心DNA 10分钟,用200μl70%乙醇,和空气干燥冲洗沉淀。
- 重悬每个DNA沉淀到25微升的dh 2 O,并汇集成一个单一的管(总共75微升)。
- 酵母共转化
- 在酵母提取物蛋白胨葡萄糖(YPD)液体培养基11准备好10毫升部分1产生的应变的过夜培养。
- 第二天早晨,接种400毫升YPD液体培养基的用8ml饱和过夜培养物,并通过摇动孵育在30℃下为4 - 5小时,以允许细胞进入对数生长期。
- 收集通过离心将细胞以〜3,220×g离心10分钟,丢弃该液体介质,和重悬细胞于1体积的10mM的Tris-HCl(pH值8.0),1mM EDTA中的,0.1M醋酸锂溶液(TE /醋酸锂) 。
- 收集在〜3,220 xg离心的细胞离心10分钟,并弃去的TE /醋酸锂。
- 悬浮细胞在1毫升的TE /醋酸锂。
- 设置以下反应鸡尾酒在一个离心管中:从步骤4.2.5 800微升的细胞,将40μl煮沸10毫克/毫升鲑鱼精子DNA的,共骨架质粒DNA 12.5微克和75微升浓PCR产物从步骤4.1.3。
注:鲑鱼精子DNA应煮沸5分钟,在反应中使用之前,在冰上放置至少5分钟。加入骨架质粒DNA的总体积应保持在最低限度(〜80微升或更小)。见代表性的成果部分骨干血浆的例子ID。 - 调匀鸡尾酒管均匀地分装成八离心管(管1 - 8)。
- 设置以下两个控制转化反应管:
- 管9(无PCR产物对照):细胞100μl的从步骤4.2.5,5微升煮沸10毫克/毫升鲑鱼精子DNA的(煮沸5分钟;参见步骤4.2.6注),总共1.56微克的骨干质粒DNA和无PCR产物加入。
- 管10(无DNA对照):细胞100μl的从步骤4.2.5煮沸10毫克/毫升鲑鱼精子DNA(见步骤4.2.6注)5微升,无骨架的质粒DNA,并将无PCR产物加入。
- 移液器向上和向下几次轻轻但调匀都管。
- 在30℃下进行30分钟孵育的十个管。
- 向每个管中,于TE /醋酸锂添加1.2毫升40%的聚乙二醇(PEG 3350)。调匀利用P-1000吸管直到溶液是均匀的。
- 孵育十管30°下进行30分钟。轻轻吹打上下混合溶液中,然后在42℃孵育管15分钟。
- 通过在〜14000 xg离心纺丝管在微量30秒收集细胞。丢弃液体和重悬细胞于1ml无菌卫生署2 O的
- 通过在〜14000 xg离心纺丝管在微量30秒收集细胞。丢弃液体和悬浮细胞在500微升无菌卫生署2 O的
- 池管1 - 8一起(为4ml总体积),并通过上下抽吸调匀。
- 板200微升上为二十完整最小丢包介质板11(板1-20)为骨架质粒的选择的上述混合物组成。
- 板从管9 200微升混合物和从管子10各200μl的混合物在其自己的选择板(板21和22,分别地)。
- 孵育22板在30℃,3 - 5天选择质粒转化。
- 孵化5天 - 3后检查转变板。大约5000菌落应该在1-21板(参见例子代表性的成果),可见没有殖民地应该存在于板22。
5.屏幕5-FOA抗性转化
- 从板传递细胞1 - 20(与转化板21作为对照)至5-氟乳清酸(5-FOA)板11由复制品镀以筛选URA3基因的损失的积分的结果,12 PCR产物在所需的位置。
- 取下盖板,按含在无菌天鹅绒殖民地板。按盘上的丝绒转移细胞从绒到5-FOA板上。在30℃下2天孵育板。
- 继为期2天的孵化,仔细检查GR 5-FOA平板owth。
注:考生整合活动将通过一个小的非对称"挤压"殖民地在5-FOA板来表示-相反,小乳头5-FOA平板上生长都是自发URA3突变可能代表菌落上生长期间出现变换板,并因此不可能代表所需的整合事件(参见图3中有关此点进一步阐述代表性的结果部分和用于一些例子)。
6.净化5-FOA抗性菌落和骨干质粒的损失
- 用无菌牙签,从在步骤5.2和条纹为单个菌落描述到YPD平板5-FOA平板挑候选菌落。在30℃C 3天 - 孵育2。
- 孵育后,副本-每个板块YPD净化板新鲜YPD板,辍学板缺乏尿嘧啶检查损失URA3基因,以及第二分出出板的监测为骨架质粒的存在或不存在。在30℃下反应2天 - 孵育1。
- 孵育后,确定从不断增长的YPD板,但没有成长在任辍学板的每个候选样本的殖民地(如殖民地有望通过重组事件已经失去了URA3基因和有丝分裂过程中丢失的骨架质粒细胞分裂)。 Restreak新鲜YPD平板这样的殖民地。这些菌落的整合候选人,将在第7步进一步分析。
7.分子分析为测定突变等位基因的适当整合
- 隔离用标准方法10所述候选样本基因组DNA。
- 扩增基因组区域包含目标站点。
- 设置每个样品的下述PCR反应:0.5微升模板DNA,5微升10μM的正向引物,5μl的10μM反向引物,0.5微升(2.5单位)Taq DNA聚合酶,5微升10×Taq DNA聚合酶缓冲液,5微升dNTP混合液(2毫各)和29微升的dh 2 O
注意:模板的DNA是从候选样本得到的基因组DNA。建议还包括两个对照反应:一种使用来自原始组蛋白geneΔ的基因组DNA :: URA3菌株作为模板,并从野生型组蛋白菌株为模板另一个使用基因组DNA。考虑到在DNA浓度和在不同基因组制备杂质水平的变化,则建议通过使用未稀释的DNA或基因组制剂的不同稀释度,以优化反应( 例如,1:10和1:100)。以确保重要的是这些引物退火到DNA序列的推定集成PCR产物所包围的区域的外侧 -这样,PCR产物在这些r中尺寸eactions可以用作在正确的基因组位置,为产品的集成的诊断工具(参见,例如代表性结果 )。 - 放置在热循环仪反应使用以下设置:94℃3分钟;的下列设置30个循环:94℃45秒,50℃45秒,72℃2分钟;和72℃10分钟。
注:可能需要特定的引物组和目标组蛋白基因的PCR参数的优化。
- 设置每个样品的下述PCR反应:0.5微升模板DNA,5微升10μM的正向引物,5μl的10μM反向引物,0.5微升(2.5单位)Taq DNA聚合酶,5微升10×Taq DNA聚合酶缓冲液,5微升dNTP混合液(2毫各)和29微升的dh 2 O
- PCR产物的加工
- 从在0.8%TBE琼脂糖凝胶上各反应运行20微升。
- 评估使用DNA标准作为参考,以确定是否URA3基因已被推定的突变蛋白的基因被成功取代PCR产物的大小(参见,例如代表性结果)。
注意:在某些情况下,所期望的突变引入到组蛋白基因创建或销毁限制性坐即如果是这样的情况下,在指示正确整合的大小的PCR产物所需突变的存在可以通过产品经受消化与相应的限制酶,随后通过凝胶电泳分析来评估(见,例如代表结果) 。 - 指示正确整合的大小,以DNA测序的主题的PCR产物以确认所期望的突变的存在,并且,以确保没有另外的突变已引入基因组中。
结果
我们描述一个hht2等位基因表达组蛋白H3突变蛋白从精氨酸作为靶向原位诱变策略的代表性例子窝藏在53位的取代为谷氨酸(H3-R53E突变体)的产生。
我们产生其中HHT2的完整ORF被URA3基因(见协议的步骤1)代替的菌株。该菌株,yAAD156,也藏着his3Δ200等位基因,这将导致细胞是营养缺陷型组氨酸。?...
讨论
序列同源性的高水平的两个非等位基因,对于每个在单倍体酿酒酵母细胞中的四个核心组蛋白的代码可以表示谁希望特异性靶向两个基因诱变之一调查一个挑战之间。先前描述的酵母诱变方法,包括Delitto普菲 ,位点特异性基因组(SSG)诱变和自由克隆基于PCR的等位基因替换方法5,6,7,以及更近的酵母基于CRISPR?...
披露声明
The authors declare that they have no competing financial interests.
致谢
We thank Reine Protacio for helpful comments during the preparation of this manuscript. We express our gratitude to the National Science Foundation (grants nos. 1243680 and 1613754) and the Hendrix College Odyssey Program for funding support.
材料
| Name | Company | Catalog Number | Comments |
| 1 kb DNA Ladder (DNA standards) | New England BioLabs | N3232L | |
| Agarose | Sigma | A5093-100G | |
| Boric Acid | Sigma | B0394-500G | |
| dNTP mix (10 mM each) | ThermoFisher Scientific | R0192 | |
| EDTA solution (0.5 M, pH 8.0) | AmericanBio | AB00502-01000 | |
| Ethanol (200 Proof) | Fisher Scientific | 16-100-824 | |
| Ethylenediaminetetraacetic acid disodium salt dihydrate (EDTA) | Sigma | E4884-500G | |
| Lithium acetate dihydrate | Sigma | L6883-250G | |
| MyCycler Thermal Cycler | BioRad | 170-9703 | |
| Poly(ethylene glycol) (PEG) | Sigma | P3640-1KG | |
| PrimeSTAR HS DNA Polymerase (high fidelity DNA polymerase) and 5x buffer | Fisher Scientific | 50-443-960 | |
| Salmon sperm DNA solution | ThermoFisher Scientific | 15632-011 | |
| Sigma 7-9 (Tris base, powder form) | Sigma | T1378-1KG | |
| Sodium acetate trihydrate | Sigma | 236500-500G | |
| Supra Sieve GPG Agarose (low metling temperature agarose) | AmericanBio | AB00985-00100 | |
| Taq Polymerase and 10x Buffer | New England BioLabs | M0273X | |
| Toothpicks | Fisher Scientific | S67859 | |
| Tris-HCl (1 M, pH 8.0) | AmericanBio | AB14043-01000 | |
| a-D(+)-Glucose | Fisher Scientific | AC170080025 | for yeast media |
| Agar | Fisher Scientific | DF0140-01-0 | for yeast media |
| Peptone | Fisher Scientific | DF0118-07-2 | for YPD medium |
| Yeast Extract | Fisher Scientific | DF0127-17-9 | for YPD medium |
| 4-aminobenzoic acid | Sigma | A9878-100G | for complete minimal dropout medium |
| Adenine | Sigma | A8626-100G | for complete minimal dropout medium |
| Glycine hydrochloride | Sigma | G2879-100G | for complete minimal dropout medium |
| L-Alanine | Sigma | A7627-100G | for complete minimal dropout medium |
| L-Arginine monohydrochloride | Sigma | A5131-100G | for complete minimal dropout medium |
| L-Asparagine monohydrate | Sigma | A8381-100G | for complete minimal dropout medium |
| L-Aspartic acid sodium salt monohydrate | Sigma | A6683-100G | for complete minimal dropout medium |
| L-Cysteine hydrochloride monohydrate | Sigma | C7880-100G | for complete minimal dropout medium |
| L-Glutamic acid hydrochloride | Sigma | G2128-100G | for complete minimal dropout medium |
| L-Glutamine | Sigma | G3126-100G | for complete minimal dropout medium |
| L-Histidine monohydrochloride monohydrate | Sigma | H8125-100G | for complete minimal dropout medium |
| L-Isoleucine | Sigma | I2752-100G | for complete minimal dropout medium |
| L-Leucine | Sigma | L8000-100G | for complete minimal dropout medium |
| L-Lysine monohydrochloride | Sigma | L5626-100G | for complete minimal dropout medium |
| L-Methionine | Sigma | M9625-100G | for complete minimal dropout medium |
| L-Phenylalanine | Sigma | P2126-100G | for complete minimal dropout medium |
| L-Proline | Sigma | P0380-100G | for complete minimal dropout medium |
| L-Serine | Sigma | S4500-100G | for complete minimal dropout medium |
| L-Threonine | Sigma | T8625-100G | for complete minimal dropout medium |
| L-Tryptophan | Sigma | T0254-100G | for complete minimal dropout medium |
| L-Tyrosine | Sigma | T3754-100G | for complete minimal dropout medium |
| L-Valine | Sigma | V0500-100G | for complete minimal dropout medium |
| myo-Inositol | Sigma | I5125-100G | for complete minimal dropout medium |
| Uracil | Sigma | U0750-100G | for complete minimal dropout medium |
| Ammonium Sulfate | Fisher Scientific | A702-500 | for complete minimal dropout medium |
| Yeast Nitrogen Base | Fisher Scientific | DF0919-07-3 | for complete minimal dropout medium |
| 5-Fluoroorotic acid (5-FOA) | AmericanBio | AB04067-00005 | for 5-FOA medium |
参考文献
- Luger, K., Mader, A. W., Richmond, R. K., Sargent, D. F., Richmond, T. J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 389 (6648), 251-260 (1997).
- Campos, E. I., Reinberg, D. Histones: annotating chromatin. Annu Rev Genet. 43, 559-599 (2009).
- Rando, O. J., Winston, F. Chromatin and transcription in yeast. Genetics. 190 (2), 351-387 (2012).
- Duina, A. A., Miller, M. E., Keeney, J. B. Budding yeast for budding geneticists: a primer on the Saccharomyces cerevisiae model system. Genetics. 197 (1), 33-48 (2014).
- Storici, F., Resnick, M. A. Delitto perfetto targeted mutagenesis in yeast with oligonucleotides. Genet Eng (N Y). 25, 189-207 (2003).
- Gray, M., Kupiec, M., Honigberg, S. M. Site-specific genomic (SSG) and random domain-localized (RDL) mutagenesis in yeast. BMC Biotechnol. 4, 7 (2004).
- Erdeniz, N., Mortensen, U. H., Rothstein, R. Cloning-free PCR-based allele replacement methods. Genome Res. 7 (12), 1174-1183 (1997).
- Brachmann, C. B., et al. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 14 (2), 115-132 (1998).
- Lundblad, V., Hartzog, G., Moqtaderi, Z. Manipulation of cloned yeast DNA. Curr Protoc Mol Biol. Chapter 13, (2001).
- Hoffman, C. S. Preparation of yeast DNA. Curr Protoc Mol Biol. Chapter 13, (2001).
- Treco, D. A., Lundblad, V. Preparation of yeast media. Curr Protoc Mol Biol. Chapter 13, (2001).
- Lederberg, J., Lederberg, E. M. Replica plating and indirect selection of bacterial mutants. J Bacteriol. 63 (3), 399-406 (1952).
- Sikorski, R. S., Hieter, P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 122 (1), 19-27 (1989).
- Johnson, P., et al. A systematic mutational analysis of a histone H3 residue in budding yeast provides insights into chromatin dynamics. G3 (Bethesda). 5 (5), 741-749 (2015).
- DiCarlo, J. E., et al. Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res. 41 (7), 4336-4343 (2013).
- Cross, S. L., Smith, M. M. Comparison of the structure and cell cycle expression of mRNAs encoded by two histone H3-H4 loci in Saccharomyces cerevisiae. Mol Cell Biol. 8 (2), 945-954 (1988).
- Libuda, D. E., Winston, F. Amplification of histone genes by circular chromosome formation in Saccharomyces cerevisiae. Nature. 443 (7114), 1003-1007 (2006).
转载和许可
请求许可使用此 JoVE 文章的文本或图形
请求许可探索更多文章
This article has been published
Video Coming Soon
版权所属 © 2025 MyJoVE 公司版权所有,本公司不涉及任何医疗业务和医疗服务。