
Se requiere una suscripción a JoVE para ver este contenido. Inicie sesión o comience su prueba gratuita.
Method Article
Targeted
En este artículo
Resumen
A strategy for generating mutations in histone genes at their endogenous location in Saccharomyces cerevisiae is presented.
Resumen
We describe a PCR- and homologous recombination-based system for generating targeted mutations in histone genes in budding yeast cells. The resulting mutant alleles reside at their endogenous genomic sites and no exogenous DNA sequences are left in the genome following the procedure. Since in haploid yeast cells each of the four core histone proteins is encoded by two non-allelic genes with highly homologous open reading frames (ORFs), targeting mutagenesis specifically to one of two genes encoding a particular histone protein can be problematic. The strategy we describe here bypasses this problem by utilizing sequences outside, rather than within, the ORF of the target genes for the homologous recombination step. Another feature of this system is that the regions of DNA driving the homologous recombination steps can be made to be very extensive, thus increasing the likelihood of successful integration events. These features make this strategy particularly well-suited for histone gene mutagenesis, but can also be adapted for mutagenesis of other genes in the yeast genome.
Introducción
Las cuatro proteínas histonas H2A, H2B, H3, H4 y juegan un papel central en la compactación, la organización y función de los cromosomas eucarióticos. Dos juegos de cada una de estas histonas forman el octámero de histona, un carrete molecular que dirige la envoltura de ~ 147 pares de bases de ADN en torno a sí mismo, en última instancia, resulta en la formación de un nucleosoma 1. Nucleosomas son participantes activos en una variedad de procesos basados en los cromosomas, tales como la regulación de la transcripción de genes y la formación de eucromatina y heterocromatina en todos los cromosomas, y como tales han sido el foco de intensa investigación en el transcurso de las últimas décadas. Varios mecanismos han sido descritos por el cual los nucleosomas se pueden manipular de manera que puedan facilitar la ejecución de los procesos específicos - Estos mecanismos incluyen la modificación postraduccional de residuos de histonas, nucleosoma remodelación dependiente de ATP, y la reorganización de nucleosomas independiente de ATPy montaje / desmontaje 2, 3.
La levadura de gemación Saccharomyces cerevisiae es un organismo modelo particularmente poderosa para la comprensión de la función de la histona en eucariotas. Esto puede atribuirse en gran parte al alto grado de conservación evolutiva de las proteínas histonas en todo el dominio Eukarya, y la flexibilidad de la levadura a una variedad de genética y bioquímica experimental se acerca 4. enfoques inversa genéticos en levadura se han usado ampliamente para estudiar los efectos de las mutaciones de histona específicas sobre diversos aspectos de la biología de la cromatina. Para estos tipos de experimentos a menudo es preferible usar células en las que las histonas mutantes se expresan a partir de sus loci genómico nativo, como la expresión de plásmidos autónomos puede conducir a niveles anormales intracelulares de proteínas histonas (debido a un número variable de plásmidos en las células) y alteración concomitante de la cromatina environments, que en última instancia pueden confundir la interpretación de los resultados.
A continuación, describimos una técnica basada en PCR que permite la mutagénesis dirigida de los genes de histonas en sus ubicaciones genómicas nativas que no requiere una etapa de clonación y los resultados en la generación de la mutación (s) deseado sin secuencias de ADN exógeno sobrantes en el genoma. Esta técnica se aprovecha del sistema de recombinación homóloga eficiente en la levadura y tiene varias características en común con otras técnicas similares desarrollados por otros grupos - sobre todo la Delitto Perfetto, mutagénesis genómica específica del sitio (SSG), y la clonación de libre alelo basado en la PCR métodos de reemplazo de 5, 6, 7. Sin embargo, la técnica descrita tiene un aspecto que hace que sea especialmente adecuado para la mutagénesis de genes de histonas. En células de levadura haploides, cada una de las cuatro histonas del núcleo está codificada por dos no unagenes llelic y altamente homólogas: por ejemplo, la histona H3 se codifican por los genes HHT1 y HHT2, y los marcos de lectura abierta (ORFs) de los dos genes son más del 90% idénticos en secuencia. Este alto grado de homología puede complicar los experimentos diseñados para dirigirse específicamente a uno de los dos genes de histonas que codifica para la mutagénesis. Considerando que los métodos antes mencionados a menudo requieren el uso de al menos algunas secuencias dentro de la ORF del gen diana para conducir la recombinación homóloga, la técnica que describimos aquí hace uso de secuencias que flanquean el ORF de los genes de histonas (que comparten mucho menos homología de secuencia) para la etapa de recombinación, aumentando así la probabilidad de que la orientación con éxito de mutagénesis para el locus deseado. Por otra parte, las regiones homólogas que dirigen la recombinación puede ser muy extensa, lo que contribuye a la eficiente recombinación homóloga dirigida.
Protocolo
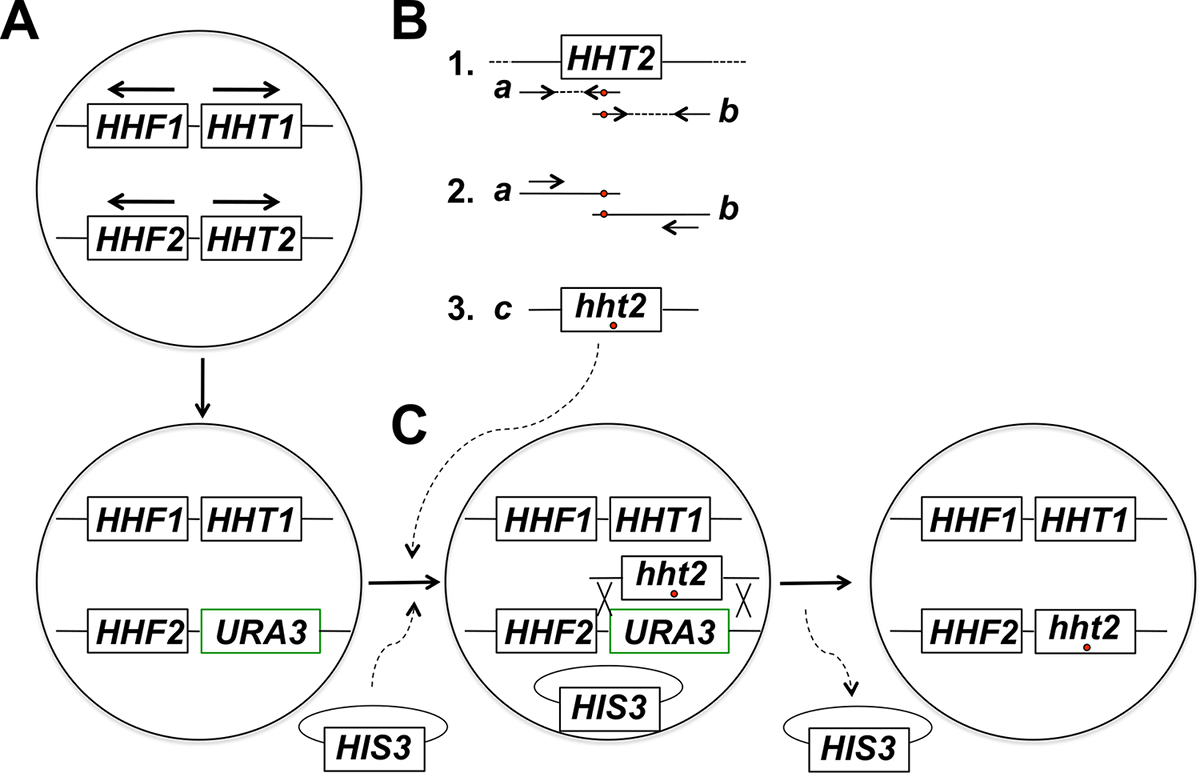
NOTA: La estrategia experimental para objetivo in situ mutagénesis de genes histona incluye varias etapas (que se resumen en la Figura 1). Estos pasos incluyen: (1) Sustitución del gen de la histona de destino con el gen URA3, (2) Generación y purificación de productos de PCR correspondientes a dos fragmentos que se superponen parcialmente del gen de la histona objetivo usando cebadores que albergan la mutación (s) deseado, (3 ) Fusión de PCR de los dos fragmentos que se superponen parcialmente para obtener productos de PCR de tamaño completo para la integración, (4) Co-transformación de los productos de PCR de tamaño completo y el plásmido columna vertebral, y la selección para el marcador en el plásmido, (5) de pantalla para el 5-FOA resistentes transformantes, (6) purificación de las colonias de 5-FOA-resistentes y la pérdida de plásmido columna vertebral, y (7) los análisis moleculares para el ensayo de integración apropiada del alelo mutante.

Figura 1: Visión general de la Estrategia para Targeted in situ Mutagénesis de genes histona levadura en ciernes. En este ejemplo el gen diana es HHT2, pero cualquier otro gen de la histona núcleo también puede ser mutagenizado usando esta estrategia. Células de levadura haploides (A) albergan dos genes de las histonas H3-codificación (HHT1 y HHT2) y dos genes H4 que codifica la histona (HHF1 y HHF2) dispuestos como se muestra en la figura (los genes HHT1 y HHF1 se encuentran en el cromosoma II y la HHT2 y genes HHF2 se encuentran en el cromosoma XIV - en cada caso, las flechas apuntan en la dirección de la transcripción). En la primera etapa del procedimiento, el ORF del gen HHT2 se sustituye con el gen URA3, dando lugar a una cepa hht2Δ :: URA3. (B) En la parte 1, una copia de tipo salvaje del gen HHT2 a partir de una muestra de ADN genómico se utilizó como molde para dos reacciones de PCR a géneroste La dos fragmentos que se superponen parcialmente del gen. El cebador inverso para la primera reacción incluye uno o más nucleótidos no coincidentes (indicadas con un círculo rojo) que se corresponden con la mutación deseada (s) para ser introducido en el genoma. El cebador directo para la segunda reacción tiene la falta de correspondencia equivalente en una configuración complementaria inversa (también se indica con un círculo rojo). Los dos productos de PCR generados en la parte 1 (productos A y B) se utilizan entonces como plantillas para la fusión PCR usando dos cebadores que hibridan con los productos A y B en la forma indicada en la parte 2. Esto se traduce en la generación de PCR de tamaño completo productos producto (c) en la parte 3 que albergan la mutación deseada (s). (C) El hht2Δ :: URA3 cepa es entonces co-transformaron con los productos de PCR de tamaño completo y un plásmido columna vertebral (a HIS3- marcado plásmido en este ejemplo), y las células se seleccionan para la presencia del plásmido (en medios que carecen de maridoistidine en este ejemplo). Los transformantes se seleccionaron a continuación para 5-FOA resistencia - células resistentes son candidatos para haber sufrido un evento de recombinación homóloga que conduce a la integración del producto de PCR y la escisión del gen URA3, como se muestra. pérdida posterior de la columna vertebral plásmido por división celular mitótico conduce a la histona cepa mutante deseada final. Hemos encontrado que la selección del plásmido columna vertebral, seguido de la detección de los resultados de resistencia a 5-FOA en una frecuencia mucho más alta de la identificación de los eventos de integración correctas en comparación con la selección directa en placas de 5-FOA, que en su mayoría identifica las células que han adquirido mutaciones URA3 espontáneas. (Esta figura se ha modificado de referencia 14). Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
1. Sustitución del objetivo de la histona génica con el gen URA3Gene
- Realizar mediada por PCR interrupción de genes de un solo paso estándar reemplazando el ORF del gen de la histona de destino con el gen URA3 8, 9.
NOTA: Se recomienda el uso de células de levadura que portan el ura3Δ0 como esta mutación elimina todo el URA3 endógeno ORF, evitando así la integración del producto PCR en el locus URA3 8. Alternativamente, el gen lactis URA3 K. puede ser utilizado eficazmente para la generación de la sustitución de la histona en cualquier fondo ura3 ya que es funcional en S. cerevisiae, pero sólo tiene una homología de secuencia parcial con el gen URA3 de S. cerevisiae. La cepa también debe ser auxotrófico para al menos un compuesto que permita la selección del plásmido columna vertebral en el experimento de transformación (véase la etapa 4 de este protocolo). Este paso no es necesario si un geneΔ histonas objetivo :: URA3cepa ya está disponible.
2. Generación y purificación de productos de PCR correspondientes a dos fragmentos que se superponen parcialmente del objetivo de la histona de genes usando cebadores portadoras de la mutación deseada (s)
- Generar productos de PCR correspondientes a dos fragmentos que se superponen parcialmente del gen de la histona de destino.
- Preparar dos reacciones de PCR como sigue:
- Para generar productos de PCR correspondientes a la primera mitad del gen (producto A en la Figura 1B), configurar la siguiente reacción: 1 l de ADN plantilla, cebador directo 5 μl10 M, 5 μl10 M cebador inverso, 0,5 l (1,25 U) termoestable ADN polimerasa, 10 l de tampón de ADN polimerasa 5x, 5 l mezcla dNTP (2 mM cada uno), y 23,5 l dH 2 O.
NOTA: La plantilla de ADN puede ser ADN genómico derivado de una cepa de tipo salvaje para el gen de la histona diana aislado utilizando Pro Estándar10 procedimientos. Para tener en cuenta las variaciones en la concentración de ADN y el nivel de impurezas en diferentes preparaciones genómicas, se recomienda para optimizar las reacciones mediante el uso de cualquiera de ADN sin diluir o diferentes diluciones de las preparaciones genómicas (por ejemplo, 1:10 y 1: 100). El cebador directo debe hibridar con una región aguas arriba del gen diana. El cebador inverso se hibrida dentro de la ORF, ser ~ 40 nucleótidos de longitud, y contiene la mutación deseada (s) en algún lugar en el medio de ella (véase la figura 1B-1 y la sección de resultados representativos para los ejemplos). El uso de una ADN polimerasa de alta fidelidad se recomienda con el fin de reducir las tasas de mutaciones no deseadas durante la síntesis de los productos de PCR. - Para generar productos de PCR correspondientes a la segunda mitad del gen (producto B en la Figura 1B), configure una reacción como se indica en 2.1.1.1, pero con diferentes cebadores.
NOTA: El PRI hacia adelantemer debe recocer dentro del ORF, ser ~ 40 nucleótidos de longitud, y contiene la mutación (s) deseado en algún lugar en el medio de ella. Tenga en cuenta que la mutación (s) en este cebador es el complemento inverso de la mutación (s) en el cebador inverso en la etapa 2.1.1.1. El cebador inverso debe hibridar con una región aguas abajo del gen diana (véase la Figura sección 1B-1 y resultados representativos para los ejemplos).
- Para generar productos de PCR correspondientes a la primera mitad del gen (producto A en la Figura 1B), configurar la siguiente reacción: 1 l de ADN plantilla, cebador directo 5 μl10 M, 5 μl10 M cebador inverso, 0,5 l (1,25 U) termoestable ADN polimerasa, 10 l de tampón de ADN polimerasa 5x, 5 l mezcla dNTP (2 mM cada uno), y 23,5 l dH 2 O.
- Colocar las reacciones en un termociclador con los siguientes ajustes: 94 ° C 30 seg; 30 ciclos de los siguientes valores: 98 ° C 10 seg, 60 ° C 5 seg, 72 ° C 1,5 min; y 72 ° C 10 min.
Nota: Optimización de los parámetros de PCR puede ser necesaria para los conjuntos de cebadores específicos y el objetivo de genes histona.
- Preparar dos reacciones de PCR como sigue:
- Ejecutar 20 - 50 l de la material de las reacciones de PCR en un gel de bajo punto de fusión de agarosa de 0,9% en la base de 89 mM Tris, 89 mM ácido bórico, EDTA mM de tampón (TBE) 2.5.
- secciones cortadas en gel de agarosa que contienen el pr PCRoductos de gel utilizando un bisturí o cuchilla de afeitar limpia y transfieren cada una a un tubo de microcentrífuga de 1,5 ml. Almacenar secciones de agarosa que contienen productos de PCR a -20 ° C hasta que esté listo para su uso.
3. La fusión por PCR de los dos fragmentos que se superponen parcialmente por conseguir el pleno tamaño de los productos de PCR para la Integración
- Preparar plantilla para reacciones de PCR
- Derretir secciones gel de agarosa del paso 2.3 mediante la colocación de los tubos de microcentrífuga en un conjunto de bloque de calor a 65 ° C durante 5 minutos (o hasta que esté completamente derretida). tubos Vortex cada 1 - 2 min para facilitar el proceso de fusión.
- Transferir una cantidad fija de agarosa derretida de cada muestra (por ejemplo, 50 l cada uno, para un total de 100 l) en un solo tubo de microcentrífuga y mezclar mediante agitación. Use esto como la plantilla en las reacciones de PCR de fusión. Se coloca el tubo a -20 ° C hasta que esté listo para su uso.
- Amplificar una gran cantidad de tamaño completo producto de PCR (producto cen la Figura 1B)
- Configurar seis reacciones de PCR, cada uno con los siguientes componentes: 2 l de ADN plantilla, cebador directo 10 l 10 mM, 10 l 10 mM cebador inverso, 1 l (2,5 U) de ADN polimerasa termoestable, 20 l de 5x tampón de ADN polimerasa, 10 l mezcla de dNTP (2 mM cada uno), y 47 l de dH2O
NOTA: El número de reacciones puede ser alterado en función de la eficiencia de la PCR. La plantilla de ADN (véase 3.1.2) debe ser calentada a 65 ° C hasta que se derrita, mezclado por agitación, y se añade la última a la mezcla de reacción de PCR. Una vez añadido, Mezclar suavemente con la pipeta la solución arriba y abajo varias veces. Para tener en cuenta las variaciones en la concentración de ADN en las diferentes muestras, se recomienda para optimizar primero las reacciones mediante el uso de cualquiera de plantilla sin diluir o diferentes diluciones de la plantilla (por ejemplo, 1:10 y 1: 100). Los dos cebadores utilizados deben hibridar con los dos fragmentos que se superponen parcialmente del gen diana como enfermosustrated en la Figura 1B-2 y ser diseñado de tal manera que los productos de PCR finales tendrán al menos 40 pares de bases a cada lado homóloga a las regiones que flanquean el ORF URA3 que impulsarán la etapa de recombinación homóloga (véase la sección resultados representativos para los ejemplos). El uso de una ADN polimerasa de alta fidelidad se recomienda con el fin de reducir las tasas de mutaciones no deseadas durante la síntesis de los productos de PCR. - Colocar los tubos en un termociclador con los siguientes ajustes: 94 ° C 30 seg; 30 ciclos de los siguientes valores: 98 ° C 10 seg, 50ºC 15 s, 72 ° C 1,5 min; y 72 ° C 10 min.
NOTA: Optimización de los parámetros de la PCR puede ser necesaria para conjuntos de cebadores específicos y gen de la histona de destino.
- Configurar seis reacciones de PCR, cada uno con los siguientes componentes: 2 l de ADN plantilla, cebador directo 10 l 10 mM, 10 l 10 mM cebador inverso, 1 l (2,5 U) de ADN polimerasa termoestable, 20 l de 5x tampón de ADN polimerasa, 10 l mezcla de dNTP (2 mM cada uno), y 47 l de dH2O
4. Co-transformación de los productos de PCR de tamaño completo y esqueleto del plásmido, y selección para el marcador en el plásmido
- La concentración de los productos de PCR
- Se combinan las six reacciones de PCR (600 en total l) de la etapa 3.2.2 en un solo tubo de microcentrífuga y mezclar mediante agitación.
- Dividir la muestra en tres 200 ml de alícuotas en tubos de microcentrífuga. Precipitar el ADN en cada tubo mediante la adición de 20 l de acetato de sodio 3 M (pH 5,2) y 550 l de etanol al 100%. Mezcle bien la solución y colocar en hielo durante al menos 15 minutos. Recoger el ADN por centrifugación a 14.000 ~ g durante 10 min, enjuagar el sedimento con 200 l de etanol al 70%, y secar al aire.
- Resuspender cada sedimento de ADN en 25 l de dH2O, y aunar en un solo tubo (para un total de 75 l).
- La levadura co-transformación
- Preparar 10 ml de cultivo de una noche de la cepa generada en la sección 1 de extracto de levadura peptona dextrosa (YPD) medio líquido 11.
- A la mañana siguiente, inocular 400 ml de medio líquido YPD con 8 ml de cultivo de una noche saturado e incubar agitandoa 30 ° C durante 4 - 5 h para permitir que las células entran en fase logarítmica de crecimiento.
- Recoger las células por centrifugación a ~ 3.220 xg durante 10 min, descartar el medio líquido, y resuspender las células en 1 volumen de 10 mM Tris-HCl (pH 8,0), EDTA 1 mM, solución 0,1 M de litio acetato (TE / LiAc) .
- Recoger las células por centrifugación a ~ 3.220 xg durante 10 minutos, y desechar el TE / AcLi.
- Resuspender las células en 1 ml de TE / LiAc.
- Configurar el siguiente cóctel de reacción en un tubo de microcentrífuga: 800 l de células de la etapa 4.2.5, 40 l de ADN de esperma / ml de salmón 10 mg hervida, un total de 12,5 g de ADN plásmido columna vertebral, y 75 l de producto de PCR concentrado de la etapa 4.1.3.
NOTA: DNA de esperma de salmón se debe hervir durante 5 min y se coloca en hielo durante al menos 5 min antes de su uso en la reacción. Volumen total de ADN de plásmido columna vertebral añadido debe mantenerse a un mínimo (~ 80 l o menos). Véase la sección Resultados Representante para un ejemplo de un plasma de columna vertebralcarné de identidad. - Mezclar el tubo de cóctel de fondo y alícuota uniforme en ocho tubos de microcentrífuga (tubos 1 - 8).
- Configurar las siguientes dos tubos de reacción de transformación de control:
- Tubo 9 (sin control del producto PCR): 100 l de células de la etapa 4.2.5, 5 l de ADN / ml de esperma de salmón 10 mg hervida (hervido durante 5 min; véase la etapa 4.2.6 Nota), un total de 1,56 g de esqueleto del plásmido ADN y ningún producto de PCR añadido.
- El tubo 10 (sin control de ADN): 100 l de células de la etapa 4.2.5, 5 l de ADN hervida 10 mg / ml de esperma de salmón (ver paso 4.2.6 Nota), no plásmido columna vertebral del ADN añadido, y ningún producto PCR añaden.
- Mezclar ambos tubos suavemente pero a fondo pipeteando arriba y abajo varias veces.
- Incubar los diez tubos a 30 ° C durante 30 min.
- A cada tubo, añadir 1,2 ml de polietilenglicol 40% (PEG 3350) en TE / LiAc. Mezclar a fondo usando una pipeta P-1000 hasta que la solución es homogénea.
- Incubar los tubos a 30 diez° C durante 30 min. Mezclar suavemente la solución de la pipeta hacia arriba y hacia abajo y luego incubar los tubos a 42 ° C durante 15 minutos.
- Recoger las células por girar los tubos en una microcentrífuga a ~ 14.000 xg durante 30 seg. Desechar el líquido y resuspender las células en 1 ml de dH 2 O. estéril
- Recoger las células por girar los tubos en una microcentrífuga a ~ 14.000 xg durante 30 seg. Desechar el líquido y resuspender las células en 500 l de dH 2 O. estéril
- piscina de los tubos 1 - 8 juntos (volumen total de 4 ml) y mezclar bien con la pipeta hacia arriba y abajo.
- Placa 200 l de la mezcla anterior en cada uno de veinte placas completas medio mínimo de abandono 11 (placas 1-20) para la selección del plásmido columna vertebral.
- Placa de 200 l de la mezcla de tubo 9 y 200 l de mezcla de tubo de 10 cada uno en su propia placa de selección (láminas 21 y 22, respectivamente).
- Se incuban las placas 22 a 30 ° C durante 3 - 5 días aseleccionar los transformantes de plásmidos.
- Inspeccionar placas de transformación después de 3 - 5 días de incubación. Aproximadamente 5.000 colonias deben ser visibles en las placas de 1-21 (ver resultados representativos para un ejemplo) y no hay colonias deben estar presentes en la placa 22.
5. Pantalla de transformantes 5-FOA-resistentes
- La transferencia de células a partir de placas de 1 - 20 (y la placa de transformación 21 como control) a 5-fluoroorótico ácido (5-FOA) placas de 11 por réplica-plating 12 con el fin de la detección de la pérdida del gen URA3 como un resultado de la integración de la productos de la PCR en el lugar deseado.
- Retire la tapa del plato y presione la placa que contiene las colonias en un terciopelo estéril. La transferencia de las células del terciopelo a una placa 5-FOA presionando la placa sobre el terciopelo. Incubar las placas a 30ºC durante 2 días.
- Después de la incubación de 2 días, es importante verificar las placas 5-FOA para growth.
NOTA: Un evento de integración candidato estará representado por una pequeña asimétrica colonia "aplastado" en una placa de 5-FOA - a la inversa, papilas pequeñas que crecen en placas de 5-FOA es probable representante de mutaciones URA3 espontáneas que surgieron durante el crecimiento de las colonias en el placas de transformación, y por lo tanto poco probable que represente el evento de integración deseada (véase la Figura 3 en la sección de resultados representativos para la elaboración adicional sobre este punto y para algunos ejemplos).
6. Purificación de Colonias 5-FOA-resistentes y pérdida de la estructura del plásmido
- El uso de palillos de dientes estériles, recoger las colonias candidatos de las placas 5-FOA descritos en el paso 5.2 y la racha de colonias individuales en placas de YPD. Incubar durante 2 - 3 días a 30 ° C.
- Después de la incubación, réplica - placa de cada placa de purificación de YPD a una placa YPD fresco, una placa de deserción que carece de uracilo para comprobar si hay pérdidasdel gen URA3, y una segunda placa de abandono para controlar la presencia o ausencia del plásmido de cadena principal. Incubar durante 1 - 2 días a 30 ° C.
- Después de la incubación, identificar una colonia de cada muestra candidato que está creciendo en la placa de YPD, pero no crece en cualquiera de las placas de deserción (se espera una colonia como haber perdido el gen URA3 a través del proceso de recombinación y perdió el plásmido columna vertebral durante la mitosis división celular). Restreak tales colonias en placas de YPD frescas. Estas colonias son los candidatos de integración y se analizarán nuevamente en el paso 7.
7. Los análisis moleculares para ensayar la integración adecuada del alelo mutante
- Aislar el ADN genómico de las muestras candidatos utilizando procedimientos estándar 10.
- Amplificar región genómica que abarca el sitio de destino.
- Configure la siguiente reacción de PCR para cada muestra: 0,5 l de ADN plantilla, 5 l10 M cebador directo, 5 l 10 mM de cebador inverso, 0,5 l (2,5 unidades) de ADN polimerasa Taq, 5 l de 10x tampón de ADN polimerasa Taq, 5 l mezcla dNTP (2 mM cada uno), y 29 l dH 2 O.
NOTA: El ADN molde es el DNA genómico derivado de las muestras de candidatos. Se recomienda incluir también dos reacciones de control: uno utilizando ADN genómico derivado de las histonas geneΔ originales :: cepa URA3 como plantilla y otro utilizando DNA genómico de una cepa de la histona de tipo salvaje como molde. Para tener en cuenta las variaciones en la concentración de ADN y el nivel de impurezas en diferentes preparaciones genómicas, se recomienda para optimizar las reacciones mediante el uso de cualquiera de ADN sin diluir o diferentes diluciones de las preparaciones genómicas (por ejemplo, 1:10 y 1: 100). Es importante asegurarse de que estos cebadores hibridan con secuencias de ADN fuera de la región abarcada por el producto de PCR supuestamente integrada - de esta manera, el tamaño de los productos de PCR en estos reactions se pueden utilizar como una herramienta de diagnóstico para la integración de los productos en la localización genómica correcta (ver resultados representativos para un ejemplo). - Coloque las reacciones en un termociclador con los siguientes valores: 94 ° C 3 min; 30 ciclos de los siguientes valores: 94 ° C 45 s, 50ºC 45 s, 72ºC 2 min; y 72 ° C 10 min.
NOTA: Optimización de los parámetros de la PCR puede ser necesaria para conjuntos de cebadores específicos y gen de la histona de destino.
- Configure la siguiente reacción de PCR para cada muestra: 0,5 l de ADN plantilla, 5 l10 M cebador directo, 5 l 10 mM de cebador inverso, 0,5 l (2,5 unidades) de ADN polimerasa Taq, 5 l de 10x tampón de ADN polimerasa Taq, 5 l mezcla dNTP (2 mM cada uno), y 29 l dH 2 O.
- Transformación de los productos de PCR
- Ejecutar 20 l de cada reacción en un gel de agarosa TBE 0,8%.
- Evaluar el tamaño de los productos de PCR usando patrones de ADN como una referencia para determinar si el gen URA3 se ha sustituido con éxito por el gen de la histona putativamente mutado (ver resultados representativos para un ejemplo).
NOTA: En ciertos casos, la mutación (s) deseada introducida en los genes de las histonas o bien crear o destruir una restricción sentarsemi. Si este es el caso, la presencia de la mutación deseada en los productos de PCR del tamaño indicativo de la integración correcta puede ser evaluada sometiendo los productos a la digestión con la enzima de restricción correspondiente seguido por análisis de electroforesis en gel (ver resultados representativos para un ejemplo) . - Asunto productos de PCR del tamaño indicativo de la integración correcta de la secuenciación del ADN para confirmar la presencia de la mutación (s) deseada y para asegurar que no hay mutaciones adicionales se han introducido en el genoma.
Resultados
Se describe la generación de un alelo HHT2 que expresa una proteína mutante de la histona H3 que alberga una sustitución en la posición 53 de una arginina a un ácido glutámico (H3-R53E mutante) como un ejemplo representativo del objetivo de la estrategia de mutagénesis situ.
Hemos generado una cepa en la que todo el ORF de HHT2 se sustituye por el gen URA3 (véase la...
Discusión
El alto grado de homología de secuencia entre los dos genes no alélicos que codifican para cada una de las cuatro proteínas histonas en células haploides de S. cerevisiae puede representar un reto para los investigadores que desean dirigirse específicamente a uno de los dos genes para la mutagénesis. Anteriormente se ha descrito metodologías de mutagénesis de levadura, incluyendo la Delitto Perfetto, mutagénesis genómica específica del sitio (SSG), y métodos de reemplazo de alelo basados en...
Divulgaciones
The authors declare that they have no competing financial interests.
Agradecimientos
We thank Reine Protacio for helpful comments during the preparation of this manuscript. We express our gratitude to the National Science Foundation (grants nos. 1243680 and 1613754) and the Hendrix College Odyssey Program for funding support.
Materiales
| Name | Company | Catalog Number | Comments |
| 1 kb DNA Ladder (DNA standards) | New England BioLabs | N3232L | |
| Agarose | Sigma | A5093-100G | |
| Boric Acid | Sigma | B0394-500G | |
| dNTP mix (10 mM each) | ThermoFisher Scientific | R0192 | |
| EDTA solution (0.5 M, pH 8.0) | AmericanBio | AB00502-01000 | |
| Ethanol (200 Proof) | Fisher Scientific | 16-100-824 | |
| Ethylenediaminetetraacetic acid disodium salt dihydrate (EDTA) | Sigma | E4884-500G | |
| Lithium acetate dihydrate | Sigma | L6883-250G | |
| MyCycler Thermal Cycler | BioRad | 170-9703 | |
| Poly(ethylene glycol) (PEG) | Sigma | P3640-1KG | |
| PrimeSTAR HS DNA Polymerase (high fidelity DNA polymerase) and 5x buffer | Fisher Scientific | 50-443-960 | |
| Salmon sperm DNA solution | ThermoFisher Scientific | 15632-011 | |
| Sigma 7-9 (Tris base, powder form) | Sigma | T1378-1KG | |
| Sodium acetate trihydrate | Sigma | 236500-500G | |
| Supra Sieve GPG Agarose (low metling temperature agarose) | AmericanBio | AB00985-00100 | |
| Taq Polymerase and 10x Buffer | New England BioLabs | M0273X | |
| Toothpicks | Fisher Scientific | S67859 | |
| Tris-HCl (1 M, pH 8.0) | AmericanBio | AB14043-01000 | |
| a-D(+)-Glucose | Fisher Scientific | AC170080025 | for yeast media |
| Agar | Fisher Scientific | DF0140-01-0 | for yeast media |
| Peptone | Fisher Scientific | DF0118-07-2 | for YPD medium |
| Yeast Extract | Fisher Scientific | DF0127-17-9 | for YPD medium |
| 4-aminobenzoic acid | Sigma | A9878-100G | for complete minimal dropout medium |
| Adenine | Sigma | A8626-100G | for complete minimal dropout medium |
| Glycine hydrochloride | Sigma | G2879-100G | for complete minimal dropout medium |
| L-Alanine | Sigma | A7627-100G | for complete minimal dropout medium |
| L-Arginine monohydrochloride | Sigma | A5131-100G | for complete minimal dropout medium |
| L-Asparagine monohydrate | Sigma | A8381-100G | for complete minimal dropout medium |
| L-Aspartic acid sodium salt monohydrate | Sigma | A6683-100G | for complete minimal dropout medium |
| L-Cysteine hydrochloride monohydrate | Sigma | C7880-100G | for complete minimal dropout medium |
| L-Glutamic acid hydrochloride | Sigma | G2128-100G | for complete minimal dropout medium |
| L-Glutamine | Sigma | G3126-100G | for complete minimal dropout medium |
| L-Histidine monohydrochloride monohydrate | Sigma | H8125-100G | for complete minimal dropout medium |
| L-Isoleucine | Sigma | I2752-100G | for complete minimal dropout medium |
| L-Leucine | Sigma | L8000-100G | for complete minimal dropout medium |
| L-Lysine monohydrochloride | Sigma | L5626-100G | for complete minimal dropout medium |
| L-Methionine | Sigma | M9625-100G | for complete minimal dropout medium |
| L-Phenylalanine | Sigma | P2126-100G | for complete minimal dropout medium |
| L-Proline | Sigma | P0380-100G | for complete minimal dropout medium |
| L-Serine | Sigma | S4500-100G | for complete minimal dropout medium |
| L-Threonine | Sigma | T8625-100G | for complete minimal dropout medium |
| L-Tryptophan | Sigma | T0254-100G | for complete minimal dropout medium |
| L-Tyrosine | Sigma | T3754-100G | for complete minimal dropout medium |
| L-Valine | Sigma | V0500-100G | for complete minimal dropout medium |
| myo-Inositol | Sigma | I5125-100G | for complete minimal dropout medium |
| Uracil | Sigma | U0750-100G | for complete minimal dropout medium |
| Ammonium Sulfate | Fisher Scientific | A702-500 | for complete minimal dropout medium |
| Yeast Nitrogen Base | Fisher Scientific | DF0919-07-3 | for complete minimal dropout medium |
| 5-Fluoroorotic acid (5-FOA) | AmericanBio | AB04067-00005 | for 5-FOA medium |
Referencias
- Luger, K., Mader, A. W., Richmond, R. K., Sargent, D. F., Richmond, T. J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 389 (6648), 251-260 (1997).
- Campos, E. I., Reinberg, D. Histones: annotating chromatin. Annu Rev Genet. 43, 559-599 (2009).
- Rando, O. J., Winston, F. Chromatin and transcription in yeast. Genetics. 190 (2), 351-387 (2012).
- Duina, A. A., Miller, M. E., Keeney, J. B. Budding yeast for budding geneticists: a primer on the Saccharomyces cerevisiae model system. Genetics. 197 (1), 33-48 (2014).
- Storici, F., Resnick, M. A. Delitto perfetto targeted mutagenesis in yeast with oligonucleotides. Genet Eng (N Y). 25, 189-207 (2003).
- Gray, M., Kupiec, M., Honigberg, S. M. Site-specific genomic (SSG) and random domain-localized (RDL) mutagenesis in yeast. BMC Biotechnol. 4, 7 (2004).
- Erdeniz, N., Mortensen, U. H., Rothstein, R. Cloning-free PCR-based allele replacement methods. Genome Res. 7 (12), 1174-1183 (1997).
- Brachmann, C. B., et al. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 14 (2), 115-132 (1998).
- Lundblad, V., Hartzog, G., Moqtaderi, Z. Manipulation of cloned yeast DNA. Curr Protoc Mol Biol. Chapter 13, (2001).
- Hoffman, C. S. Preparation of yeast DNA. Curr Protoc Mol Biol. Chapter 13, (2001).
- Treco, D. A., Lundblad, V. Preparation of yeast media. Curr Protoc Mol Biol. Chapter 13, (2001).
- Lederberg, J., Lederberg, E. M. Replica plating and indirect selection of bacterial mutants. J Bacteriol. 63 (3), 399-406 (1952).
- Sikorski, R. S., Hieter, P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 122 (1), 19-27 (1989).
- Johnson, P., et al. A systematic mutational analysis of a histone H3 residue in budding yeast provides insights into chromatin dynamics. G3 (Bethesda). 5 (5), 741-749 (2015).
- DiCarlo, J. E., et al. Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res. 41 (7), 4336-4343 (2013).
- Cross, S. L., Smith, M. M. Comparison of the structure and cell cycle expression of mRNAs encoded by two histone H3-H4 loci in Saccharomyces cerevisiae. Mol Cell Biol. 8 (2), 945-954 (1988).
- Libuda, D. E., Winston, F. Amplification of histone genes by circular chromosome formation in Saccharomyces cerevisiae. Nature. 443 (7114), 1003-1007 (2006).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados